


Combined High—Throughput Proteomics and Random Forest Machine-Learning Approach Differentiates and Classifies Metabolic, Immune, Signaling and ECM Intra-Tumor Heterogeneity of Colorectal Cancer
 , , ,
, , ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
- Reagents and Instruments
- Study subjects
- Sample Preparation
- HPLC–HR–MS/MS analysis
- Statistical analysis
- Pathway/process enrichment analysis
- Protein–Protein interaction network and topological analysis
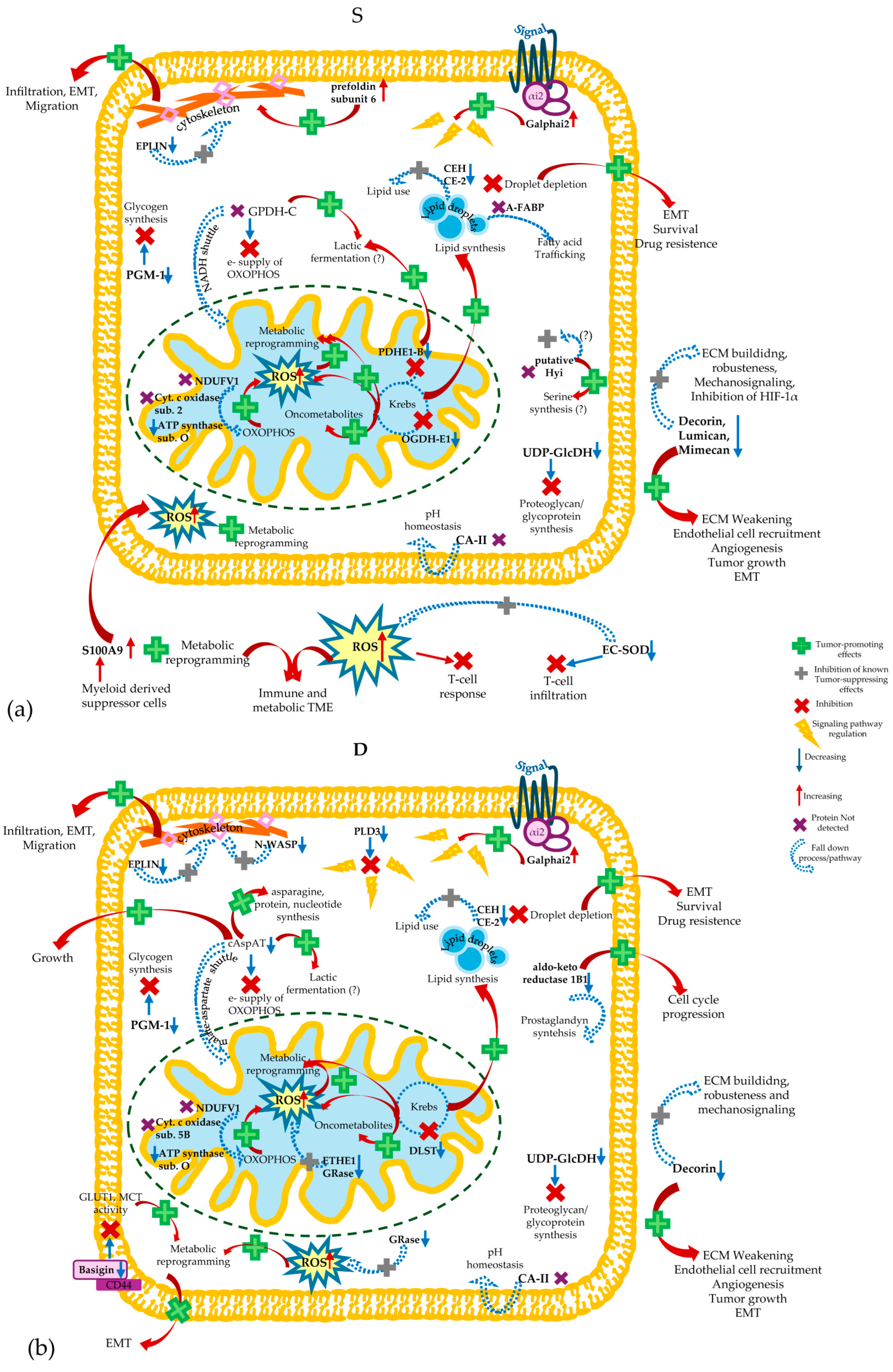
3. Results
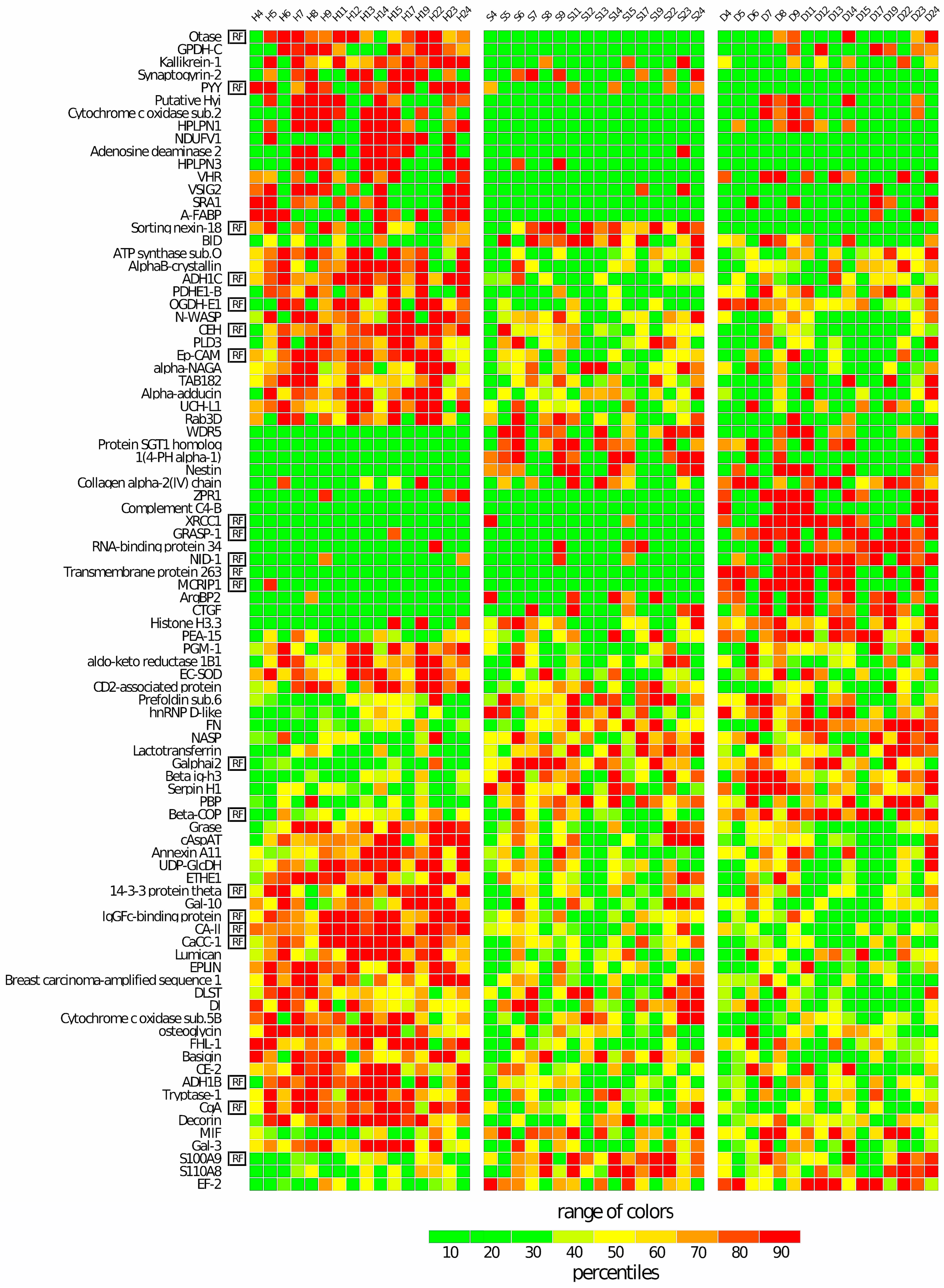
3.1. Statistical Analysis of LFQ Abundances Measured in S, D, and H Samples
Differential Statistical Analysis
- 30 in the S vs. H comparison (MW test), with 19 less-abundant and 11 more-abundant proteins in the S tumor region;
- 31 in the D vs. H comparison (MW test), with 16 less-abundant and 15 more-abundant proteins in the D tumor region.
- 20 in both S vs. H and D vs. H comparisons (MW test), with 18 less-abundant and 2 more-abundant proteins in both tumor regions;
- 2 in both S vs. D and D vs. H comparisons (MW test).
