
Spike Protein of SARS-CoV-2 Activates Cardiac Fibrogenesis through NLRP3 Inflammasomes and NF-κB Signaling


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human CFs Cultures
2.2. Scratch Assay
2.3. MTS Proliferation Assay
2.4. Immunoblotting
2.5. TGF-β1 Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. TGF-β1 Real-Time Polymerase Chain Reaction (RT-PCR)
2.7. Statistical Analysis
3. Results
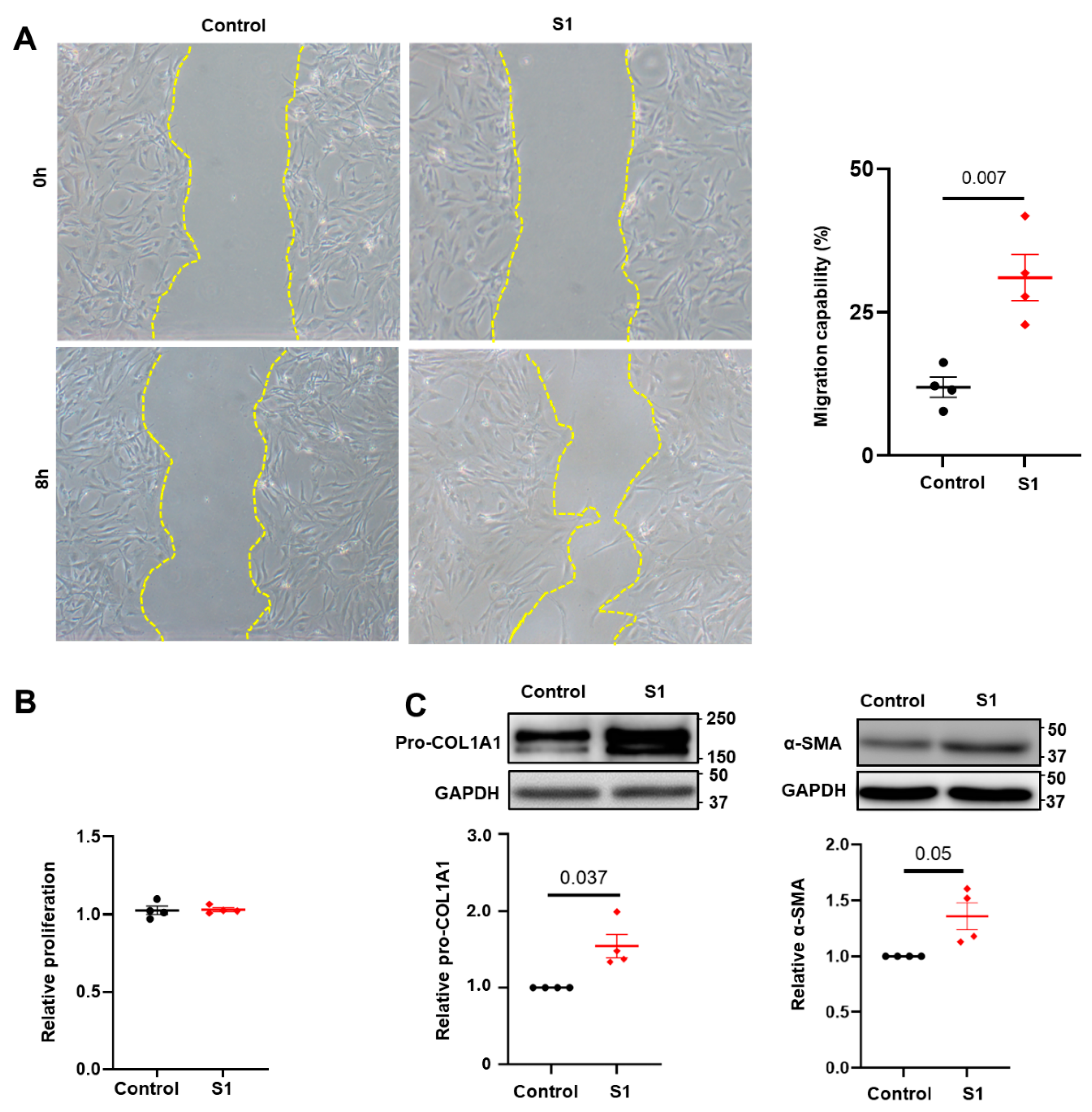
3.1. Effects of S1 Protein on CFs Migration and Proliferation
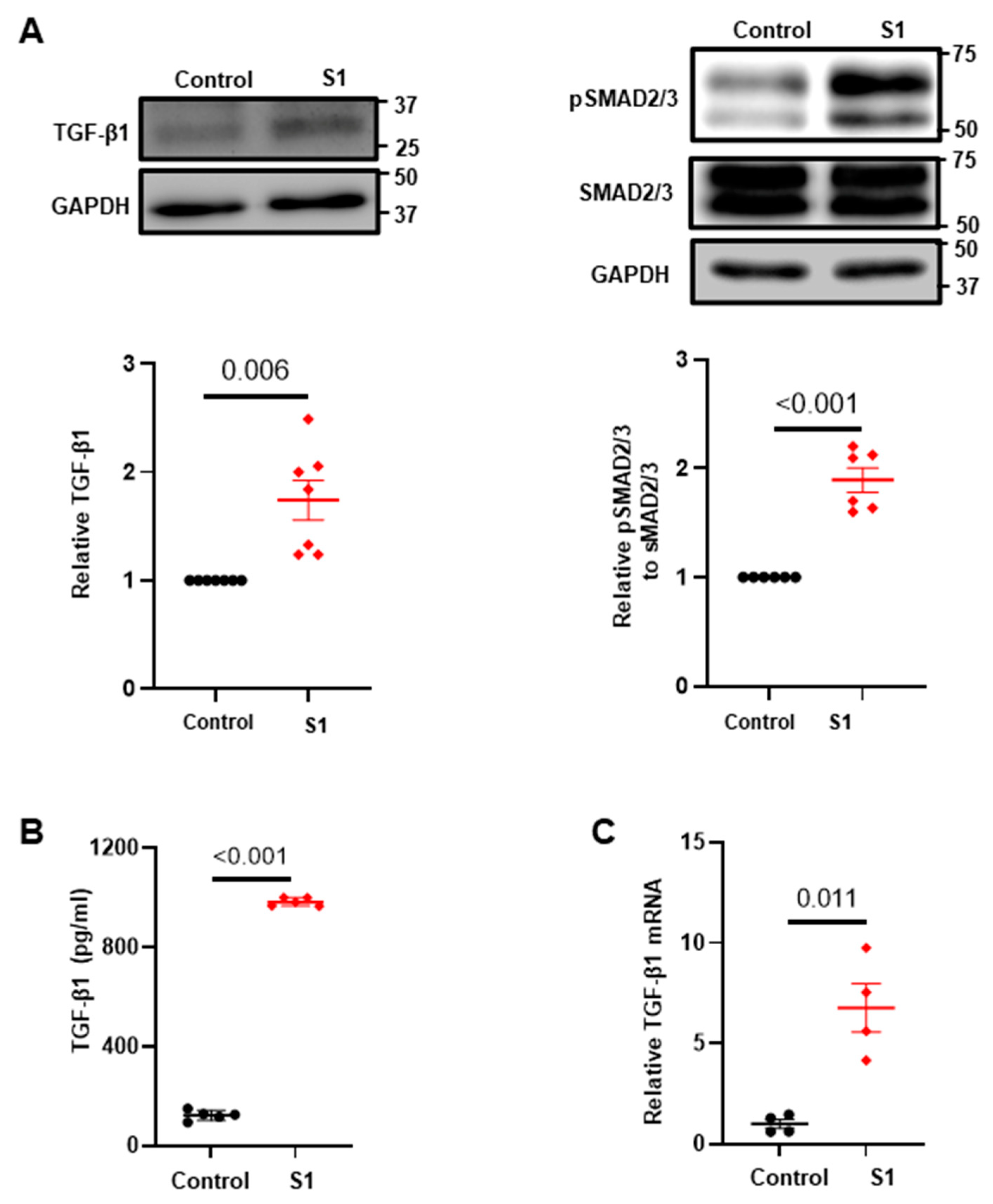
3.2. Effects of S1 Protein on CFs Fibrosis Signaling
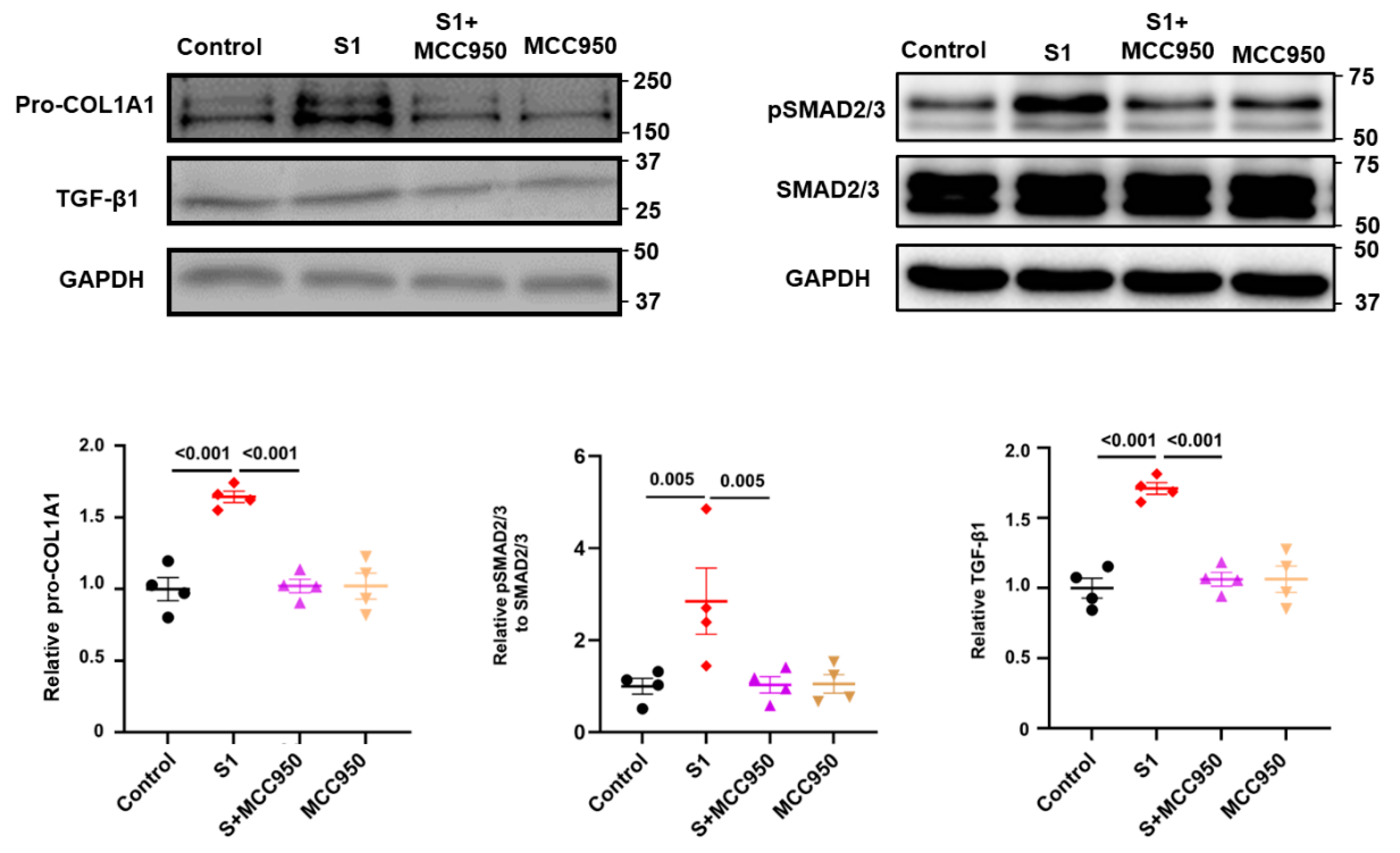
3.3. Role of NLRP3 and TLR4 in S1 Protein-Induced CFs Activation
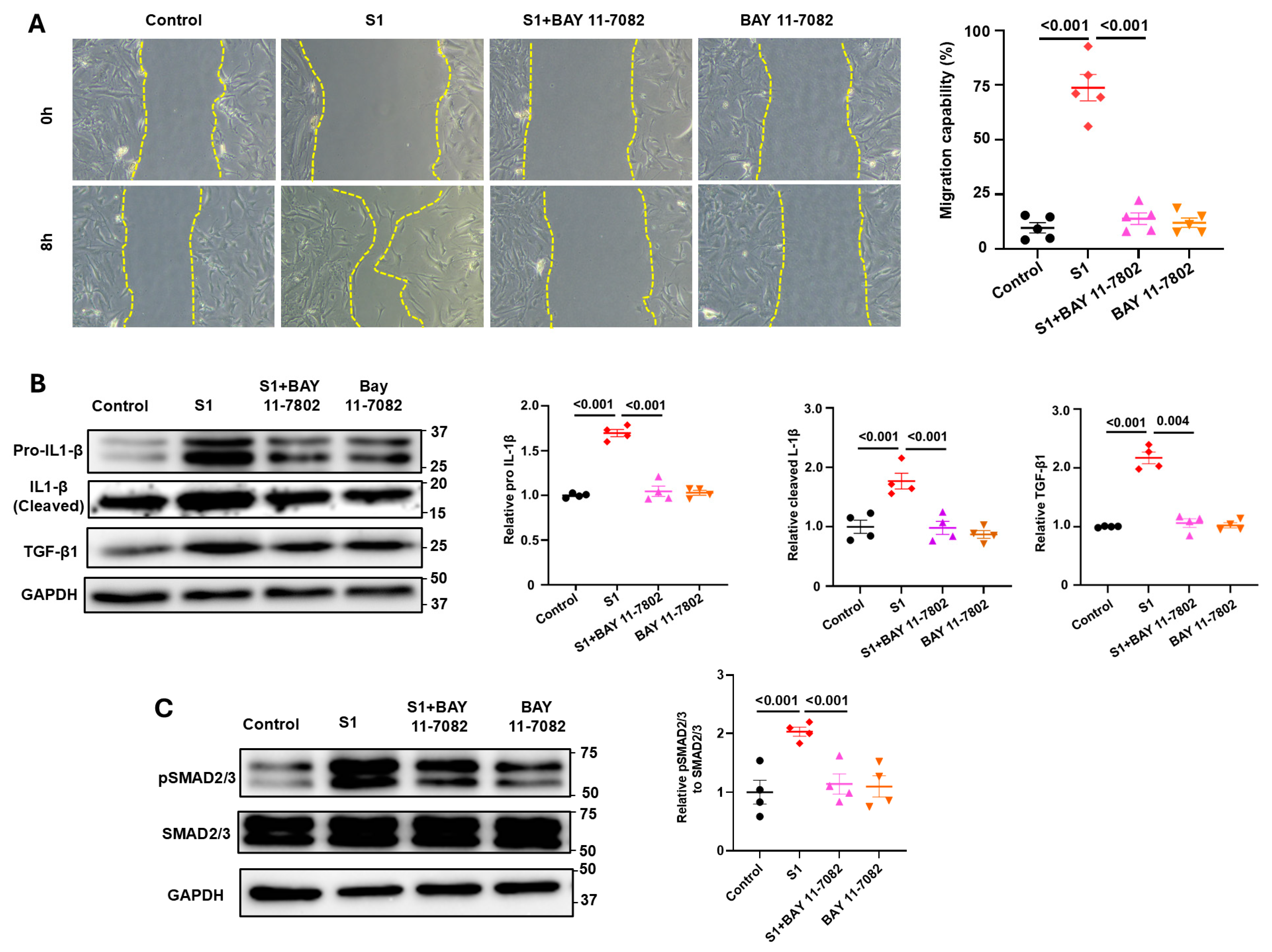
3.4. Role of NF-kB Signaling in S1 Protein-Induced CFs Activation
3.5. S1 Protein Activates CFs in an ACE2-Dependent Manner
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zeng, H.; Jiang, H.; Yang, Y.; Yuan, Z.; Cheng, X.; Jing, Z.; Liu, B.; Chen, J.; Nie, S. CSC expert consensus on principles of clinical management of patients with severe emergent cardiovascular diseases during the COVID-19 epidemic. Circulation 2020, 141, e810–e816. [Google Scholar] [CrossRef] [PubMed]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential effects of coronaviruses on the cardiovascular system: A review. JAMA Cardiol. 