
Gastrointestinal Cancer Patient Derived Organoids at the Frontier of Personalized Medicine and Drug Screening

Abstract
:1. Introduction
2. Establishment of Gastrointestinal Cancer PDOs
3. Advances in Gastrointestinal Cancer Patient-Derived Organoids (PDOs)
3.1. Esophageal Cancer
3.2. Gastric Cancer
3.3. Liver Cancer
3.4. Pancreatic Cancer
3.5. Colorectal Cancer
3.6. Propagation of PDOs
3.7. Technological Advances in Gastrointestinal PDOs
4. Gastrointestinal Cancer PDOs in Personalized Medicine and Drug Screening
4.1. Clinical Implication of PDO-Based Personalized Medicine and Drug Screening
4.2. PDOs for Precision Medicine and Personalized Treatment
4.3. PDOs for Large-Scale Drug Screening
5. Limitations of PDOs in Personalized Medicine and Drug Screening
6. Clinical Application of Organoid Technologies: From Bench to Bedside
7. Outlooks and Perspectives
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Jain, R.K.; Duda, D.G. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma: A Review. JAMA Oncol. 2020, 7, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-M.; Hong, P.; Xu, W.W.; He, Q.-Y.; Li, B. Advances in targeted therapy for esophageal cancer. Signal Transduct. Target. Ther. 2020, 5, 229. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.T.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Mousavi Shaegh, S.A.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef]
- Achberger, K.; Probst, C.; Haderspeck, J.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. eLife 2019, 8, e46188. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.C.H.; Kranenburg, O.; Xiao, H.; Yu, J. Organoid models of gastrointestinal cancers in basic and translational research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Kopper, O.; De Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- Sasai, Y. Next-generation regenerative medicine: Organogenesis from stem cells in 3D culture. Cell Stem Cell 2013, 12, 520–530. [Google Scholar] [CrossRef]
- McCauley, H.A.; Wells, J.M. Pluripotent stem cell-derived organoids: Using principles of developmental biology to grow human tissues in a dish. Development 2017, 144, 958–962. [Google Scholar] [CrossRef]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering Stem Cell Organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; Van Gerwen, B.; Van Der Ven, M.; Van De Wetering, M.; Egan, D.A.; et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. eLife 2016, 5, e18489. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.J.; Shin, T.H.; Kim, M.; Sung, C.O.; Jang, S.J.; Jeong, G.S. A one-stop microfluidic-based lung cancer organoid culture platform for testing drug sensitivity. Lab Chip 2019, 19, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Knutsdottir, H.; Hui, K.; Weiss, M.J.; He, J.; Philosophe, B.; Cameron, A.M.; Wolfgang, C.L.; Pawlik, T.M.; Ghiaur, G.; et al. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 2019, 4, e121490. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef]