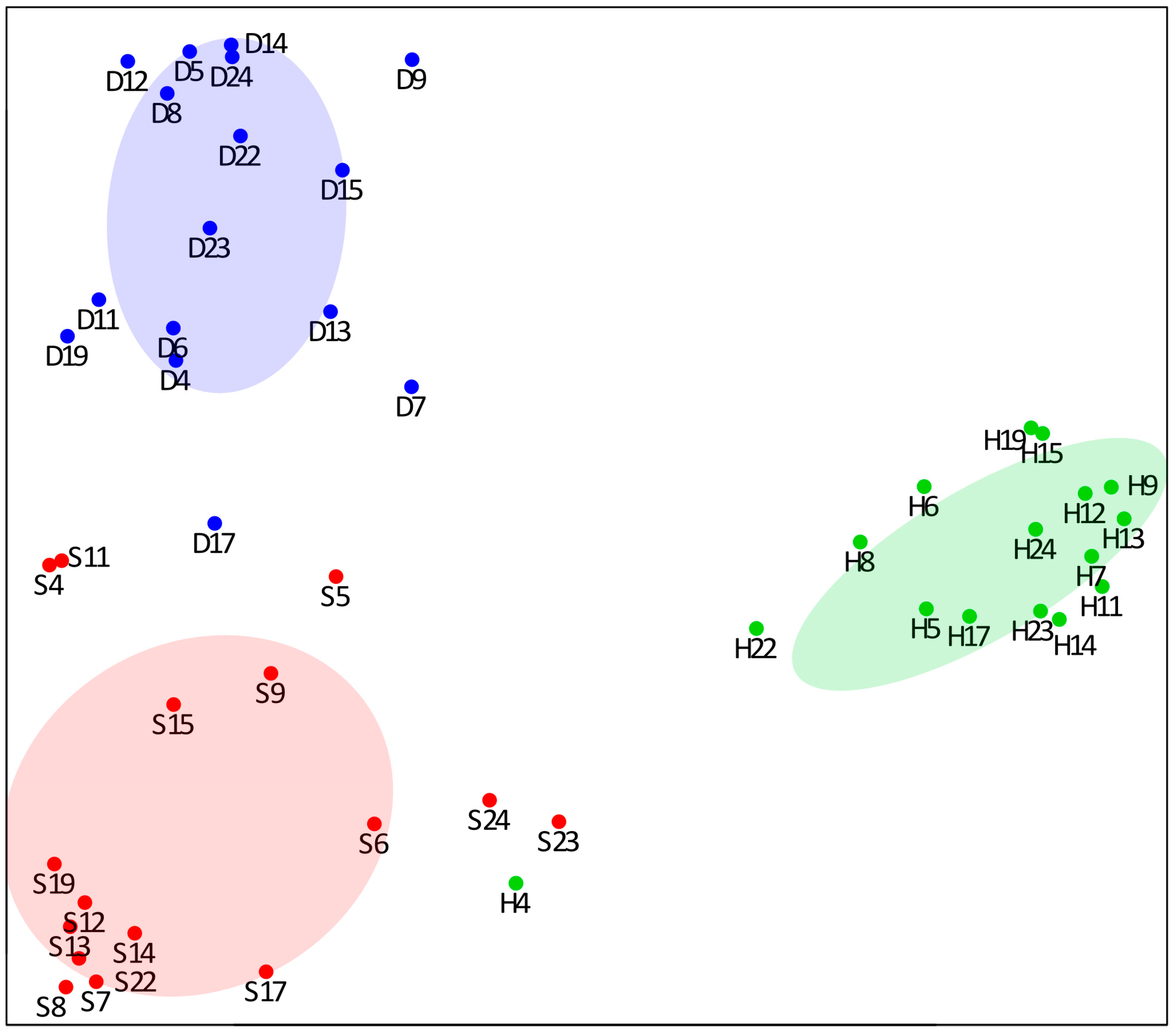
3.2. RF Classification Analysis
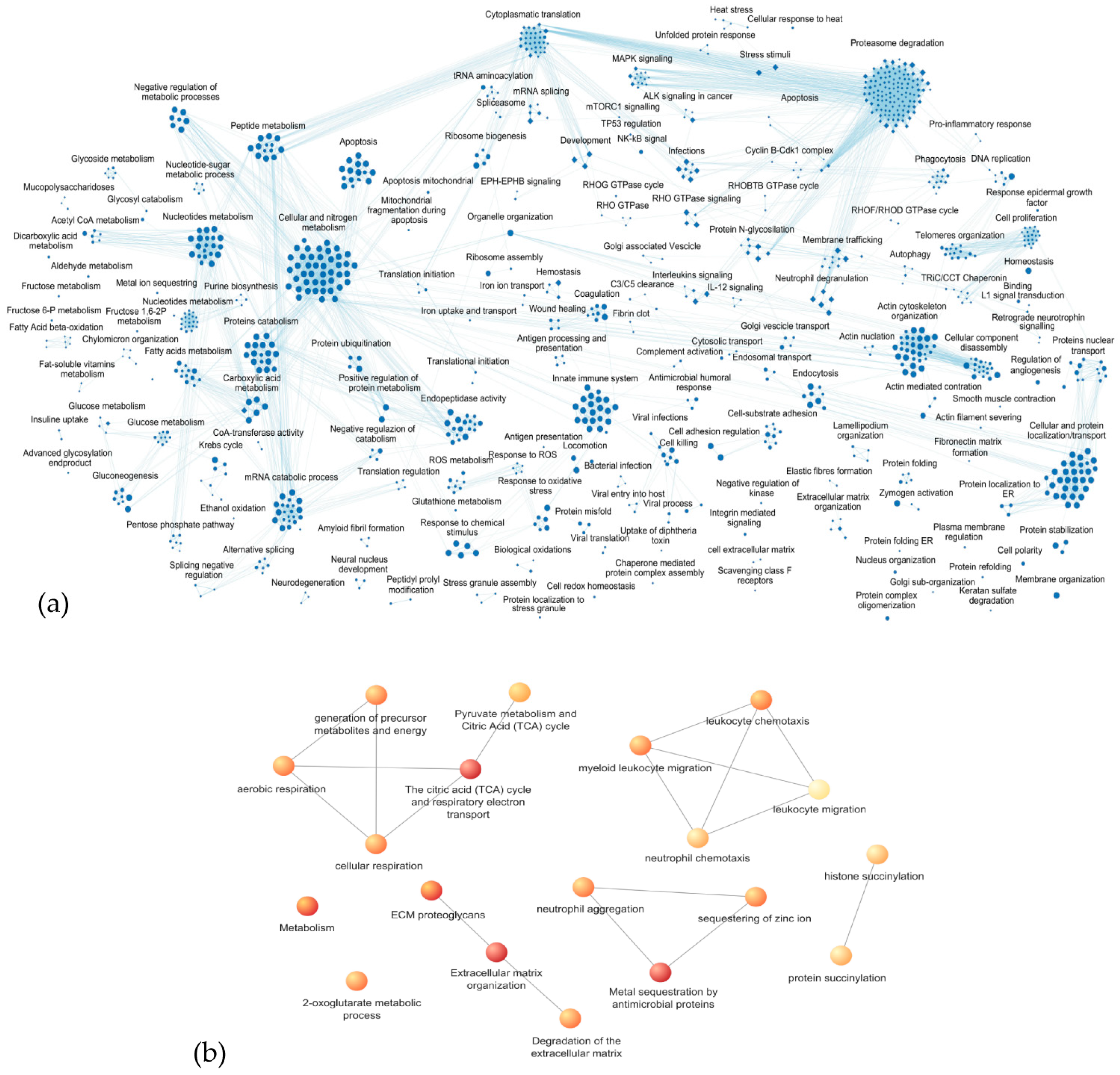
3.3. Enrichment Analysis of Biological Processes and Pathways
3.3.1. Proteoglycans and Extracellular Matrix Dynamics
3.3.2. Metabolic Processes
3.3.3. Leukocyte Migration and Aggregation
3.4. PPI Network and Topological Analysis
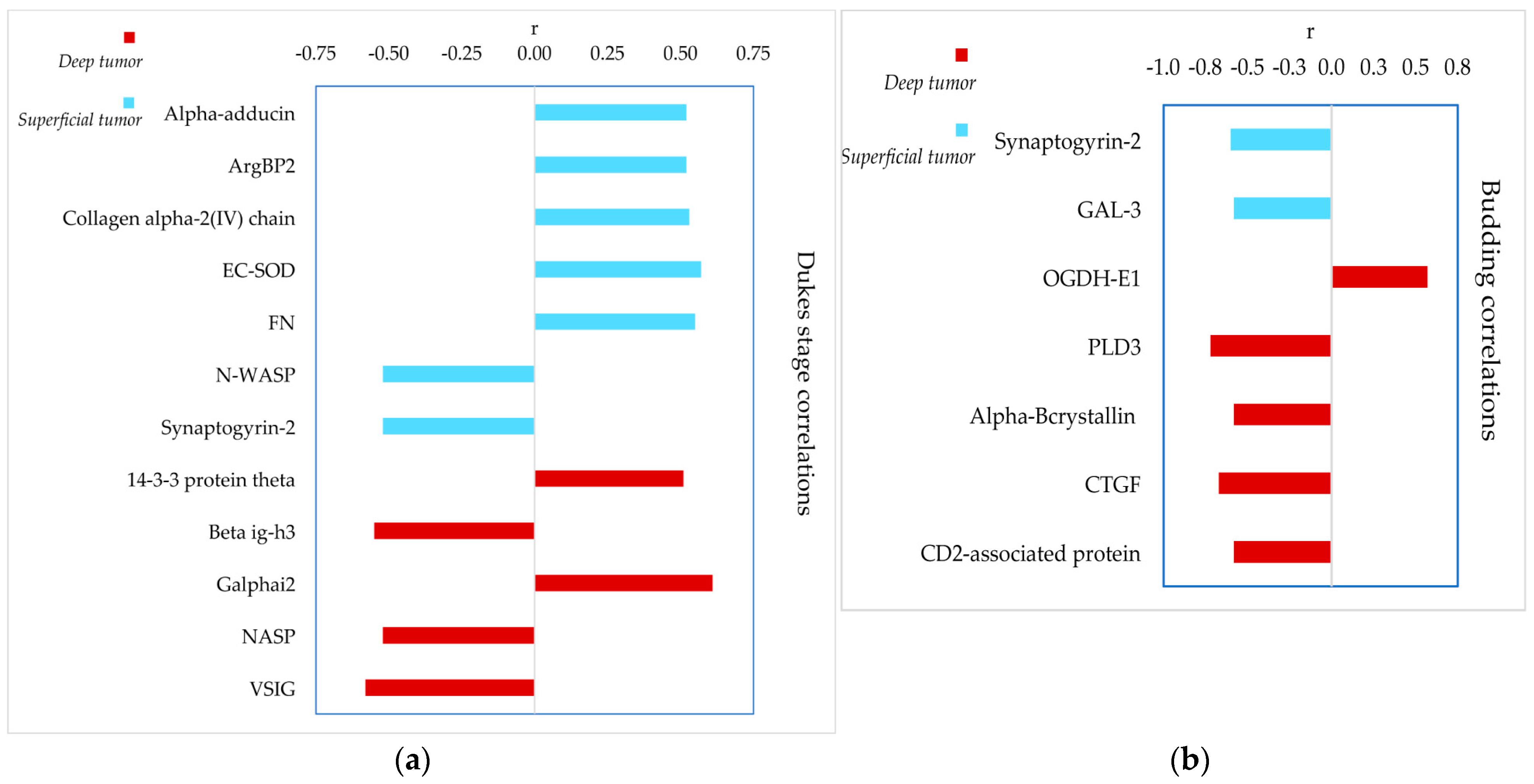
3.5. Correlation between Proteomic and Clinical Data
4. Discussion
4.1. Quantitative Differences in Protein Profiles in Tumor and Healthy Tissues and the Implication of Biological Processes/Pathways
4.1.1. Proteins Implicated in Processes Related to Proteoglycans and ECM
4.1.2. Proteins Implicated in Leukocyte Migration and Aggregation
4.1.3. Proteins Implicated in Metabolic Processes
4.1.4. Other Interesting Proteins Potentially Hallmarks of CRC
Hallmarks Implicated in Nucleic Acid Processing
Hallmarks Implicated in Protein Trafficking
Hallmarks Implicated in Regulation of Intra-/Extracellular Signaling Paths
4.2. Identification of Highly Sensitive and Specific Classifying Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- WHO. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 20 March 2024).
- Lim, L.C.; Lim, Y.M. Proteome Heterogeneity in Colorectal Cancer. Proteomics 2018, 18, 1700169. [Google Scholar] [CrossRef] [PubMed]
- Árnadóttir, S.S.; Mattesen, T.B.; Vang, S.; Madsen, M.R.; Madsen, A.H.; Birkbak, N.J.; Bramsen, J.B.; Andersen, C.L. Transcriptomic and Proteomic Intra-Tumor Heterogeneity of Colorectal Cancer Varies Depending on Tumor Location within the Colorectum. PLoS ONE 2020, 15, e0241148. [Google Scholar] [CrossRef] [PubMed]
- Nenkov, M.; Ma, Y.; Gaßler, N.; Chen, Y. Metabolic Reprogramming of Colorectal Cancer Cells and the Microenvironment: Implication for Therapy. Int. J. Mol. Sci. 2021, 22, 6262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zou, S.; Fang, L. Metabolic Reprogramming in Colorectal Cancer: Regulatory Networks and Therapy. Cell Biosci. 2023, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Olianas, A.; Serrao, S.; Piras, V.; Manconi, B.; Contini, C.; Iavarone, F.; Pichiri, G.; Coni, P.; Zorcolo, L.; Orrù, G.; et al. Thymosin Β4 and Β10 Are Highly Expressed at the Deep Infiltrative Margins of Colorectal Cancer—A Mass Spectrometry Analysis. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 7285–7296. [Google Scholar] [CrossRef] [PubMed]
- Nemolato, S.; Restivo, A.; Cabras, T.; Coni, P.; Zorcolo, L.; Orrù, G.; Fanari, M.; Cau, F.; Gerosa, C.; Fanni, D.; et al. Thymosin β-4 in colorectal cancer is localized predominantly at the invasion front in tumor cells undergoing epithelial mesenchymal transition. Cancer Biol. Ther. 2012, 13, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Piao, Z.; Hong, C.S.; Jung, M.R.; Choi, C.; Park, Y.K. Thymosin β4 induces invasion and migration of human colorectal cancer cells through the ILK/AKT/β-catenin signaling pathway. Biochem. Biophys. Res. Commun. 2014, 452, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, A.T.; Stepanov, A.A.; Malsagova, K.A.; Soni, D.; Kushlinsky, N.E.; Enikeev, D.V.; Potoldykova, N.V.; Lisitsa, A.V.; Kaysheva, A.L. Revelation of Proteomic Indicators for Colorectal Cancer in Initial Stages of Development. Molecules 2020, 25, 619. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Chaver, P. Proteomics for Discovery of Candidate Colorectal Cancer Biomarkers. World J. Gastroenterol. 2014, 20, 3804. [Google Scholar] [CrossRef]
- Martínez-Aguilar, J.; Chik, J.; Nicholson, J.; Semaan, C.; McKay, M.J.; Molloy, M.P. Quantitative Mass Spectrometry for Colorectal Cancer Proteomics. Proteom. Clin. Appl. 2013, 7, 42–54. [Google Scholar] [CrossRef]
- Akkoca, A.N.; Yanık, S.; Özdemir, Z.T.; Cihan, F.G.; Sayar, S.; Cincin, G.; Çam, A.; Özer, C. TNM and Modified Dukes Staging along with the Demographic Characteristics of Patients with Colorectal Carcinoma. Int. J. Clin. Exp. Med. 2014, 7, 2828–2835. [Google Scholar]
- Koelzer, V.H.; Zlobec, I.; Lugli, A. Tumor Budding in Colorectal Cancer. Ready for Diagnostic Practice? Human Pathol. 2016, 47, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kursa, M.B.; Jankowski, A.; Rudnicki, W.R. Boruta—A System for Feature Selection. Fundam. Informaticae 2010, 101, 271–285. [Google Scholar] [CrossRef]
- R CoreTeam. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway Enrichment Analysis and Visualization of Omics Data Using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The Importance of Bottlenecks in Protein Networks: Correlation with Gene Essentiality and Expression Dynamics. PLoS Comput. Biol. 2007, 3, e59. [Google Scholar] [CrossRef]