2020, 5, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ma, Y.; Zhang, J.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Parhizgar, P.; Yazdankhah, N.; Rzepka, A.M.; Chung, K.Y.C.; Ali, I.; Lai Fat Fur, R.; Russell, V.; Cheung, A.M. Beyond Acute COVID-19: A Review of Long-term Cardiovascular Outcomes. Can. J. Cardiol. 2023, 39, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Raafs, A.G.; Ghossein, M.A.; Brandt, Y.; Henkens, M.; Kooi, M.E.; Vernooy, K.; Spaanderman, M.E.A.; Gerretsen, S.; van Santen, S.; Driessen, R.G.H.; et al. Cardiovascular outcome 6 months after severe coronavirus disease 2019 infection. J. Hypertens. 2022, 40, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Ellenberger, K.A.; Phetsouphanh, C.; Kelleher, A.P.; Matthews, G.V.; Darley, D.R.; Holloway, C.J. Myocardial fibrosis occurs in non-hospitalised patients with chronic symptoms after COVID-19. Int. J. Cardiol. Heart Vasc. 2022, 39, 100964. [Google Scholar] [CrossRef] [PubMed]
- Boretti, A. PQQ Supplementation and SARS-CoV-2 Spike Protein-Induced Heart Inflammation. Nat. Prod. Commun. 2022, 17, 1934578x221080929. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Nguyen, V.; Tsai, J.; Gao, C.; Tian, Y.; Zhang, Y.; Carver, W.; Kiaris, H.; Cui, T.; Tan, W. The SARS-CoV-2 spike protein induces long-term transcriptional perturbations of mitochondrial metabolic genes, causes cardiac fibrosis, and reduces myocardial contractile in obese mice. Mol. Metab. 2023, 74, 101756. [Google Scholar] [CrossRef]
- Huynh, T.V.; Rethi, L.; Lee, T.W.; Higa, S.; Kao, Y.H.; Chen, Y.J. Spike Protein Impairs Mitochondrial Function in Human Cardiomyocytes: Mechanisms Underlying Cardiac Injury in COVID-19. Cells 2023, 12, 877. [Google Scholar] [CrossRef]
- Clemens, D.J.; Ye, D.; Zhou, W.; Kim, C.S.J.; Pease, D.R.; Navaratnarajah, C.K.; Barkhymer, A.; Tester, D.J.; Nelson, T.J.; Cattaneo, R.; et al. SARS-CoV-2 spike protein-mediated cardiomyocyte fusion may contribute to increased arrhythmic risk in COVID-19. PLoS ONE 2023, 18, e0282151. [Google Scholar] [CrossRef]
- Liu, H.; Gai, S.; Wang, X.; Zeng, J.; Sun, C.; Zhao, Y.; Zheng, Z. Single-cell analysis of SARS-CoV-2 receptor ACE2 and spike protein priming expression of proteases in the human heart. Cardiovasc. Res. 2020, 116, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Avolio, E.; Srivastava, P.K.; Ji, J.; Carrabba, M.; Tsang, C.T.W.; Gu, Y.; Thomas, A.C.; Gupta, K.; Berger, I.; Emanueli, C.; et