- Driehuis, E.; Spelier, S.; Beltrán Hernández, I.; de Bree, R.; Willems, S.M.; Clevers, H.; Oliveira, S. Patient-Derived Head and Neck Cancer Organoids Recapitulate EGFR Expression Levels of Respective Tissues and Are Responsive to EGFR-Targeted Photodynamic Therapy. J. Clin. Med. 2019, 8, 1880. [Google Scholar] [CrossRef]
- Bossi, P.; Resteghini, C.; Paielli, N.; Licitra, L.; Pilotti, S.; Perrone, F. Prognostic and predictive value of EGFR in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 74362–74379. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lõhmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Seidlitz, T.; Stange, D.E. Gastrointestinal cancer organoids-applications in basic and translational cancer research. Exp. Mol. Med. 2021, 53, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018, 9, 2983. [Google Scholar] [CrossRef] [PubMed]
- Derouet, M.F.; Allen, J.; Wilson, G.W.; Ng, C.; Radulovich, N.; Kalimuthu, S.; Tsao, M.-S.; Darling, G.E.; Yeung, J.C. Towards personalized induction therapy for esophageal adenocarcinoma: Organoids derived from endoscopic biopsy recapitulate the pre-treatment tumor. Sci. Rep. 2020, 10, 14514. [Google Scholar] [CrossRef] [PubMed]
- Kijima, T.; Nakagawa, H.; Shimonosono, M.; Chandramouleeswaran, P.M.; Hara, T.; Sahu, V.; Kasagi, Y.; Kikuchi, O.; Tanaka, K.; Giroux, V.; et al. Three-Dimensional Organoids Reveal Therapy Resistance of Esophageal and Oropharyngeal Squamous Cell Carcinoma Cells. Cell. Mol. Gastroenterol. Hepatol. 2018, 7, 73–91. [Google Scholar] [CrossRef]
- Seidlitz, T.; Merker, S.R.; Rothe, A.; Zakrzewski, F.; von Neubeck, C.; Grützmann, K.; Sommer, U.; Schweitzer, C.; Schölch, S.; Uhlemann, H.; et al. Human gastric cancer modelling using organoids. Gut 2019, 68, 207–217. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e11. [Google Scholar] [CrossRef] [PubMed]
- Nanki, K.; Toshimitsu, K.; Takano, A.; Fujii, M.; Shimokawa, M.; Ohta, Y.; Matano, M.; Seino, T.; Nishikori, S.; Ishikawa, K.; et al. Divergent Routes toward Wnt and R-spondin Niche Independency during Human Gastric Carcinogenesis. Cell 2018, 174, 856–869.e17. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e6. [Google Scholar] [CrossRef]
- Weeber, F.; van de Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-van Hooijdonk, C.G.M.; van der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.J.G.; et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015, 112, 13308–13311. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; Van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; Van De Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.e6. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Wu, C.; O’Rourke, K.P.; Szeglin, B.C.; Zheng, Y.; Sauvé, C.-E.G.; Adileh, M.; Wasserman, I.; Marco, M.R.; Kim, A.S.; et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019, 25, 1607–1614. [Google Scholar] [CrossRef]