- Nithya, C.; Kiran, M.; Nagarajaram, H.A. Dissection of Hubs and Bottlenecks in a Protein-Protein Interaction Network. Comput. Biol. Chem. 2023, 102, 107802. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Bronsert, P.; Fichtner-Feigl, S.; Jud, A.; Schilling, O. Proteome Biology of Primary Colorectal Carcinoma and Corresponding Liver Metastases. Neoplasia 2021, 23, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Yang, X.; Bu, X.; Hou, N.; Chen, P. Alpha B-Crystallin Promotes the Invasion and Metastasis of Colorectal Cancer via Epithelial-Mesenchymal Transition. Biochem. Biophys. Res. Commun. 2017, 489, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Navarra, G.; Gazzerro, P.; Vitale, M.; Notarnicola, M.; Caruso, M.G.; Cavalcanti, E.; Armentano, R.; Laezza, C.; Bifulco, M. Association of Alpha B-Crystallin Expression with Tumor Differentiation Grade in Colorectal Cancer Patients. Diagnostics 2021, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Mussunoor, S.; Murray, G. The Role of Annexins in Tumour Development and Progression. J. Pathol. 2008, 216, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Dong, D.W.; Whelan, K.A.; Ruthel, G.; Uchikado, Y.; Natsugoe, S.; Nakagawa, H.; Avadhani, N.G. Disruption of Cytochrome c Oxidase Function Induces the Warburg Effect and Metabolic Reprogramming. Oncogene 2016, 35, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, C.; Cheng, P.; Zhang, S.; Zhou, W.; Xu, Y.; Xu, H.; Ji, G. FHL1 Inhibits the Progression of Colorectal Cancer by Regulating the Wnt/β-Catenin Signaling Pathway. J. Cancer 2021, 12, 5345–5354. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Oparina, N.Y.; Hankin, S.L.; Mashkova, T.D.; Ershov, A.N.; Zatsepina, O.G.; Karpov, V.L.; Beresten, S.F. Identification of Proteins with Altered Expression in Colorectal Cancer by Means of 2D-Proteomics. Mol. Biol. 2009, 43, 321–328. [Google Scholar] [CrossRef]
- Yi, J.M.; Dhir, M.; Van Neste, L.; Downing, S.R.; Jeschke, J.; Glöckner, S.C.; De Freitas Calmon, M.; Hooker, C.M.; Funes, J.M.; Boshoff, C.; et al. Genomic and Epigenomic Integration Identifies a Prognostic Signature in Colon Cancer. Clin. Cancer Res. 2011, 17, 1535–1545. [Google Scholar] [CrossRef]
- Lu, Y.P.; Ishiwata, T.; Kawahara, K.; Watanabe, M.; Naito, Z.; Moriyama, Y.; Sugisaki, Y.; Asano, G. Expression of Lumican in Human Colorectal Cancer Cells. Pathol. Int. 2002, 52, 519–526. [Google Scholar] [CrossRef]
- Hu, X.; Li, Y.Q.; Li, Q.G.; Ma, Y.L.; Peng, J.J.; Cai, S.J. Osteoglycin-Induced VEGF Inhibition Enhances T Lymphocytes Infiltrating in Colorectal Cancer. eBioMedicine 2018, 34, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, X.; Bai, J.; Long, L.; Liu, D.; Zhou, Y. PGM1 Suppresses Colorectal Cancer Cell Migration and Invasion by Regulating the PI3K/AKT Pathway. Cancer Cell Int. 2022, 22, 201. [Google Scholar] [CrossRef] [PubMed]
- Mahdevar, M.; Vatandoost, J.; Seyed Forootan, F.; Kiani-Esfahani, A.; Esmaeili, M.; Peymani, M.; Tavakkoli, H.; Osmay Gure, A.; Nasr Esfahani, M.H.; Ghaedi, K. Steroid Receptor RNA Activator Gene Footprint in the Progression and Drug Resistance of Colorectal Cancer through Oxidative Phosphorylation Pathway. Life Sci. 2021, 285, 119950. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, N.; Li, M.; Hong, T.; Meng, W.; Ouyang, T. Ubiquitin C-terminal hydrolase-L1: A New Cancer Marker and Therapeutic Target with Dual Effects (Review). Oncol. Lett. 2023, 25, 123. [Google Scholar] [CrossRef]
- Torres, S.; Bartolomé, R.A.; Mendes, M.; Barderas, R.; Fernandez-Aceñero, M.J.; Peláez-García, A.; Peña, C.; Lopez-Lucendo, M.; Villar-Vázquez, R.; De Herreros, A.G.; et al. Proteome Profiling of Cancer-Associated Fibroblasts Identifies Novel Proinflammatory Signatures and Prognostic Markers for Colorectal Cancer. Clin. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Chen, X.; Cai, Y.; Wang, X.; Xing, C. miR-20a-Directed Regulation of BID Is Associated with the TRAIL Sensitivity in Colorectal Cancer. Oncol. Rep. 2017, 37, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zheng, Y.; Fan, S.; Qin, Z.; Li, N.; Tang, A.; Ai, F.; Zhang, X.; Bian, Y.; Dang, W.; et al. Lactoferrin Deficiency Promotes Colitis-Associated Colorectal Dysplasia in Mice. PLoS ONE 2014, 9, e103298. [Google Scholar] [CrossRef] [PubMed]
- Szymańska-Chabowska, A.; Świątkowski, F.; Jankowska-Polańska, B.; Mazur, G.; Chabowski, M. Nestin Expression as a Diagnostic and Prognostic Marker in Colorectal Cancer and Other Tumors. Clin. Med. Insights Oncol. 2021, 15, 117955492110382. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Duan, L.; Wu, R.; Ye, L.; Wang, H.; Yang, X.; Zhang, Y.; Chen, X.; Zuo, G.; Zhang, Y.; Weng, Y.; et al. S100A8 and S100A9 Are Associated with Colorectal Carcinoma Progression and Contribute to Colorectal Carcinoma Cell Survival and Migration via Wnt/β-Catenin Pathway. PLoS ONE 2013, 8, e62092. [Google Scholar] [CrossRef]
- Neilsen, B.K.; Chakraborty, B.; McCall, J.L.; Frodyma, D.E.; Sleightholm, R.L.; Fisher, K.W.; Lewis, R.E. WDR5 Supports Colon Cancer Cells by Promoting Methylation of H3K4 and Suppressing DNA Damage. BMC Cancer 2018, 18, 673. [Google Scholar] [CrossRef]
- Khayami, R.; Hashemi, S.R.; Kerachian, M.A. Role of Aldo-Keto Reductase Family 1 Member B1 (AKR1B1) in the Cancer Process and Its Therapeutic Potential. J. Cell. Mol. Med. 2020, 24, 8890–8902. [Google Scholar] [CrossRef]
- Gu, C.; Shang, A.; Liu, G.; Zhu, J.; Zhang, W.; Jin, L.; Sun, Z.; Li, D. Identification of CD147-Positive Extracellular Vesicles as Novel Non-Invasive Biomarkers for the Diagnosis and Prognosis of Colorectal Cancer. Clin. Chim. Acta 2023, 548, 117510. [Google Scholar] [CrossRef] [PubMed]
- Graboń, W.; Otto-Ślusarczyk, D.; Chrzanowska, A.; Mielczarek-Puta, M.; Joniec-Maciejak, I.; Słabik, K.; Barańczyk-Kuźma, A. Lactate Formation in Primary and Metastatic Colon Cancer Cells at Hypoxia and Normoxia. Cell Biochem. Funct. 2016, 34, 483–490. [Google Scholar] [CrossRef]
