

Molecular Mechanisms of Autophagy Decline during Aging


Abstract
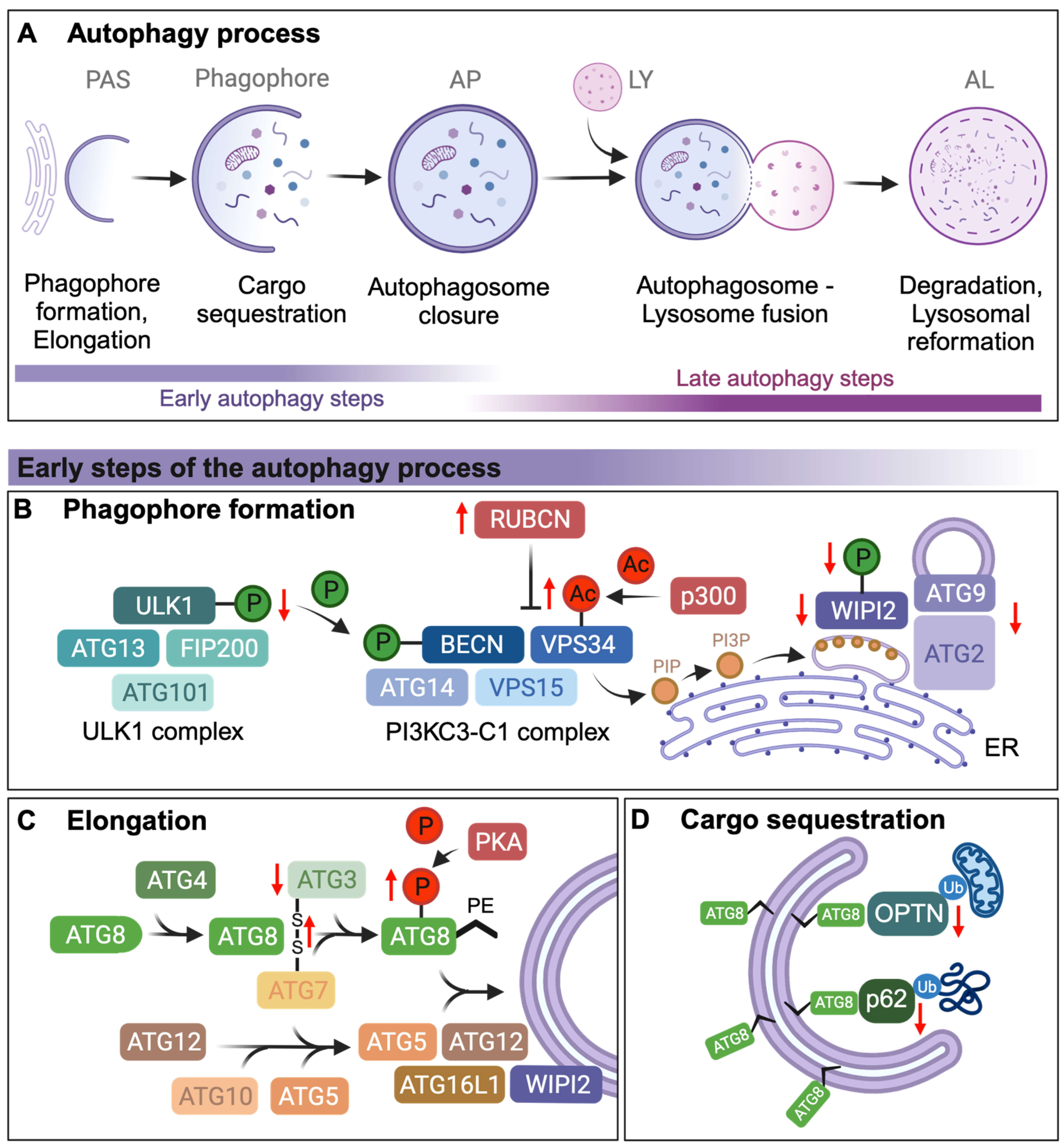
:1. Introduction
2. Autophagosome Formation and Elongation
3. Cargo Sequestration via Autophagy Receptors
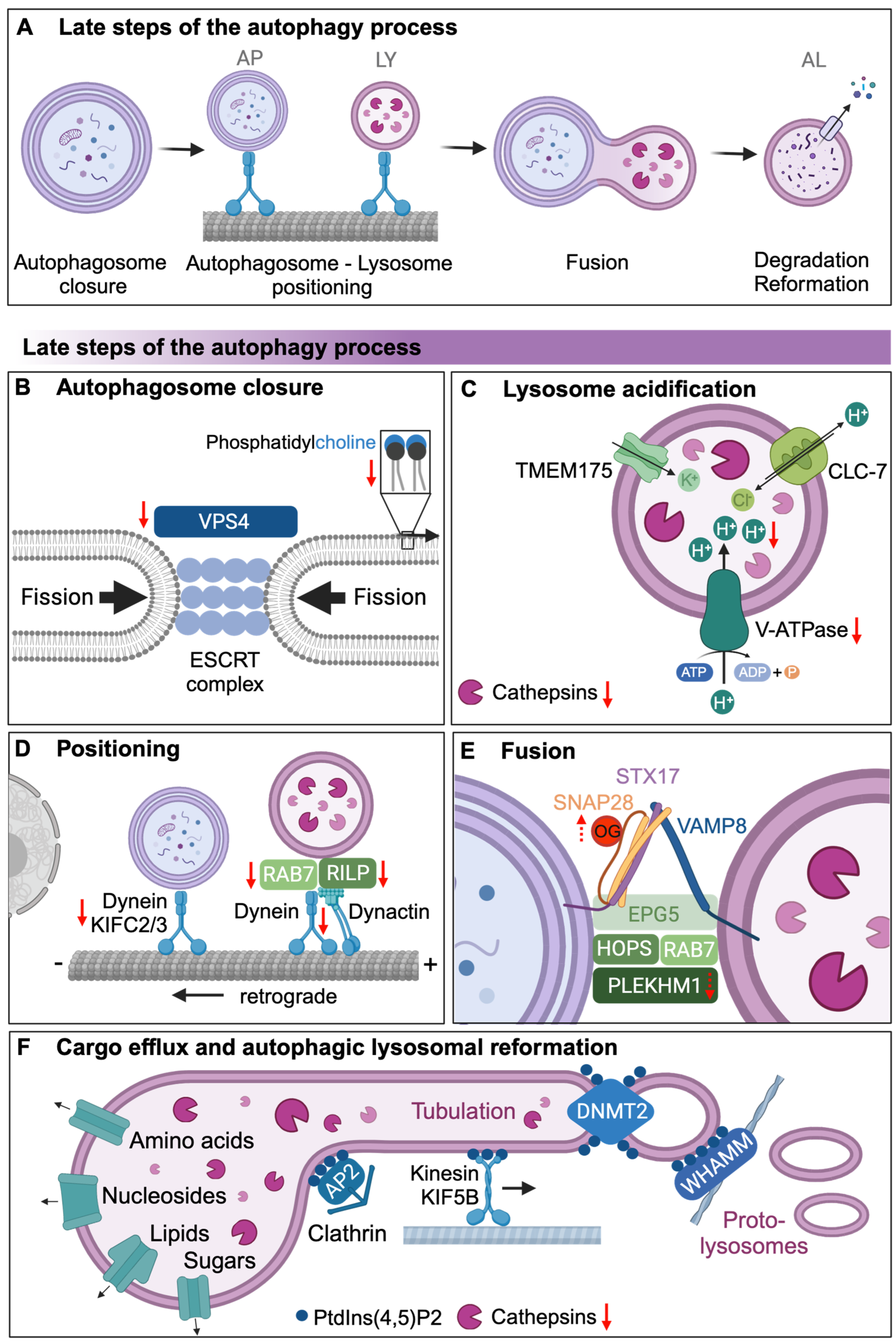
4. Autophagosomal Closure and Autophagosomal Lysosomal Fusion
5. Lysosomal Degradation of Autophagic Cargo and Autophagic Lysosomal Reformation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, M.; Zhao, D.; Li, X.; Yang, C.; Wang, X. Lysosome activity is modulated by multiple longevity pathways and is important for lifespan extension in C. elegans. eLife 2020, 9, e55745. [Google Scholar] [CrossRef]
- Gelino, S.; Chang, J.T.; Kumsta, C.; She, X.; Davis, A.; Nguyen, C.; Panowski, S.; Hansen, M. Intestinal Autophagy Improves Healthspan and Longevity in C. elegans during Dietary Restriction. PLoS Genet. 2016, 12, e1006135. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.F.; Mazzotti, D.R.; Cendoroglo, M.S.; Ramos, L.R.; Tufik, S.; Silva, V.C.D.; D’Almeida, V. Genes related to maintenance of autophagy and successful aging. Arq. Neuro-Psiquiatr. 2018, 76, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Ott, C.; Konig, J.; Hohn, A.; Jung, T.; Grune, T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016, 10, 266–273. [Google Scholar] [CrossRef]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef]
- Chang, J.T.; Kumsta, C.; Hellman, A.B.; Adams, L.M.; Hansen, M. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife 2017, 6, e18459. [Google Scholar] [CrossRef]
- Jahanian, S.; Pareja-Cajiao, M.; Gransee, H.M.; Sieck, G.C.; Mantilla, C.B. Autophagy markers LC3 and p62 in aging lumbar motor neurons. Exp. Gerontol. 2024, 194, 112483. [Google Scholar] [CrossRef]
- Stavoe, A.K.; Gopal, P.P.; Gubas, A.; Tooze, S.A.; Holzbaur, E.L. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. eLife 2019, 8, e44219. [Google Scholar] [CrossRef]
- Pitcairn, C.; Murata, N.; Zalon, A.J.; Stojkovska, I.; Mazzulli, J.R. Impaired Autophagic-Lysosomal Fusion in Parkinson’s Patient Midbrain Neurons Occurs through Loss of ykt6 and Is Rescued by Farnesyltransferase Inhibition. J. Neurosci. 2023, 43, 2615–2629. [Google Scholar] [CrossRef]
- Nichenko, A.S.; Sorensen, J.R.; Southern, W.M.; Qualls, A.E.; Schifino, A.G.; McFaline-Figueroa, J.; Blum, J.E.; Tehrani, K.F.; Yin, H.; Mortensen, L.J.; et al. Lifelong Ulk1-Mediated Autophagy Deficiency in Muscle Induces Mitochondrial Dysfunction and Contractile Weakness. Int. J. Mol. Sci. 2021, 22, 1937. [Google Scholar] [CrossRef]