- Kodack, D.P.; Farago, A.F.; Dastur, A.; Held, M.A.; Dardaei, L.; Friboulet, L.; von Flotow, F.; Damon, L.J.; Lee, D.; Parks, M.; et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep. 2017, 21, 3298–3309. [Google Scholar] [CrossRef]
- Wang, E.; Xiang, K.; Zhang, Y.; Wang, X.-F. Patient-derived organoids (PDOs) and PDO-derived xenografts (PDOXs): New opportunities in establishing faithful pre-clinical cancer models. J. Natl. Cancer Cent. 2022, 2, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Ivanics, T.; Bergquist, J.R.; Liu, G.; Kim, M.P.; Kang, Y.; Katz, M.H.; Perez, M.V.R.; Thomas, R.M.; Fleming, J.B.; Truty, M.J. Patient-derived xenograft cryopreservation and reanimation outcomes are dependent on cryoprotectant type. Lab. Investig. 2018, 98, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Zanella, E.R.; Grassi, E.; Trusolino, L. Towards precision oncology with patient-derived xenografts. Nat. Rev. Clin. Oncol. 2022, 19, 719–732. [Google Scholar] [CrossRef]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Dao, V.; Yuki, K.; Lo, Y.-H.; Nakano, M.; Kuo, C.J. Immune organoids: From tumor modeling to precision oncology. Trends Cancer 2022, 8, 870–880. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Steele, N.G.; Chakrabarti, J.; Wang, J.; Biesiada, J.; Holokai, L.; Chang, J.; Nowacki, L.M.; Hawkins, J.; Mahe, M.; Sundaram, N.; et al. An Organoid-Based Preclinical Model of Human Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 161–184. [Google Scholar] [CrossRef]
- Ukai, S.; Honma, R.; Sakamoto, N.; Yamamoto, Y.; Pham, Q.T.; Harada, K.; Takashima, T.; Taniyama, D.; Asai, R.; Fukada, K.; et al. Molecular biological analysis of 5-FU-resistant gastric cancer organoids; KHDRBS3 contributes to the attainment of features of cancer stem cell. Oncogene 2020, 39, 7265–7278. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Gao, M.; Cavnar, M.J.; Kim, J. Utilizing gastric cancer organoids to assess tumor biology and personalize medicine. World J. Gastrointest. Oncol. 2019, 11, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Lin, M.; Rao, M.; Thompson, H.; Hirai, K.; Choi, M.; Georgakis, G.V.; Sasson, A.R.; Bucobo, J.C.; Tzimas, D.; et al. Development of Patient-Derived Gastric Cancer Organoids from Endoscopic Biopsies and Surgical Tissues. Ann. Surg. Oncol. 2018, 25, 2767–2775. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S.; Bayram, T.; van de Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In Vitro Expansion of Human Gastric Epithelial Stem Cells and Their Responses to Bacterial Infection. Gastroenterology 2015, 148, 126–136.e6. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.M.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef]