- Kurilla, A.; László, L.; Takács, T.; Tilajka, Á.; Lukács, L.; Novák, J.; Pancsa, R.; Buday, L.; Vas, V. Studying the Association of TKS4 and CD2AP Scaffold Proteins and Their Implications in the Partial Epithelial–Mesenchymal Transition (EMT) Process. Int. J. Mol. Sci. 2023, 24, 15136. [Google Scholar] [CrossRef]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen Sulfide Perturbs Mitochondrial Bioenergetics and Triggers Metabolic Reprogramming in Colon Cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef] [PubMed]
- Ågesen, T.H.; Berg, M.; Clancy, T.; Thiis-Evensen, E.; Cekaite, L.; Lind, G.E.; Nesland, J.M.; Bakka, A.; Mala, T.; Hauss, H.J.; et al. CLC and IFNAR1 Are Differentially Expressed and a Global Immunity Score Is Distinct between Early- and Late-Onset Colorectal Cancer. Genes Immun. 2011, 12, 653–662. [Google Scholar] [CrossRef]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Romaniuk, W.; Markowski, A.; Kędra, B.; Zalewska, A.; Pryczynicz, A.; Matowicka-Karna, J.; Guzińska-Ustymowicz, K. Antioxidant Barrier, Redox Status, and Oxidative Damage to Biomolecules in Patients with Colorectal Cancer. Can Malondialdehyde and Catalase Be Markers of Colorectal Cancer Advancement? Biomolecules 2019, 9, 637. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Toms, A.M.; Davies, L.M.; Cheng, S.; Jiang, W.G. The Clinical and Biological Implications of N-WASP Expression in Human Colorectal Cancer. Transl. Gastrointest. Cancer 2012, 1, 10–20. [Google Scholar] [CrossRef]
- Luo, Y.; Ye, G.Y.; Qin, S.L.; Mu, Y.F.; Zhang, L.; Qi, Y.; Qiu, Y.E.; Yu, M.H.; Zhong, M. High Expression of Rab3D Predicts Poor Prognosis and Associates with Tumor Progression in Colorectal Cancer. Int. J. Biochem. Cell Biol. 2016, 75, 53–62. [Google Scholar] [CrossRef]
- Liu, C.; Qu, Z.; Zhao, H.; Wang, P.; Zhan, C.; Zhang, Y. Pan-Cancer Analysis of SYNGR2 with a Focus on Clinical Implications and Immune Landscape in Liver Hepatocellular Carcinoma. BMC Bioinform. 2023, 24, 192. [Google Scholar] [CrossRef] [PubMed]
- Funke, V.; Lehmann-Koch, J.; Bickeböller, M.; Benner, A.; Tagscherer, K.E.; Grund, K.; Pfeifer, M.; Herpel, E.; Schirmacher, P.; Chang-Claude, J.; et al. The PEA-15/PED Protein Regulates Cellular Survival and Invasiveness in Colorectal Carcinomas. Cancer Lett. 2013, 335, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Nakurte, I.; Jekabsons, K.; Rembergs, R.; Zandberga, E.; Abols, A.; Linē, A.; Muceniece, R. Colorectal Cancer Cell Line SW480 and SW620 Released Extravascular Vesicles: Focus on Hypoxia-Induced Surface Proteome Changes. Anticancer Res. 2018, 38, 6133–6138. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Zeng, S.; Zhang, Y.; Zhang, Y.; Jin, Z.; Liu, S.; Zou, X. Bioinformatic Analyses and Experimental Verification Reveal that High FSTL3 Expression Promotes EMT via Fibronectin-1/α5β1 Interaction in Colorectal Cancer. Front. Mol. Biosci. 2021, 8, 762924. [Google Scholar] [CrossRef] [PubMed]
- Bhat, I.P.; Rather, T.B.; Maqbool, I.; Rashid, G.; Akhtar, K.; Bhat, G.A.; Parray, F.Q.; Syed, B.; Khan, I.Y.; Kazi, M.; et al. Connective Tissue Growth Factor Expression Hints at Aggressive Nature of Colorectal Cancer. World J. Gastroenterol. 2022, 28, 547–569. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Denk, S.; Wiegering, A. Targeting Protein Synthesis in Colorectal Cancer. Cancers 2020, 12, 1298. [Google Scholar] [CrossRef] [PubMed]
- Albrethsen, J.; Knol, J.C.; Piersma, S.R.; Pham, T.V.; De Wit, M.; Mongera, S.; Carvalho, B.; Verheul, H.M.W.; Fijneman, R.J.A.; Meijer, G.A.; et al. Subnuclear Proteomics in Colorectal Cancer: Identification of proteins enriched in the nuclear matrix fraction and regulation in adenoma to carcinoma progression. Mol. Cell. Proteom. 2010, 9, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Klemke, L.; De Oliveira, T.; Witt, D.; Winkler, N.; Bohnenberger, H.; Bucala, R.; Conradi, L.-C.; Schulz-Heddergott, R. Hsp90-Stabilized MIF Supports Tumor Progression via Macrophage Recruitment and Angiogenesis in Colorectal Cancer. Cell Death Dis. 2021, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Jaeckel, S.; Hermeking, H. Nidogen-1/NID1 Function and Regulation during Progression and Metastasis of Colorectal Cancer. Cancers 2023, 15, 5316. [Google Scholar] [CrossRef]
- Anant, S.; Houchen, C.W.; Pawar, V.; Ramalingam, S. Role of RNA-Binding Proteins in Colorectal Carcinogenesis. Curr. Color. Cancer Rep. 2010, 6, 68–73. [Google Scholar] [CrossRef]
- Mortezapour, M.; Tapak, L.; Bahreini, F.; Najafi, R.; Afshar, S. Identification of Key Genes in Colorectal Cancer Diagnosis by Weighted Gene Co-Expression Network Analysis. Comput. Biol. Med. 2023, 157, 106779. [Google Scholar] [CrossRef] [PubMed]
- Maurya, N.S.; Kushwaha, S.; Chawade, A.; Mani, A. Transcriptome Profiling by Combined Machine Learning and Statistical R Analysis Identifies TMEM236 as a Potential Novel Diagnostic Biomarker for Colorectal Cancer. Sci. Rep. 2021, 11, 14304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Jin, Z.; Chen, H.; Zhang, C.; Wang, W.; Jing, J.; Pan, W. Clinical Impact of X-Ray Repair Cross-Complementary 1 (XRCC1) and the Immune Environment in Colorectal Adenoma–Carcinoma Pathway Progression. J. Inflamm. Res. 2021, 14, 5403–5417. [Google Scholar] [CrossRef] [PubMed]
- Offermans, N.S.M.; Ketcham, S.M.; Van Den Brandt, P.A.; Weijenberg, M.P.; Simons, C.C.J.M. Alcohol Intake, ADH1B and ADH1C Genotypes, and the Risk of Colorectal Cancer by Sex and Subsite in the Netherlands Cohort Study. Carcinogenesis 2018, 39, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Z.; Song, J.; Wang, T.; Wang, H.; Wang, Y.; Guo, J. Identification of Down-Regulated ADH1C Is Associated With Poor Prognosis in Colorectal Cancer Using Bioinformatics Analysis. Front. Mol. Biosci. 2022, 9, 791249. [Google Scholar] [CrossRef] [PubMed]