al. Murine studies and expressional analyses of human cardiac pericytes reveal novel trajectories of SARS-CoV-2 Spike protein-induced microvascular damage. Signal Transduct. Target. Ther. 2023, 8, 232. [Google Scholar] [CrossRef]
- Kato, Y.; Nishiyama, K.; Man Lee, J.; Ibuki, Y.; Imai, Y.; Noda, T.; Kamiya, N.; Kusakabe, T.; Kanda, Y.; Nishida, M. TRPC3-Nox2 Protein Complex Formation Increases the Risk of SARS-CoV-2 Spike Protein-Induced Cardiomyocyte Dysfunction through ACE2 Upregulation. Int. J. Mol. Sci. 2022, 24, 102. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z. More than a key-the pathological roles of SARS-CoV-2 spike protein in COVID-19 related cardiac injury. Sports Med. Health Sci. 2023, in press. [Google Scholar] [CrossRef]
- Avolio, E.; Carrabba, M.; Milligan, R.; Kavanagh Williamson, M.; Beltrami, A.P.; Gupta, K.; Elvers, K.T.; Gamez, M.; Foster, R.R.; Gillespie, K.; et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: A potential non-infective mechanism of COVID-19 microvascular disease. Clin. Sci. 2021, 135, 2667–2689. [Google Scholar] [CrossRef] [PubMed]