- Fowler, S.; Chen, W.L.K.; Duignan, D.B.; Gupta, A.; Hariparsad, N.; Kenny, J.R.; Lai, W.G.; Liras, J.; Phillips, J.A.; Gan, J. Microphysiological systems for ADME-related applications: Current status and recommendations for system development and characterization. Lab Chip 2020, 20, 446–467. [Google Scholar] [CrossRef]
- Fabre, K.; Berridge, B.; Proctor, W.R.; Ralston, S.; Will, Y.; Baran, S.W.; Yoder, G.; Van Vleet, T.R. Introduction to a manuscript series on the characterization and use of microphysiological systems (MPS) in pharmaceutical safety and ADME applications. Lab Chip 2020, 20, 1049–1057. [Google Scholar] [CrossRef]
- Lo, Y.H.; Karlsson, K.; Kuo, C.J. Applications of Organoids for Cancer Biology and Precision Medicine. Nat. Cancer 2020, 1, 761–773. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Pasch, C.A.; Favreau, P.F.; Yueh, A.E.; Babiarz, C.P.; Gillette, A.A.; Sharick, J.T.; Karim, M.R.; Nickel, K.P.; DeZeeuw, A.K.; Sprackling, C.M.; et al. Patient-Derived Cancer Organoid Cultures to Predict Sensitivity to Chemotherapy and Radiation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5376–5387. [Google Scholar] [CrossRef] [PubMed]
- Ao, Z.; Cai, H.; Havert, D.J.; Wu, Z.; Gong, Z.; Beggs, J.M.; Mackie, K.; Guo, F. One-stop Microfluidic Assembly of Human Brain Organoids to Model Prenatal Cannabis Exposure. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Schuster, B.; Junkin, M.; Kashaf, S.S.; Romero-Calvo, I.; Kirby, K.; Matthews, J.; Weber, C.R.; Rzhetsky, A.; White, K.P.; Tay, S. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 2020, 11, 5271. [Google Scholar] [CrossRef] [PubMed]
- Håkanson, M.; Cukierman, E.; Charnley, M. Miniaturized pre-clinical cancer models as research and diagnostic tools. Adv. Drug Deliv. Rev. 2014, 69–70, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Mehling, M.; Tay, S. Microfluidic cell culture. Curr. Opin. Biotechnol. 2014, 25, 95–102. [Google Scholar] [CrossRef]
- Sung, K.E.; Beebe, D.J. Microfluidic 3D models of cancer. Adv. Drug Deliv. Rev. 2014, 79–80, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Valadez, A.V.; Zuo, P.; Nie, Z. Microfluidic 3D cell culture: Potential application for tissue-based bioassays. Bioanalysis 2012, 4, 1509–1525. [Google Scholar] [CrossRef]
- Au, S.H.; Chamberlain, M.D.; Mahesh, S.; Sefton, M.V.; Wheeler, A.R. Hepatic organoids for microfluidic drug screening. Lab Chip 2014, 14, 3290–3299. [Google Scholar] [CrossRef]
- Jin, B.-J.; Battula, S.; Zachos, N.; Kovbasnjuk, O.; Fawlke-Abel, J.; In, J.; Donowitz, M.; Verkman, A.S. Microfluidics platform for measurement of volume changes in immobilized intestinal enteroids. Biomicrofluidics 2014, 8, 024106. [Google Scholar] [CrossRef]