- Cheraghi, O.; Dabirmanesh, B.; Ghazi, F.; Amanlou, M.; Atabakhshi-kashi, M.; Fathollahi, Y.; Khajeh, K. The Effect of Nrf2 Deletion on the Proteomic Signature in a Human Colorectal Cancer Cell Line. BMC Cancer 2022, 22, 979. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.G.; De Carvalho, A.F.; Pinheiro, N.A.; Simpson, A.J.G.; De Souza, S.J. NABC1 (BCAS1): Alternative Splicing and Downregulation in Colorectal Tumors. Genomics 2000, 65, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, W.; Zhou, J.; Huang, Y.; Peng, J.; Yuan, Y.; Yu, J.; Zheng, S. CLCA1 Suppresses Colorectal Cancer Aggressiveness via Inhibition of the Wnt/Beta-Catenin Signaling Pathway. Cell Commun. Signal 2017, 15, 38. [Google Scholar] [CrossRef]
- Viikilä, P.; Kivelä, A.J.; Mustonen, H.; Koskensalo, S.; Waheed, A.; Sly, W.S.; Doisy, E.A.; Pastorek, J.; Pastorekova, S.; Parkkila, S.; et al. Carbonic Anhydrase Enzymes II, VII, IX and XII in Colorectal Carcinomas. World J. Gastroenterol. 2016, 22, 8168. [Google Scholar] [CrossRef]
- Tang, X.; Wu, H.; Wu, Z.; Wang, G.; Wang, Z.; Zhu, D. Carboxylesterase 2 Is Downregulated in Colorectal Cancer Following Progression of the Disease. Cancer Investig. 2008, 26, 178–181. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Guan, A.; Zhou, H.; Zhu, Y.; Wang, R.; Li, R. EPHX2 Inhibits Colon Cancer Progression by Promoting Fatty Acid Degradation. Front. Oncol. 2022, 12, 870721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Shen, B.; Sun, X.-F. Chromogranin-A Expression as a Novel Biomarker for Early Diagnosis of Colon Cancer Patients. Int. J. Mol. Sci. 2019, 20, 2919. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Nyström, H. The Extracellular Matrix in Colorectal Cancer and Its Metastatic Settling—Alterations and Biological Implications. Crit. Rev. Oncol. Hematol. 2022, 175, 103712. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, M.; Zakerzade, Z. EPCAM Expression in Colon Adenocarcinoma and Its Relationship with TNM Staging. Adv. Biomed. Res. 2017, 6, 56. [Google Scholar] [CrossRef]
- Aureli, A.; Del Cornò, M.; Marziani, B.; Gessani, S.; Conti, L. Highlights on the Role of Galectin-3 in Colorectal Cancer and the Preventive/Therapeutic Potential of Food-Derived Inhibitors. Cancers 2022, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Niu, X.; Li, Y.; Zhang, J.; Zhu, S.; Yang, Q.; Zhang, W.; Gong, L. Role of the Mucin-like Glycoprotein FCGBP in Mucosal Immunity and Cancer. Front. Immunol. 2022, 13, 863317. [Google Scholar] [CrossRef]
- Jing, F.; Liu, G.; Zhang, R.; Xue, W.; Lin, J.; Zhu, H.; Zhu, Y.; Wu, C.; Luo, Y.; Chen, T.; et al. PYY Modulates the Tumorigenesis and Progression of Colorectal Cancer Unveiled by Proteomics. Am. J. Cancer Res. 2022, 12, 5500–5515. [Google Scholar] [PubMed]
- Price, M.J.; Nguyen, A.D.; Byemerwa, J.K.; Flowers, J.; Baëta, C.D.; Goodwin, C.R. UDP-Glucose Dehydrogenase (UGDH) in Clinical Oncology and Cancer Biology. Oncotarget 2023, 14, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular Matrix Signatures of Human Primary Metastatic Colon Cancers and Their Metastases to Liver. BMC Cancer 2014, 14, 518. [Google Scholar] [CrossRef]
- Miete, C.; Solis, G.P.; Koval, A.; Brückner, M.; Katanaev, V.L.; Behrens, J.; Bernkopf, D.B. Gαi2-Induced Conductin/Axin2 Condensates Inhibit Wnt/β-Catenin Signaling and Suppress Cancer Growth. Nat. Commun. 2022, 13, 674. [Google Scholar] [CrossRef]
- Luo, C.; Shen, J. Adducin in Tumorigenesis and Metastasis. Oncotarget 2017, 8, 48453–48459. [Google Scholar] [CrossRef]
- Bastin, J.; Sroussi, M.; Nemazanyy, I.; Laurent-Puig, P.; Mouillet-Richard, S.; Djouadi, F. Downregulation of Mitochondrial Complex I Induces ROS Production in Colorectal Cancer Subtypes That Differently Controls Migration. J. Transl. Med. 2023, 21, 522. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Khan, S.; Huang, D.; Li, L. V-Set and Immunoglobulin Domain Containing (VSIG) Proteins as Emerging Immune Checkpoint Targets for Cancer Immunotherapy. Front. Immunol. 2022, 13, 938470. [Google Scholar] [CrossRef]
- Barkovskaya, A.; Buffone, A.; Žídek, M.; Weaver, V.M. Proteoglycans as Mediators of Cancer Tissue Mechanics. Front. Cell Dev. Biol. 2020, 8, 569377. [Google Scholar] [CrossRef] [PubMed]
- Spicer, A.P.; Joo, A.; Bowling, R.A. A Hyaluronan Binding Link Protein Gene Family Whose Members Are Physically Linked Adjacent to Chrondroitin Sulfate Proteoglycan Core Protein Genes. J. Biol. Chem. 2003, 278, 21083–21091. [Google Scholar] [CrossRef]
- Aimola, V.; Fanni, D.; Gerosa, C.; Cerrone, G.; Ziranu, P.; Pretta, A.; Murru, R.; Piras, M.; Cau, F.; Zorcolo, L.; et al. Balance between the Stem Cell Marker CD44 and CDX2 Expression in Colorectal Cancer. Ann. Res. Oncol. 2022, 02, 160. [Google Scholar] [CrossRef]
- Ziranu, P.; Aimola, V.; Pretta, A.; Dubois, M.; Murru, R.; Liscia, N.; Cau, F.; Persano, M.; Deias, G.; Palmas, E.; et al. New Horizons in Metastatic Colorectal Cancer: Prognostic Role of CD44 Expression. Cancers 2023, 15, 1212. [Google Scholar] [CrossRef]
- Muramatsu, T. Basigin (CD147), a Multifunctional Transmembrane Glycoprotein with Various Binding Partners. J. Biochem. 2016, 159, 481–490. [Google Scholar] [CrossRef]