- Sen, P.; Lan, Y.; Li, C.Y.; Sidoli, S.; Donahue, G.; Dou, Z.; Frederick, B.; Chen, Q.; Luense, L.J.; Garcia, B.A.; et al. Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol. Cell 2019, 73, 684–698. [Google Scholar] [CrossRef]
- Lessard-Beaudoin, M.; Laroche, M.; Loudghi, A.; Demers, M.J.; Denault, J.B.; Grenier, G.; Riechers, S.P.; Wanker, E.E.; Graham, R.K. Organ-specific alteration in caspase expression and STK3 proteolysis during the aging process. Neurobiol. Aging 2016, 47, 50–62. [Google Scholar] [CrossRef]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef]
- Cherra, S.J., 3rd; Kulich, S.M.; Uechi, G.; Balasubramani, M.; Mountzouris, J.; Day, B.W.; Chu, C.T. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 2010, 190, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, R.; Rana, A.; Walker, D.W. Upregulation of the Autophagy Adaptor p62/SQSTM1 Prolongs Health and Lifespan in Middle-Aged Drosophila. Cell Rep. 2019, 28, 1029–1040. [Google Scholar] [CrossRef]
- Kwon, J.; Han, E.; Bui, C.B.; Shin, W.; Lee, J.; Lee, S.; Choi, Y.B.; Lee, A.H.; Lee, K.H.; Park, C.; et al. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012, 13, 150–156. [Google Scholar] [CrossRef]
- Roca-Agujetas, V.; Barbero-Camps, E.; de Dios, C.; Podlesniy, P.; Abadin, X.; Morales, A.; Mari, M.; Trullas, R.; Colell, A. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 15. [Google Scholar] [CrossRef]
- Rempel, I.L.; Crane, M.M.; Thaller, D.J.; Mishra, A.; Jansen, D.P.; Janssens, G.; Popken, P.; Aksit, A.; Kaeberlein, M.; van der Giessen, E.; et al. Age-dependent deterioration of nuclear pore assembly in mitotic cells decreases transport dynamics. eLife 2019, 8, e48186. [Google Scholar] [CrossRef]
- Rahmatpanah, F.; Agrawal, S.; Scarfone, V.M.; Kapadia, S.; Mercola, D.; Agrawal, A. Transcriptional Profiling of Age-Associated Gene Expression Changes in Human Circulatory CD1c+ Myeloid Dendritic Cell Subset. J. Gerontol. Ser. A 2019, 74, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Szinyakovics, J.; Keresztes, F.; Kiss, E.A.; Falcsik, G.; Vellai, T.; Kovacs, T. Potent New Targets for Autophagy Enhancement to Delay Neuronal Ageing. Cells 2023, 12, 1753. [Google Scholar] [CrossRef]
- Kimura, N.; Samura, E.; Suzuki, K.; Okabayashi, S.; Shimozawa, N.; Yasutomi, Y. Dynein Dysfunction Reproduces Age-Dependent Retromer Deficiency: Concomitant Disruption of Retrograde Trafficking Is Required for Alteration in beta-Amyloid Precursor Protein Metabolism. Am. J. Pathol. 2016, 186, 1952–1966. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Hughes, A.L.; Gottschling, D.E. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 2012, 492, 261–265. [Google Scholar] [CrossRef]