- Narasimhan, V.; Wright, J.A.; Churchill, M.; Wang, T.; Rosati, R.; Lannagan, T.R.M.; Vrbanac, L.; Richardson, A.B.; Kobayashi, H.; Price, T.; et al. Medium-throughput Drug Screening of Patient-derived Organoids from Colorectal Peritoneal Metastases to Direct Personalized Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3662–3670. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Schipper, L.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van de Haar, J.; Prevoo, W.; van Werkhoven, E.; Snaebjornsson, P.; et al. Prospective experimental treatment of colorectal cancer patients based on organoid drug responses. ESMO Open 2021, 6, 100103. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.K.; Khawar, I.A.; Jeong, S.Y.; Chung, S.; Kuh, H.J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 4. [Google Scholar] [CrossRef]
- Ruppen, J.; Wildhaber, F.D.; Strub, C.; Hall, S.R.R.; Schmid, R.A.; Geiser, T.; Guenat, O.T. Towards personalized medicine: Chemosensitivity assays of patient lung cancer cell spheroids in a perfused microfluidic platform. Lab Chip 2015, 15, 3076–3085. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Lee, J.H.; Shin, Y.; Chung, S.; Kuh, H.J. Co-Culture of Tumor Spheroids and Fibroblasts in a Collagen Matrix-Incorporated Microfluidic Chip Mimics Reciprocal Activation in Solid Tumor Microenvironment. PLoS ONE 2016, 11, e0159013. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Iancu, E.M.; Kandalaft, L.E. Challenges and advantages of cell therapy manufacturing under Good Manufacturing Practices within the hospital setting. Curr. Opin. Biotechnol. 2020, 65, 233–241. [Google Scholar] [CrossRef]
- Gronholm, M.; Feodoroff, M.; Antignani, G.; Martins, B.; Hamdan, F.; Cerullo, V. Patient-Derived Organoids for Precision Cancer Immunotherapy. Cancer Res. 2021, 81, 3149–3155. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhang, B.; He, Y.; Bao, J. Liver Organoids: Formation Strategies and Biomedical Applications. Tissue Eng. Regen. Med. 2021, 18, 573–585. [Google Scholar] [CrossRef]
- Tatullo, M.; Marrelli, B.; Benincasa, C.; Aiello, E.; Makeeva, I.; Zavan, B.; Ballini, A.; De Vito, D.; Spagnuolo, G. Organoids in Translational Oncology. J. Clin. Med. 2020, 9, 2774. [Google Scholar] [CrossRef]
- Kasendra, M.; Troutt, M.; Broda, T.; Bacon, W.C.; Wang, T.C.; Niland, J.C.; Helmrath, M.A. Intestinal organoids: Roadmap to the clinic. Am. J. Physiol.-Gastrointest. Liver Physiol. 2021, 321, G1–G10. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Kawazoe, A.; Yañez, P.; Li, N.; Lonardi, S.; Kolesnik, O.; Barajas, O.; Bai, Y.; Shen, L.; Tang, Y.; et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 2021, 600, 727–730. [Google Scholar] [CrossRef]
- Galle, P.R.; Finn, R.S.; Qin, S.; Ikeda, M.; Zhu, A.X.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.; et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef]