- Molfetta, R.; Paolini, R. The Controversial Role of Intestinal Mast Cells in Colon Cancer. Cells 2023, 12, 459. [Google Scholar] [CrossRef]
- Levi-Galibov, O.; Lavon, H.; Wassermann-Dozorets, R.; Pevsner-Fischer, M.; Mayer, S.; Wershof, E.; Stein, Y.; Brown, L.E.; Zhang, W.; Friedman, G.; et al. Heat Shock Factor 1-Dependent Extracellular Matrix Remodeling Mediates the Transition from Chronic Intestinal Inflammation to Colon Cancer. Nat. Commun. 2020, 11, 6245. [Google Scholar] [CrossRef]
- Chen, Y.; Ouyang, Y.; Li, Z.; Wang, X.; Ma, J. S100A8 and S100A9 in Cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188891. [Google Scholar] [CrossRef] [PubMed]
- Benedyk, M.; Sopalla, C.; Nacken, W.; Bode, G.; Melkonyan, H.; Banfi, B.; Kerkhoff, C. HaCaT Keratinocytes Overexpressing the S100 Proteins S100A8 and S100A9 Show Increased NADPH Oxidase and NF-κB Activities. J. Investig. Dermatol. 2007, 127, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rey, D.; Carmona-Rodríguez, L.; Fernández-Aceñero, M.J.; Mira, E.; Mañes, S. Extracellular Superoxide Dismutase, the Endothelial Basement Membrane, and the WNT Pathway: New Players in Vascular Normalization and Tumor Infiltration by T-Cells. Front. Immunol. 2020, 11, 579552. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic Pathways and the Electron Transport Chain: A dangeROS Liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic Regulation of Glutamine Metabolism through HIF1 and SIAH2 Supports Lipid Synthesis That Is Necessary for Tumor Growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Gorgoglione, R.; Impedovo, V.; Riley, C.L.; Fratantonio, D.; Tiziani, S.; Palmieri, L.; Dolce, V.; Fiermonte, G. Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives. Cancers 2022, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Mathivanan, S.; Ji, H.; Simpson, R.J. Two Distinct Populations of Exosomes Are Released from LIM1863 Colon Carcinoma Cell-Derived Organoids. Mol. Cell. Proteom. 2013, 12, 587–598. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, S.; Liang, Q.; Liu, Y.; Liu, L. Unraveling the Multifaceted Role of EpCAM in Colorectal Cancer: An Integrated Review of Its Function and Interplay with Non-Coding RNAs. Med. Oncol. 2023, 41, 35. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Gender and Age | Dukes Stage | Budding |
|---|---|---|---|
| #4 | F, 79 yr | B | 3 |
| #5 | M, 78 yr | B | 3 |
| #6 | F, 55 yr | B | 2 |
| #7 | M, 83 yr | B | 1 |
| #8 | M, 70 yr | B | 2 |
| #9 | F, 81 yr | B | 0 |
| #11 | M, 75 yr | B | 1 |
| #12 | M, 66 yr | B | 3 |
| #13 | F, 52 yr | C | 1 |
| #14 | M, 52 yr | C | 3 |
| #15 | M, 64 yr | C | 3 |
| #17 | M, 76 yr | A | 1 |
| #19 | M, 76 yr | C | 1 |
| #22 | M, 80 yr | B | 0 |
| #23 | F, 66 yr | B | 2 |
| #24 | F, 80 yr | B | 1 |
| UniProt KB Code (Gene) | Protein | ND | MW S vs. D p Change FC | MW S vs. H p Change FC | MW D vs. H p Change FC | KW p | GDA Score | Ref. for CRC and Used Technique | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proteins changing in the comparison S–D and D–H | ||||||||||||||
| Q4V328 (GRIPAP1) | GRASP-1 | ND in S | 0.00001 | S<D | >100 | 0.0001 | D>H | >100 | 0.00001 | |||||
| Q96RF0 (SNX18) | Sorting nexin-18 | ND in D | 0.00001 | S>D | >100 | 0.0003 | D<H | >100 | 0.0001 | LC–MS/MS [24] | ||||
| Proteins changing in the comparison S–H with S↓ | ||||||||||||||
| P15090 (FABP4) | A-FABP | ND in S | 0.0007 | S<H | >100 | 0.0009 | 0.02 | qPCR a, IF b [4] | ||||||
| P02511 (CRYAB) | AlphaB-crystallin | 0.0009 | S<H | >100 | 0.0016 | 0.04 | qPCR, IHC c, WB d [25,26] | |||||||
| P50995 (ANXA11) | Annexin A11 | 0.0011 | S<H | 2.8 | 0.01 | MALDI–TOF MS e, IHC [27] | ||||||||
| P00403 (MT–CO2) | Cytochrome c oxidase subunit 2 | ND in S | 0.0007 | S<H | >100 | 0.5 | Gene expression, WB, IHC [28] | |||||||
| P08294 (SOD3) | EC–SOD | 0.0001 | S<H | 4.1 | ||||||||||
| Q13642 (FHL1) | FHL-1 | 0.0002 | S<H | 10.1 | 0.0003 | mRNA expression [29] | ||||||||
| P21695 (GPD1) | GPDH-C | ND in S | 0.0001 | S<H | >100 | 0.0004 | MALDI–TOF [30] | |||||||
| P10915 (HAPLN1) | HPLPN1 | ND in S | 0.0003 | S<H | >100 | 0.0004 | 0.5 | Gene expression, qMSP f [31] | ||||||
| P51884 (LUM) | Lumican | 0.0001 | S<H | 7.2 | 0.0004 | 0.01 | WB, IHC, PCR [32] | |||||||
| P20774 (OGN) | Mimecan (osteoglycin) | 0.0004 | S<H | 11.7 | 0.0004 | 0.03 | IHC, WB, IF [33] | |||||||
| Q02218 (OGDH) | OGDH-E1 | 0.0008 | S<H | >100 | 0.0002 | 0.01 | LC–MS/MS, WB [5] | |||||||
| Q9UIQ6 (LNPEP) | OTase | ND in S | 0.0000 | S<H | >100 | 0.00001 | ||||||||
| P11177 (PDHB) | PDHE1-B | 0.0002 | S<H | >100 | 0.0005 | |||||||||
| P36871 (PGM1) | PGM-1 | 0.0009 | S<H | 3.2 | 0.3 | qPCR, WB IHC [34] | ||||||||
| Q5T013 (HYI) | Putative Hyi | ND in S | 0.0007 | S<H | >100 | |||||||||
| Q9HD15 (SRA1) | SRA1 | ND in S | 0.0007 | S<H | >100 | WGCNA g [35] | ||||||||
| Q9C0C2 (TNKS1BP1) | TAB182 | 0.0009 | S<H | 1.6 | ||||||||||
| P09936 (UCHL1) | UCH-L1 | 0.0002 | S<H | >100 | 0.0004 | 0.04 | WB, gene expression, qMSP [36] | |||||||
| P51452 (DUSP3) | VHR | ND in S | 0.0007 | S<H | >100 | |||||||||
| Proteins changing in the comparison S–H with S↑ | ||||||||||||||
| P13674 (P4HA1) | 4-PH alpha-1 | ND in H | 0.0003 | S>H | >100 | 0.0006 | LC–MS/MS [37] | |||||||
| P55957 (BID) | BH3-interacting domain death agonist | 0.0012 | S>H | >100 | 0.01 | qPCR [38] | ||||||||
| P02788 (LTF) | Lactotransferrin | 0.0001 | S>H | 6.0 | 0.0004 | qPCR, IHC, WB [39] | ||||||||