- Baxi, K.; Ghavidel, A.; Waddell, B.; Harkness, T.A.; de Carvalho, C.E. Regulation of Lysosomal Function by the DAF-16 Forkhead Transcription Factor Couples Reproduction to Aging in Caenorhabditis elegans. Genetics 2017, 207, 83–101. [Google Scholar] [CrossRef]
- Truschel, S.T.; Clayton, D.R.; Beckel, J.M.; Yabes, J.G.; Yao, Y.; Wolf-Johnston, A.; Birder, L.A.; Apodaca, G. Age-related endolysosome dysfunction in the rat urothelium. PLoS ONE 2018, 13, e0198817. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Volinia, S.; Dhand, R.; Vanhaesebroeck, B.; MacDougall, L.K.; Stein, R.; Zvelebil, M.J.; Domin, J.; Panaretou, C.; Waterfield, M.D. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 1995, 14, 3339–3348. [Google Scholar] [CrossRef]
- Su, H.; Yang, F.; Wang, Q.; Shen, Q.; Huang, J.; Peng, C.; Zhang, Y.; Wan, W.; Wong, C.C.L.; Sun, Q.; et al. VPS34 Acetylation Controls Its Lipid Kinase Activity and the Initiation of Canonical and Non-canonical Autophagy. Mol. Cell 2017, 67, 907–921. [Google Scholar] [CrossRef]
- Tezil, T.; Chamoli, M.; Ng, C.P.; Simon, R.P.; Butler, V.J.; Jung, M.; Andersen, J.; Kao, A.W.; Verdin, E. Lifespan-increasing drug nordihydroguaiaretic acid inhibits p300 and activates autophagy. NPJ Aging Mech. Dis. 2019, 5, 7. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Allison, D.B.; Ames, B.N.; Astle, C.M.; Atamna, H.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nadon, N.L.; et al. Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 2014, 13, 273–282. [Google Scholar] [CrossRef]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.S.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an alpha-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef]
- Strong, R.; Miller, R.A.; Astle, C.M.; Floyd, R.A.; Flurkey, K.; Hensley, K.L.; Javors, M.A.; Leeuwenburgh, C.; Nelson, J.F.; Ongini, E.; et al. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell 2008, 7, 641–650. [Google Scholar] [CrossRef]
- Economos, A.C.; Ballard, R.C.; Miquel, J.; Binnard, R.; Philpott, D.E. Accelerated aging of fasted Drosophila. Preservation of physiological function and cellular fine structure by thiazolidine carboxylic acid (TCA). Exp. Gerontol. 1982, 17, 105–114. [Google Scholar] [CrossRef]