- Borkotoky, S.; Dey, D.; Hazarika, Z. Interactions of angiotensin-converting enzyme-2 (ACE2) and SARS-CoV-2 spike receptor-binding domain (RBD): A structural perspective. Mol. Biol. Rep. 2023, 50, 2713–2721. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Wang, K.; Viveiros, A.; Kellner, M.J.; Penninger, J.M. Angiotensin-converting enzyme 2-at the heart of the COVID-19 pandemic. Cell 2023, 186, 906–922. [Google Scholar] [CrossRef]
- Babadaei, M.M.N.; Hasan, A.; Bloukh, S.H.; Edis, Z.; Sharifi, M.; Kachooei, E.; Falahati, M. The expression level of angiotensin-converting enzyme 2 determines the severity of COVID-19: Lung and heart tissue as targets. J. Biomol. Struct. Dyn. 2021, 39, 3780–3786. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.L.; Lambert, D.W.; Turner, A.J.; Porter, K.E. Functional angiotensin-converting enzyme 2 is expressed in human cardiac myofibroblasts. Exp. Physiol. 2008, 93, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef] [PubMed]
- Wicherska-Pawłowska, K.; Wróbel, T.; Rybka, J. Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int. J. Mol. Sci. 2021, 22, 13397. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Gong, Z.; Zhang, S.; Cao, J.; Mao, W.; Yao, Y.; Zhao, J.; Li, Q.; Liu, K.; Liu, B.; et al. Besides TLR2 and TLR4, NLRP3 is also involved in regulating Escherichia coli infection-induced inflammatory responses in mice. Int. Immunopharmacol. 2023, 121, 110556. [Google Scholar] [CrossRef]