| P49321 (NASP) | NASP | 0.0012 | S>H | 4.3 | ||||||||||
| P48681 (NES) | Nestin | ND in H | 0.0007 | S>H | >100 | 0.01 | IHC [40] | |||||||
| P02775 (PPBP) | PBP | 0.0004 | S>H | 2.2 | 0.03 | mRNA analysis, WB, IF [41] | ||||||||
| O15212 (PFDN6) | Prefoldin subunit 6 | 0.0006 | S>H | 2.8 | ||||||||||
| Q9Y2Z0 (SUGT1) | Protein SGT1 homolog | ND in H | 0.0003 | S>H | >100 | 0.0008 | ||||||||
| P05109 (S100A8) | S100-A8 | 0.0006 | S>H | 7.7 | qPCR, WB, IHC [42] | |||||||||
| P06702 (S100A9) | S100-A9 | 0.0000 | S>H | 8.1 | 0.0001 | 0.08 | qPCR, WB, IHC [42] | |||||||
| P61964 (WDR5) | WDR5 | ND in H | 0.0003 | S>H | >100 | 0.0008 | 0.01 | qPCR, WB [43] | ||||||
| Proteins changing in the comparison D–H with D↓ | ||||||||||||||
| P15121 (AKR1B1) | Aldo-keto reductase 1B1 | 0.0008 | D<H | 3.7 | 0.05 | gene expression, qMSP [44] | ||||||||
| P17050 (NAGA) | Alpha-N-acetylgalactosaminidase | 0.0009 | D<H | 5.1 | ||||||||||
| P35613 (BSG) | Basigin | 0.0002 | D<H | 4.8 | 0.0011 | 0.06 | WB [45] | |||||||
| P17174 (GOT1) | cAspAT | 0.0002 | D<H | 2.4 | 0.01 | qPCR [46] | ||||||||
| Q9Y5K6 (CD2AP) | CD2-associated protein | 0.0012 | D<H | 3.7 | 0.02 | LC–MS/MS, IHC [47] | ||||||||
| P10606 (COX5B) | Cytochrome c oxidase sub. 5B | 0.0012 | D<H | 4.5 | ||||||||||
| Q13011 (ECH1) | DI (mitochondrial) | 0.0007 | D<H | 3.3 | ||||||||||
| P36957 (DLST) | DLST | 0.0008 | D<H | 3.4 | 0.0011 | |||||||||
| O95571 (ETHE1) | ETHE1 | 0.0000 | D<H | 4.0 | 0.0002 | 0.02 | IHC, WB, LC–MS/MS [48] | |||||||
| Q05315 (CLC) | Gal-10 | 0.0011 | D<H | 4.9 | 0.03 | RNA analysis [49] | ||||||||
| P00390 (GSR) | GRase | 0.0002 | D<H | 4.6 | 0.0012 | 0.01 | Enzymatic assay [50] | |||||||
| P06870 (KLK1) | Kallikrein-1 | 0.0006 | D<H | >100 | 0.0003 | |||||||||
| O00401 (WASL) | N-WASP | 0.0009 | D<H | >100 | 0.01 | qPCR [51] | ||||||||
| Q8IV08 (PLD3) | PLD3 | 0.0005 | D<H | >100 | 0.0012 | |||||||||
| O95716 (RAB3D) | Rab-3D | 0.0010 | D<H | >100 | IHC, WB, qPCR [52] | |||||||||
| O43760 (SYNGR2) | Synaptogyrin-2 | ND in D | 0.0007 | D<H | >100 | 0.0015 | GSEA h [53] | |||||||
| Proteins changing in the comparison D–H with D↑ | ||||||||||||||
| Q15121 (PEA15) | 15 kDa phosphoprotein enriched in astrocytes | 0.0009 | D>H | >100 | 0.0011 | 0.02 | WB, cell assays [54] | |||||||
| O94875 (SORBS2) | ArgBP2 | 0.0007 | D>H | >100 | ||||||||||
| P53618 (COPB1) | Beta-COP | 0.0002 | D>H | 5.7 | 0.0001 | 0.01 | WB, IHC, LC–TOF MS [55] | |||||||
| P08572 (COL4A2) | Collagen alpha-2(IV) chain | 0.0003 | D>H | >100 | 0.0005 | 0.1 | LC–MS/MS [37] | |||||||
| P02751 (FN1) | FN | 0.0002 | D>H | 2.9 | 0.0004 | 0.4 | RNA analysis, WB, IHC, GSEA [56] | |||||||
| P29279 (CCN2) | CTGF | ND in H | 0.0003 | D>H | >100 | 0.0012 | 0.08 | WB, qPCR [57] | ||||||
| P13639 (EEF2) | EF-2 | 0.0002 | D>H | 10.7 | 0.01 | MALDI–TOF, IHC, WB, ELISA [58] | ||||||||
| P84243 (H3–3A) | Histone H3.3 | 0.0012 | D>H | >100 | 0.01 | RNA analysis [49] | ||||||||
| O14979 (HNRNPDL) | hnRNP D-like | 0.0001 | D>H | 2.0 | 0.0009 | 0.03 | LC–MS/MS [59] | |||||||
| P14174 (MIF) | MIF | 0.0007 | D>H | 2.4 | 0.03 | IHC, WB, qPCR [60] | ||||||||
| P14543 (NID1) | NID-1 | 0.0002 | D>H | >100 | 0.000001 | 0.3 | qPCR [61] | |||||||
| P42696 (RBM34) | RNA-binding protein 34 | 0.0009 | D>H | >100 | 0.0013 | qPCR, WB, IHC [62] | ||||||||
| P50454 (SERPINH1) | Serpin H1 | 0.0004 | D>H | 3.4 | 0.01 | DEGs i, WGCNA [63] | ||||||||
| Q8WUH6 (TMEM263) | Transmembrane protein 263 | ND in S/H | 0.0003 | D>H | >100 | 0.000001 | 0.02 | DEGs, ML j [64] | ||||||
| P18887 (XRCC1) | XRCC1 | ND in H | 0.0001 | D>H | >100 | 0.00001 | 0.4 | Gene expression, IHC [65] | ||||||
| Proteins changing in the comparison D–H and S–H with D↓ and S↓ | ||||||||||||||
| P27348 (YWHAQ) | 14-3-3 protein theta | 0.0001 | S<H | 8.2 | 0.0003 | D<H | 8.3 | 0.0001 | ||||||
| P00325 (ADH1B) | ADH1B | 0.0010 | S<H | 9.5 | 0.0013 | D<H | 8.3 | 0.0006 | 0.4 | Gene expression [66] | ||||
| P00326 (ADH1C) | ADH1C | 0.00001 | S<H | 22.3 | 0.00001 | D<H | 26.3 | 0.00001 | 0.06 | DEGs, ML, IHC, WB, qPCR [67] | ||||
| P48047 (ATP5PO) | ATP synthase subunit O | 0.0008 | S<H | 16.5 | 0.0013 | D<H | 3.5 | 0.0004 | LC–MS/MS, qPCR, WB [68] | |||||
| O75363 (BCAS1) | Breast carcinoma-amplified sequence 1 | 0.0011 | S<H | 3.5 | 0.00001 | D<H | 7.4 | 0.0001 | qPCR [69] | |||||
| A8H7I4 (CLCA1) | CaCC-1 | 0.00001 | S<H | 9.2 | 0.00001 | D<H | 22.1 | 0.00001 | 0.06 | ELISA, IHC, WB [70] | ||||
| P00918 (CA2) | CA-II | 0.00001 | S<H | 5.7 | 0.00001 | D<H | 8.0 | 0.00001 | 0.02 | IHC [71] | ||||
| O00748 (SULT2) | CE-2 | 0.0006 | S<H | 9.2 | 0.0004 | D<H | 10.9 | 0.0004 | 0.03 | WB [72] | ||||
| P34913 (EPHX2) | CEH | 0.0002 | S<H | 4.8 | 0.00001 | D<H | >100 | 0.00001 | IHC, qPCR, WB [73] | |||||
| P10645 (CHGA) | CgA, pro-protein | 0.0001 | S<H | 8.7 | 0.00001 | D<H | 18.2 | 0.00001 | 0.03 | ML of gene data, qPCR [74] | ||||
| P07585 (DCN) | Decorin | 0.0002 | S<H | 9.7 | 0.0008 | D<H | 5.7 | 0.0001 | 0.08 | IF [75] | ||||
| P16422 (EPCAM) | Ep-CAM | 0.0001 | S<H | >100 | 0.0001 | D<H | >100 | 0.00001 | 0.1 | IHC [76] | ||||
| Q9UHB6 (LIMA1) | EPLIN | 0.0010 | S<H | 4.0 | 0.0000 | D<H | 5.6 | 0.0001 | 0.1 | LC–MS/MS [59] | ||||
| P17931 (LGALS3) | Gal-3 | 0.0008 | S<H | 2.2 | 0.0007 | D<H | 2.5 | 0.0008 | 0.1 | IHC [77] | ||||
| Q9Y6R7 (FCGBP) | IgGFc-binding protein | 0.00001 | S<H | 9.8 | 0.00001 | D<H | 35.7 | 0.00001 | DEGs and ML [78] | |||||