- Lannagan, T.R.M.; Lee, Y.K.; Wang, T.; Roper, J.; Bettington, M.L.; Fennell, L.; Vrbanac, L.; Jonavicius, L.; Somashekar, R.; Gieniec, K.; et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 2019, 68, 684–692. [Google Scholar] [CrossRef]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Fritz, J.V.; Glaab, E.; Desai, M.S.; Greenhalgh, K.; Frachet, A.; Niegowska, M.; Estes, M.; Jäger, C.; Seguin-Devaux, C.; et al. A microfluidics-based in vitro model of the gastrointestinal human–microbe interface. Nat. Commun. 2016, 7, 11535. [Google Scholar] [CrossRef] [PubMed]
- Rauth, S.; Karmakar, S.; Batra, S.K.; Ponnusamy, M.P. Recent advances in organoid development and applications in disease modeling. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188527. [Google Scholar] [CrossRef]
- Williamson, I.A.; Arnold, J.W.; Samsa, L.A.; Gaynor, L.; DiSalvo, M.; Cocchiaro, J.L.; Carroll, I.; Azcarate-Peril, M.A.; Rawls, J.F.; Allbritton, N.L.; et al. A High-Throughput Organoid Microinjection Platform to Study Gastrointestinal Microbiota and Luminal Physiology. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 301–319. [Google Scholar] [CrossRef]
- Bozzetti, V.; Senger, S. Organoid technologies for the study of intestinal microbiota–host interactions. Trends Mol. Med. 2022, 28, 290–303. [Google Scholar] [CrossRef] [PubMed]
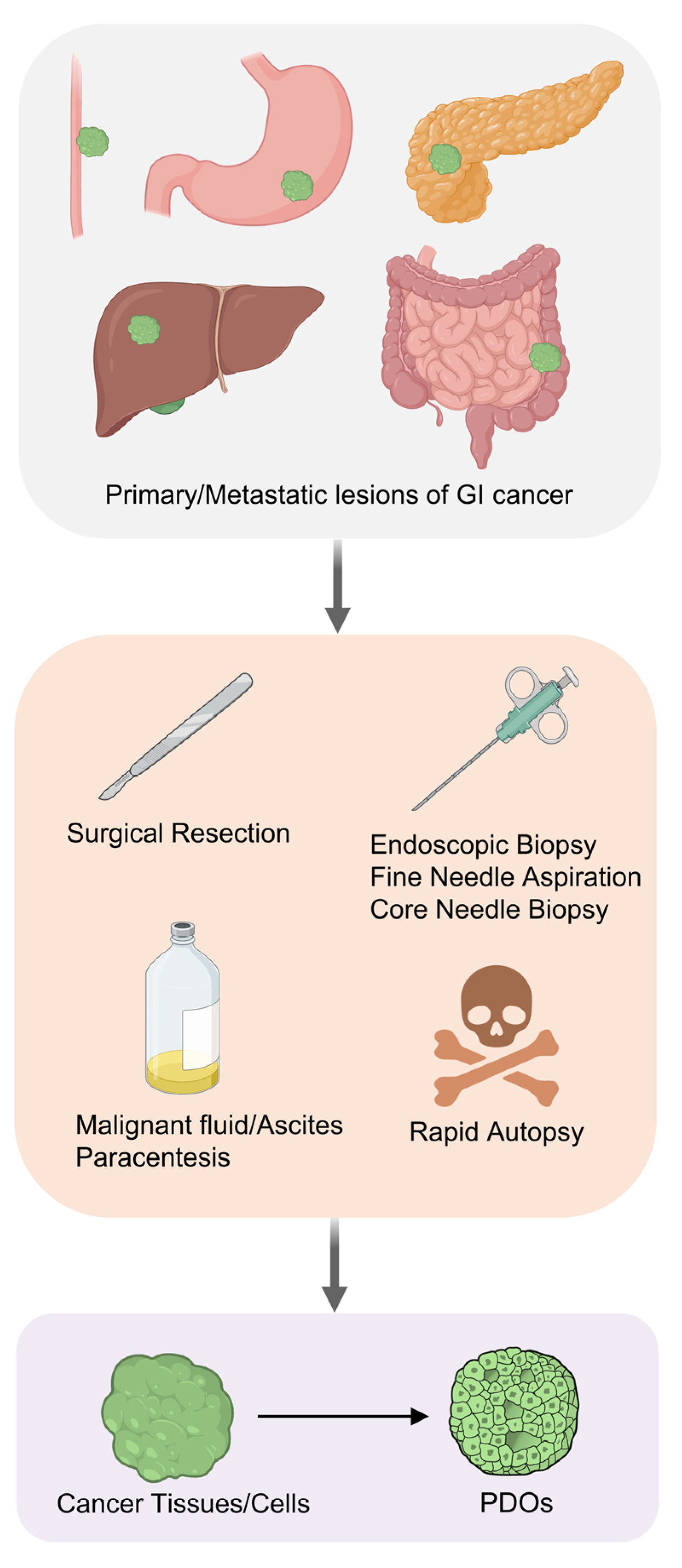
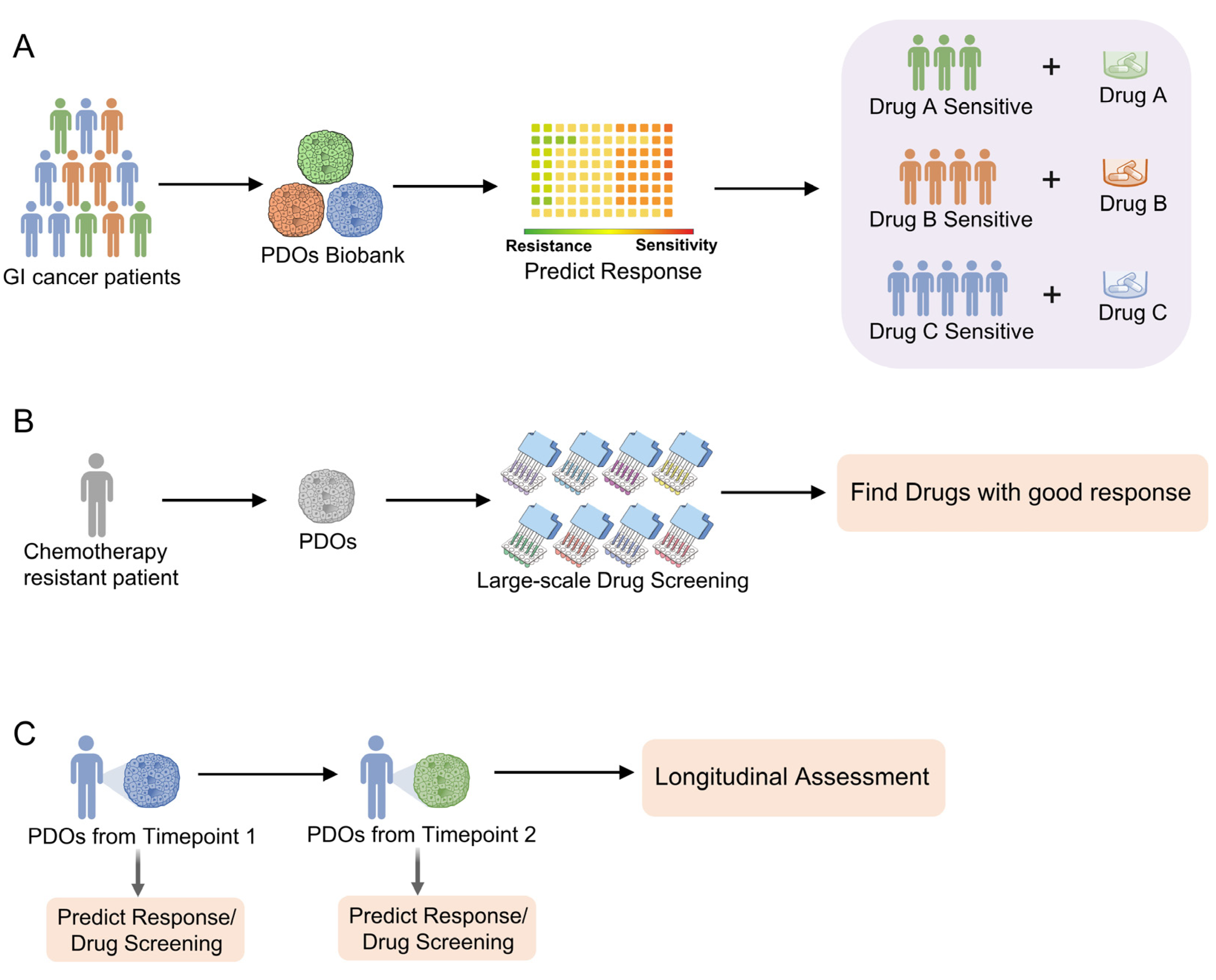
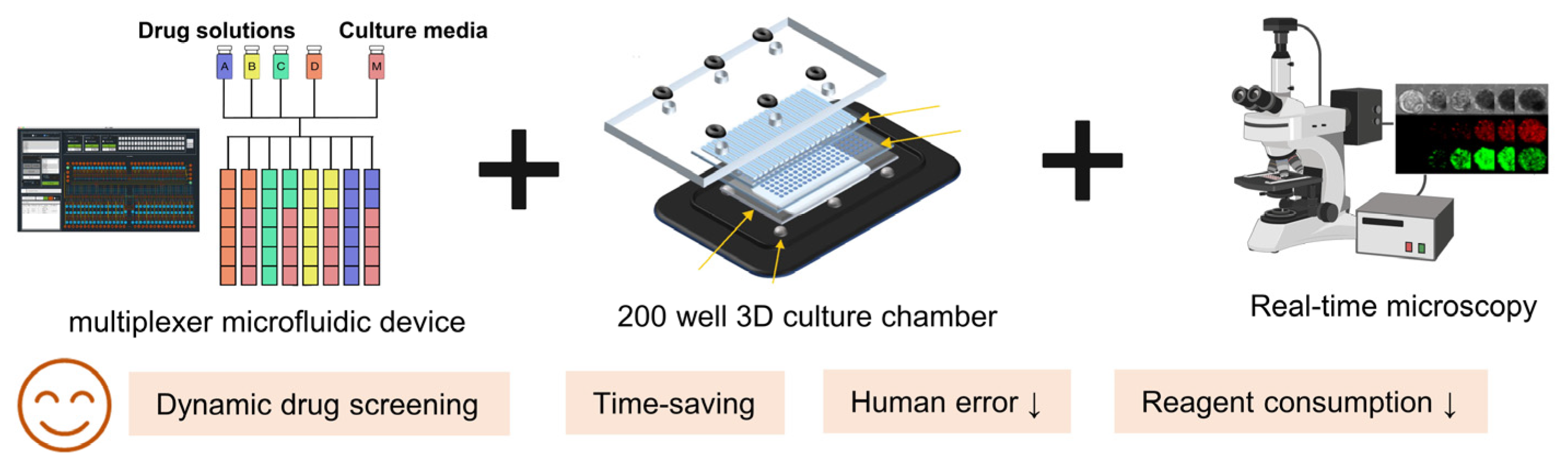

{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Sample | Cell Type | Location | Success Rate | Maintenance | Ref. |
|---|---|---|---|---|---|---|
| Esophageal cancer | SR | EADC | PT | 31% (10/32) | ≥25 passages | [37] |
| Esophageal cancer | EB | EADC | PT | 57.2% (16/28) | N/A | [38] |
| Esophageal cancer | EB | ESCC | PT | 71.4% (15/21) | N/A | [39] |
| Gastric cancer | SR | Adenocarcinoma | PT f | N/A | ≥1 year | [40] |
| Gastric cancer | SR | MSI; EBV; CIN; GS | PT; M | over 50% | ≥6 months | [41] |
| Gastric cancer | SR; EB; Paracentesis | MSI; CIN; GS | PT; M; Ascites | 74.6% (44/59) | ≥3 months | [42] |
| Gastric cancer | EB; US-guided NB; CT-guided NB | N/A | M | N/A | N/A | [43] |
| Liver cancer | SR | HCC; CC; CHC | PT | 44% (8/18) | ≈1year | [44] |
| Liver cancer | SR | HCC; CC | PT | N/A | N/A | [27] |
| Liver cancer | US-guided NB | HCC; CC; LEL-CC | PT | 26% (10/38) | ≥32 weeks | [28] |
| Pancreatic cancer | SR; FNA | PDAC | PT; M | 80% a; 100% b | ≈20 passages | [45] |
| Pancreatic cancer | SR; FNA; Rapid autopsy | PDAC | PT; M g; Ascites | 78.2% c; 71.6% d; 45% e | ≥5 passages | [46] |
| Pancreatic cancer | SR; FNA | ACC; PDAC; Adenosquamous PDAC; IPMN-derived PDAC | PT | 62.7% (52/83) | N/A | [47] |
| Pancreatic cancer | SR; FNA; Paracentesis | PDAC | PT; M | N/A | N/A | [48] |
| Colorectal cancer | SR | N/A | PT | 81.5% (22/27) | N/A | [24] |
| Colorectal cancer | CT-guided NB | N/A | M | 71% (10/14) | N/A | [49] |