- Richie, J.P., Jr.; Mills, B.J.; Lang, C.A. Dietary nordihydroguaiaretic acid increases the life span of the mosquito. Proc. Soc. Exp. Biol. Med. 1986, 183, 81–85. [Google Scholar] [CrossRef]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Leidal, A.M.; Debnath, J.; Hansen, M. Beyond Autophagy: The Expanding Roles of ATG8 Proteins. Trends Biochem. Sci. 2021, 46, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.S.; Jariwala, J.S.; Anderson, E.; Mitra, K.; Meisenhelder, J.; Chang, J.T.; Ideker, T.; Hunter, T.; Nizet, V.; Dillin, A.; et al. Phosphorylation of LC3 by the Hippo kinases STK3/STK4 is essential for autophagy. Mol. Cell 2015, 57, 55–68. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Shanahan, S.L.; Chassefeyre, R.; Chaiamarit, T.; Zaretski, S.; Landeras-Bueno, S.; Verhelle, A.; Encalada, S.E.; Hansen, M. LC3B phosphorylation regulates FYCO1 binding and directional transport of autophagosomes. Curr. Biol. 2021, 31, 3440–3449. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villen, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Nah, J.; Zablocki, D.; Sadoshima, J. The roles of the inhibitory autophagy regulator Rubicon in the heart: A new therapeutic target to prevent cardiac cell death. Exp. Mol. Med. 2021, 53, 528–536. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, J.; Fan, W.; Wong, K.N.; Ding, X.; Chen, S.; Zhong, Q. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J. Biol. Chem. 2011, 286, 185–191. [Google Scholar] [CrossRef]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef]
- Li, L.; Tong, M.; Fu, Y.; Chen, F.; Zhang, S.; Chen, H.; Ma, X.; Li, D.; Liu, X.; Zhong, Q. Lipids and membrane-associated proteins in autophagy. Protein Cell 2021, 12, 520–544. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Nishimura, T.; Sakamaki, Y.; Koyama-Honda, I.; Yamamoto, H.; Mizushima, N. Differential requirement for ATG2A domains for localization to autophagic membranes and lipid droplets. FEBS Lett. 2017, 591, 3819–3830. [Google Scholar] [CrossRef]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef]
- Nagy, P.; Hegedus, K.; Pircs, K.; Varga, A.; Juhasz, G. Different effects of Atg2 and Atg18 mutations on Atg8a and Atg9 trafficking during starvation in Drosophila. FEBS Lett. 2014, 588, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Z.; Hu, W.; Ren, H.; Tian, E.; Zhao, Y.; Lu, Q.; Huang, X.; Yang, P.; Li, X.; et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010, 141, 1042–1055. [Google Scholar] [CrossRef]
- Xu, P.; Damschroder, D.; Zhang, M.; Ryall, K.A.; Adler, P.N.; Saucerman, J.J.; Wessells, R.J.; Yan, Z. Atg2, Atg9 and Atg18 in mitochondrial integrity, cardiac function and healthspan in Drosophila. J. Mol. Cell Cardiol. 2019, 127, 116–124. [Google Scholar] [CrossRef]
- Liang, W.; Moyzis, A.G.; Lampert, M.A.; Diao, R.Y.; Najor, R.H.; Gustafsson, A.B. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell 2020, 19, e13187. [Google Scholar] [CrossRef]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Sou, Y.S.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef]