- Albornoz, E.A.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Aguado, J.; Zamora, A.P.; McMillan, C.L.D.; Liang, B.; et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol. Psychiatry 2023, 28, 2878–2893. [Google Scholar] [CrossRef]
- Del Re, A.; Corpetti, C.; Pesce, M.; Seguella, L.; Steardo, L.; Palenca, I.; Rurgo, S.; De Conno, B.; Sarnelli, G.; Esposito, G. Ultramicronized Palmitoylethanolamide Inhibits NLRP3 Inflammasome Expression and Pro-Inflammatory Response Activated by SARS-CoV-2 Spike Protein in Cultured Murine Alveolar Macrophages. Metabolites 2021, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef]
- Corpetti, C.; Del Re, A.; Seguella, L.; Palenca, I.; Rurgo, S.; De Conno, B.; Pesce, M.; Sarnelli, G.; Esposito, G. Cannabidiol inhibits SARS-Cov-2 spike (S) protein-induced cytotoxicity and inflammation through a PPARγ-dependent TLR4/NLRP3/Caspase-1 signaling suppression in Caco-2 cell line. Phytother. Res. 2021, 35, 6893–6903. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- World Health Organization. Data. Who. int, WHO Coronavirus (COVID-19) Dashboard Cases [Dashboard]. Available online: https://data.who.int/dashboards/covid19/cases (accessed on 24 March 2024).