| Colorectal cancer | SR; EB | N/A | PT; M | N/A | ≥3 months | [18] |
| Colorectal cancer | EB; US-guided NB; CT-guided NB | N/A | M | N/A | N/A | [43] |
| Colorectal cancer | CT-guided NB | N/A | M h | 63% (40/63) | N/A | [50] |
| Colon adenoma | EB | Tubular adenoma; Tubulovillous adenoma; Sessilie serrated adenoma/polyp | PT | N/A | ≥3 months | [18] |
| Rectal cancer | EB | Adenocarcinoma; Mucinous adenocarcinoma; Signet ring cell carcinoma | PT | 85.7% (96/112) | N/A | [51] |
| Rectal cancer | SR; EB | N/A | PT; M i | 77% (65/84) | N/A | [52] |
| Cancer Type | Embed | Base | Elements | Ref. |
|---|---|---|---|---|
| Esophageal cancer | BME-2 | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-acetylcysteine, Nicotinamide, Noggin, EGF, A83-01, FGF10, Wnt-3A, R-Spondin1, SB202190 | [37] |
| Esophageal cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, Neomycin, Antibiotic-antimycotic, Primocin, HEPES, GlutaMAX, B27, N-acetylcysteine, Noggin, EGF, Gastrin, A83-01, CHIR 99021, Wnt-3A, Rspondin1, SB202190 | [38] |
| Esophageal cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, HEPES, GlutaMAX, B27, N-2, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Wnt-3A, R-Spondin1, SB202190, Y-27632 | [39] |
| Gastric cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, HEPES, GlutaMAX, B27, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A-83-01, Y-27632, FGF10, Wnt-3A, R-Spondin1 | [40] |
| Gastric cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-acetylcysteine, Noggin, EGF, Gastrin, A-83-01, Y-27632, FGF10, Wnt-3A, R-Spondin1, Nutlin3a b | [41] |
| Gastric cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-acetylcysteine, EGF, Gastrin, A83-01, FGF10, Y-27632, Wnt-3A, R-Spondin1 c | [42] |
| Gastric cancer | Matrigel | Advanced DMEM/F12 | Penicillin-streptomycin, L-Glutamine, B27, N-2, Nicotinamide, Noggin, Gastrin, A83-01, R-Spondin1, Y-27632, PGE2, Wnt-3A, R-Spondin1, SB202190, BSA, EGF, FGF10, FGF-basic | [43] |
| Liver cancer | BME-2 | Advanced DMEM/F12 | Penicillin/streptomycin, HEPES, GlutaMAX, B27, N-2, N-acetylcysteine, Nicotinamide, EGF, Gastrin, A83-01, FGF10, Y-27632, FGF10, HGF, Forskolin, Dexamethasone | [44] |
| Liver cancer | Matrigel BME-2 | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-2, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Y-27632, FGF10, HGF, Forskolin, Wnt-3A, R-Spondin1 | [27,28] |
| Pancreatic cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Y-27632, FGF10, Wnt-3A, R-Spondin1 | [45] |
| Pancreatic cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Y-27632, FGF10, Wnt-3A, R-Spondin1, PGE2 | [46] |
| Pancreatic cancer | BME-2 | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Y-27632, FGF10, Wnt-3A, R-spondin1 d | [47] |
| Pancreatic cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, HEPES, GlutaMAX, B27, N-acetylcysteine, Noggin, Gastrin, A83-01, Y27632, Wnt-3A, R-spondin1, SB202190 | [48] |