| P10082 (PYY) | PYY | ND in D | 0.0010 | S<H | >100 | 0.0000 | D<H | >100 | 0.000001 | 0.01 | LC–MS/MS, qPCR, IHC [79] | |||
| Q15661 (TPSAB1) | Tryptase-1 | 0.0003 | S<H | 10.1 | 0.0002 | D<H | 6.5 | 0.0003 | ||||||
| O60701 (UGDH) | UDP-GlcDH | 0.0001 | S<H | 3.4 | 0.0000 | D<H | 5.0 | 0.0001 | qPCR, WB [80] | |||||
| Proteins changing in the comparison D–H and S–H with D↑ and S↑ | ||||||||||||||
| Q15582 (TGFBI) | Beta ig-h3 | 0.0008 | S>H | 3.1 | 0.0000 | D>H | 2.7 | 0.0001 | 0.02 | LC–MSMS, GSEA [81] | ||||
| P04899 (GNAI2) | Galphai2 | 0.00001 | S>H | 6.3 | 0.0003 | D>H | 5.4 | 0.00001 | IF, WB, qPCR [82] | |||||
| Proteins changing in KW comparisons among all three groups but not in MW pairwise comparisons | ||||||||||||||
| Q9NZK5 (ADA2) | Adenosine deaminase 2 | N.D in D | 0.0006 | |||||||||||
| P35611 (ADD1) | Alpha-adducin | 0.0012 | 0.01 | Gene expression [83] | ||||||||||
| P0C0L5 (C4B) | Complement C4-B | ND in S/H | 0.0013 | |||||||||||
| Q96S86 (HAPLN3) | HPLPN3 | ND in D | 0.0009 | |||||||||||
| C9JLW8 (MCRIP1) | MCRIP1 | ND in S | 0.0001 | |||||||||||
| P49821 (NDUFV1) | NDUFV1 | ND in S/D | 0.0003 | WB, enzyme assay [84] | ||||||||||
| Q96IQ7 (VSIG2) | VSIG2 | 0.0015 | WGCNA [85] | |||||||||||
| O75312 (ZPR1) | Zinc finger protein ZPR1 (ZPR1) | ND in S | 0.0005 | |||||||||||
| Biological Processes/ Pathways | GO/Reactome Entry (FDR) | Proteins |
|---|---|---|
| Degradation of the ECM | REAC:R-HSA-3560782 (0.02) | Decorin; lumican; mimecan; basigin; collagen alpha-2(IV) chain; FN; NID-1, tryptase-1 |
| ECM proteoglycans | REAC:R-HSA-3000178 (0.01) | Decorin; lumican; HPLPN1; collagen alpha-2(IV) chain; FN |
| ECM organization | REAC:R-HSA-1474244 (0.003) | Decorin; lumican; HPLPN1; basigin; collagen alpha-2(IV) chain; FN; tryptase-1; 4-PH alpha-1; NID-1; serpin H1 |
| Aerobic respiration/ Cellular respiration | GO:0009060/GO:0045333 (0.02) | PDHE1-B; OGDH-E1; ATP synthase subunit O; cytochrome c oxidase subunit 2; cytochrome c oxidase sub. 5B; DLST; NDUFV1 |
| Generation of precursor metabolites and energy | GO:0006091 (0.02) | PDHE1-B; OGDH-E1; DLST; ATP synthase subunit O; cytochrome c oxidase subunit 2; cytochrome c oxidase sub. 5B; NDUFV1; PGM-1; ADH1C; ADH1B |
| Citric acid cycle and respiratory electron transport | REAC:R-HSA-1428517 (0.003) | PDHE1-B; OGDH-E1; DLST; ATP synthase subunit O; cytochrome c oxidase subunit 2; cytochrome c oxidase sub. 5B; NDUFV1; basigin |
| 2-oxoglutarate metabolic process | GO:0006103 (0.02) | OGDH-E1; DLST; cAspAT |
| Histone succinylation/ Protein succinylation | GO:0106077/GO:0018335 (0.03)/(0.04) | OGDH-E1; DLST |
| Metabolism | REAC:R-HSA-1430728 (0.01) | PDHE1-B; OGDH-E1; DLST; cAspAT; ATP synthase subunit O; cytochrome c oxidase subunit 2; cytochrome c oxidase sub. 5B; NDUFV1; PGM-1; GPDH-C; ETHE1; ADH1C; ADH1B; CEH; CE-2; A-FABP; aldo-keto reductase 1B1; UDP-GlcDH; decorin; lumican; mimecan; CA-II; basigin; Galphai2; PLD3; N-WASP |
| Neutrophil chemotaxis | GO:0030593 (0.03) | S100A8; S100A9; Gal-3; PBP; basigin |
| Leukocyte chemotaxis/ Myeloid leukocyte migration | GO:0030595/GO:0097529 (0.02) | S100A8; S100A9; Gal-3; PBP; basigin; CgA; MIF |
| Leuckocyte migration | GO:0050900 (0.04) | S100A8; S100A9; Gal-3; PBP; basigin; CgA; MIF; N-WASP |
| Neutrophil aggregation | GO:0070488 (0.02) | S100A8; S100A9 |
| Metal sequestration by antimicrobial proteins | REAC:R-HSA-6799990 (0.002) | Lactotransferrin; S100A8; S100A9 |
| Sequestering of zinc ion | GO:0032119 (0.02) | S100A8; S100A9 |
| Gene | Closeness Centrality | Degree | Betweenness Centrality | Protein |
|---|---|---|---|---|
| COPB2 * | 0.36 | 6 | 0.002 | Beta-COP |
| DPP4 * | 0.37 | 5 | 0.0001 | ADABP |
| EEF2 | 0.52 | 44 | 0.022 | EF-2 |
| FN1 | 0.51 | 37 | 0.025 | FN |
| GOT1 * | 0.32 | 5 | 0.0002 | cAspAT |
| IGHV3–43D * | 0.31 | 2 | 0.0001 | Immunoglobulin heavy variable 3–43D |
| LGALS3 * | 0.43 | 12 | 0.004 | Gal-3 |
| LTF * | 0.38 | 10 | 0.003 | Lactotransferrin |
| PDHB * | 0.41 | 9 | 0.0003 | PDHE1-B |
| WASL * | 0.41 | 14 | 0.003 | N-WASP |
| WDR5 * | 0.41 | 9 | 0.002 | WD repeat-containing protein 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contini, C.; Manconi, B.; Olianas, A.; Guadalupi, G.; Schirru, A.; Zorcolo, L.; Castagnola, M.; Messana, I.; Faa, G.; Diaz, G.; et al. Combined High—Throughput Proteomics and Random Forest Machine-Learning Approach Differentiates and Classifies Metabolic, Immune, Signaling and ECM Intra-Tumor Heterogeneity of Colorectal Cancer. Cells 2024, 13, 1311. https://doi.org/10.3390/cells13161311
Contini C, Manconi B, Olianas A, Guadalupi G, Schirru A, Zorcolo L, Castagnola M, Messana I, Faa G, Diaz G, et al. Combined High—Throughput Proteomics and Random Forest Machine-Learning Approach Differentiates and Classifies Metabolic, Immune, Signaling and ECM Intra-Tumor Heterogeneity of Colorectal Cancer. Cells. 2024; 13(16):1311. https://doi.org/10.3390/cells13161311
Chicago/Turabian StyleContini, Cristina, Barbara Manconi, Alessandra Olianas, Giulia Guadalupi, Alessandra Schirru, Luigi Zorcolo, Massimo Castagnola, Irene Messana, Gavino Faa, Giacomo Diaz, and et al. 2024. "Combined High—Throughput Proteomics and Random Forest Machine-Learning Approach Differentiates and Classifies Metabolic, Immune, Signaling and ECM Intra-Tumor Heterogeneity of Colorectal Cancer" Cells 13, no. 16: 1311. https://doi.org/10.3390/cells13161311