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Liu, F.; Liu, F.; Wang, L. COVID-19 and cardiovascular diseases. J. Mol. Cell Biol. 2021, 13, 161–167. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Cai, Y.; Liu, T.; Shen, B.; Yang, F.; Cao, S.; Liu, X.; Xiang, Y.; Zhao, Q. Characteristics and clinical significance of myocardial injury in patients with severe coronavirus disease 2019. Eur. Heart J. 2020, 41, 2070–2079. [Google Scholar] [CrossRef]
- Kole, C.; Stefanou, Ε.; Karvelas, N.; Schizas, D.; Toutouzas, K.P. Acute and Post-Acute COVID-19 Cardiovascular Complications: A Comprehensive Review. Cardiovasc. Drugs Ther. 2023, 1–16. [Google Scholar] [CrossRef]
- Imig, J.D. SARS-CoV-2 spike protein causes cardiovascular disease independent of viral infection. Clin. Sci. 2022, 136, 431–434. [Google Scholar] [CrossRef]
- Huang, X.; Huang, B.; He, Y.; Feng, L.; Shi, J.; Wang, L.; Peng, J.; Chen, Y. Sars-Cov-2 Spike Protein-Induced Damage of hiPSC-Derived Cardiomyocytes. Adv. Biol. 2022, 6, e2101327. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Turner, N.A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef]
- Bai, Y.J.; Li, Z.G.; Liu, W.H.; Gao, D.; Zhang, P.Y.; Liu, M. Effects of IL-1β and IL-18 induced by NLRP3 inflammasome activation on myocardial reperfusion injury after PCI. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10101–10106. [Google Scholar] [CrossRef] [PubMed]
- Segura-Villalobos, D.; Roa-Velázquez, D.; Zavala-Vargas, D.I.; Filisola-Villaseñor, J.G.; Castillo Arellano, J.I.; Morales Ríos, E.; Reyes-Chilpa, R.; González-Espinosa, C. Jacareubin inhibits TLR4-induced lung inflammatory response caused by the RBD domain of SARS-CoV-2 Spike protein. Pharmacol. Rep. 2022, 74, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Dantas, F.L.; Fernandes, G.G.; Gutman, E.G.; De Lima, E.V.; Antonio, L.S.; Hammerle, M.B.; Mota-Araujo, H.P.; Colodeti, L.C.; Araújo, S.M.B.; Froz, G.M.; et al. SARS-CoV-2 Spike protein induces TLR4-mediated long-term cognitive dysfunction recapitulating post-COVID-19 syndrome in mice. Cell Rep. 2023, 42, 112189. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Bao, C.; Yang, Z.; Liu, S.; Sun, Y.; Cao, W.; Wang, T.; Schwantes-An, T.-H.; Choy, J.S.; Naidu, S.; et al. SARS-CoV-2 spike protein induces IL-18-mediated cardiopulmonary inflammation via reduced mitophagy. Signal Transduct. Target. Ther. 2023, 8, 108. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Tin, H.; Rethi, L.; Higa, S.; Kao, Y.-H.; Chen, Y.-J. Spike Protein of SARS-CoV-2 Activates Cardiac Fibrogenesis through NLRP3 Inflammasomes and NF-κB Signaling. Cells 2024, 13, 1331. https://doi.org/10.3390/cells13161331
Van Tin H, Rethi L, Higa S, Kao Y-H, Chen Y-J. Spike Protein of SARS-CoV-2 Activates Cardiac Fibrogenesis through NLRP3 Inflammasomes and NF-κB Signaling. Cells. 2024; 13(16):1331. https://doi.org/10.3390/cells13161331
Chicago/Turabian StyleVan Tin, Huynh, Lekha Rethi, Satoshi Higa, Yu-Hsun Kao, and Yi-Jen Chen. 2024. "Spike Protein of SARS-CoV-2 Activates Cardiac Fibrogenesis through NLRP3 Inflammasomes and NF-κB Signaling" Cells 13, no. 16: 1331. https://doi.org/10.3390/cells13161331
APA StyleVan Tin, H., Rethi, L., Higa, S., Kao, Y.-H., & Chen, Y.-J. (2024). Spike Protein of SARS-CoV-2 Activates Cardiac Fibrogenesis through NLRP3 Inflammasomes and NF-κB Signaling. Cells, 13(16), 1331. https://doi.org/10.3390/cells13161331