| Colorectal cancer | BME-2 Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, Primocin, HEPES, GlutaMAX, B27, N-2, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Y-27632, PGE2, R-Spondin1, SB202190 | [24,49] |
| Colorectal cancer and colon adenoma | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, HEPES, GlutaMAX, B27, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Y-27632, Wnt-3A, R-Spondin1, SB202190 a | [18] |
| Colorectal cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, L-Glutamine, B27, N-2, Nicotinamide, Noggin, Gastrin, A83-01, R-Spondin1, Y-27632, PGE2, Wnt-3A, R-Spondin1, SB202190, BSA, EGF, FGF10, FGF-basic | [43] |
| Colorectal cancer | Matrigel | Advanced DMEM/F12 | Penicillin/streptomycin, HEPES, GlutaMAX, B27, N-2, N-acetylcysteine, EGF, A-83-01, Y-27632, SB202190 | [50] |
| Rectal cancer | Matrigel | Advanced DMEM/F12 | Gentamicin/amphoteritin B, Normocin, HEPES, Glutamax B27, N-2, N-acetylcysteine, Nicotinamide, Noggin, EGF, Gastrin, A83-01, Y-27632, PGE2, R-Spondin1, SB202190 | [51] |
| Rectal cancer | Matrigel | Advanced DMEM/F12 | Antibiotic-antimycotic, HEPES, GlutaMAX, B27, N-2, N-acetylcysteine, Nicotinamide, EGF, Gastrin, A83-01, SB202190 | [52] |
| Features | Cell Lines | Patient-Derived Xenografts | Patient-Derived Organoids |
|---|---|---|---|
| Cost | Low | Highest | Moderate |
| Ease of maintenance | Easy to culture | Must be maintained in immunocompromised mice | Variable success rate, easy to culture after establishment |
| Time demand | Low | Highest, serial passages in mice required | Moderate, faster than PDXs |
| Long-term stability | Moderate | High | High |
| In vivo tumor phenotype | Limited predictive power | Highest predictive power with modelling of stromal interactions | High predictive power |
| Tumor genomic spectrum | Low | High degree of tumor heterogeneity | Some degree of tumor heterogeneity |
| High throughput | Readily adopted for high-throughput assays | Not suitable for high-throughput assays | Can be adopted for high-throughput assays |
| Genetic manipulation | Easy | Difficult | Moderate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Yu, J.; Wong, C.C. Gastrointestinal Cancer Patient Derived Organoids at the Frontier of Personalized Medicine and Drug Screening. Cells 2024, 13, 1312. https://doi.org/10.3390/cells13161312
Yang Z, Yu J, Wong CC. Gastrointestinal Cancer Patient Derived Organoids at the Frontier of Personalized Medicine and Drug Screening. Cells. 2024; 13(16):1312. https://doi.org/10.3390/cells13161312
Chicago/Turabian StyleYang, Zhenjie, Jun Yu, and Chi Chun Wong. 2024. "Gastrointestinal Cancer Patient Derived Organoids at the Frontier of Personalized Medicine and Drug Screening" Cells 13, no. 16: 1312. https://doi.org/10.3390/cells13161312