- Ramos, B.P.; Birnbaum, S.G.; Lindenmayer, I.; Newton, S.S.; Duman, R.S.; Arnsten, A.F. Dysregulation of protein kinase a signaling in the aged prefrontal cortex: New strategy for treating age-related cognitive decline. Neuron 2003, 40, 835–845. [Google Scholar] [CrossRef]
- Enns, L.C.; Ladiges, W. Protein kinase A signaling as an anti-aging target. Ageing Res. Rev. 2010, 9, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Enns, L.C.; Morton, J.F.; Treuting, P.R.; Emond, M.J.; Wolf, N.S.; Dai, D.F.; McKnight, G.S.; Rabinovitch, P.S.; Ladiges, W.C. Disruption of protein kinase A in mice enhances healthy aging. PLoS ONE 2009, 4, e5963. [Google Scholar] [CrossRef]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Mills, J. Non-canonical autophagy in aging and age-related diseases. Front. Cell Dev. Biol. 2023, 11, 1137870. [Google Scholar] [CrossRef]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef]
- Yang, Y.; Arnold, M.L.; Lange, C.M.; Sun, L.H.; Broussalian, M.; Doroodian, S.; Ebata, H.; Choy, E.H.; Poon, K.; Moreno, T.M.; et al. Autophagy protein ATG-16.2 and its WD40 domain mediate the beneficial effects of inhibiting early-acting autophagy genes in C. elegans neurons. Nat. Aging 2024, 4, 198–212. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Papandreou, M.E.; Tavernarakis, N. Selective Autophagy as a Potential Therapeutic Target in Age-Associated Pathologies. Metabolites 2021, 11, 588. [Google Scholar] [CrossRef]
- Du, Y.; Wooten, M.C.; Gearing, M.; Wooten, M.W. Age-associated oxidative damage to the p62 promoter: Implications for Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 492–501. [Google Scholar] [CrossRef]
- Du, Y.; Wooten, M.C.; Wooten, M.W. Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol. Dis. 2009, 35, 302–310. [Google Scholar] [CrossRef]
- Kumsta, C.; Chang, J.T.; Lee, R.; Tan, E.P.; Yang, Y.; Loureiro, R.; Choy, E.H.; Lim, S.H.Y.; Saez, I.; Springhorn, A.; et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat. Commun. 2019, 10, 5648. [Google Scholar] [CrossRef]
- Schmid, E.T.; Pyo, J.H.; Walker, D.W. Neuronal induction of BNIP3-mediated mitophagy slows systemic aging in Drosophila. Nat. Aging 2022, 2, 494–507. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Polyansky, A.; Shatz, O.; Fraiberg, M.; Shimoni, E.; Dadosh, T.; Mari, M.; Reggiori, F.M.; Qin, C.; Han, X.; Elazar, Z. Phospholipid imbalance impairs autophagosome completion. EMBO J. 2022, 41, e110771. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Ruiz-Gutierrez, V.; Sobrino, F.; Santa-Maria, C. Age-related changes in membrane lipid composition, fluidity and respiratory burst in rat peritoneal neutrophils. Clin. Exp. Immunol. 2001, 124, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, W.; Shin, N.K.; Bae, Y.M.; Wie, J. Unveiling the impact of lysosomal ion channels: Balancing ion signaling and disease pathogenesis. Korean J. Physiol. Pharmacol. 2023, 27, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Sarkis, G.J.; Ashcom, J.D.; Hawdon, J.M.; Jacobson, L.A. Decline in protease activities with age in the nematode Caenorhabditis elegans. Mech. Ageing Dev. 1988, 45, 191–201. [Google Scholar] [CrossRef]
- Schiffer, I.; Gerisch, B.; Kawamura, K.; Laboy, R.; Hewitt, J.; Denzel, M.S.; Mori, M.A.; Vanapalli, S.; Shen, Y.; Symmons, O.; et al. miR-1 coordinately regulates lysosomal v-ATPase and biogenesis to impact proteotoxicity and muscle function during aging. eLife 2021, 10, e66768. [Google Scholar] [CrossRef]
- Kasper, D.; Planells-Cases, R.; Fuhrmann, J.C.; Scheel, O.; Zeitz, O.; Ruether, K.; Schmitt, A.; Poet, M.; Steinfeld, R.; Schweizer, M.; et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005, 24, 1079–1091. [Google Scholar] [CrossRef]
- Wang, F.; Gomez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Lorincz, P.; Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Murray, J.W.; Wang, X.; Pampliega, O.; Yin, D.; Patel, B.; Yuste, A.; Wolkoff, A.W.; Cuervo, A.M. Defective recruitment of motor proteins to autophagic compartments contributes to autophagic failure in aging. Aging Cell 2018, 17, e12777. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Wang, K.; Wang, L.; Liu, W.; Zhang, C.; Qiu, Y.; Liu, W.; Zhang, H.; Zhang, D.; Yang, Z.; et al. RAB7 activity is required for the regulation of mitophagy in oocyte meiosis and oocyte quality control during ovarian aging. Autophagy 2022, 18, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Teng, J.; Chen, J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 2021, 17, 2680–2688. [Google Scholar] [CrossRef]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Liang, Q.; Li, L.; Hu, Z.; Wu, F.; Zhang, P.; Ma, Y.; Zhao, B.; Kovacs, A.L.; Zhang, Z.; et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol. 2014, 16, 1215–1226. [Google Scholar] [CrossRef]
- Fulop, N.; Feng, W.; Xing, D.; He, K.; Not, L.G.; Brocks, C.A.; Marchase, R.B.; Miller, A.P.; Chatham, J.C. Aging leads to increased levels of protein O-linked N-acetylglucosamine in heart, aorta, brain and skeletal muscle in Brown-Norway rats. Biogerontology 2008, 9, 139. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Keppler, D.; Walter, R.; Perez, C.; Sierra, F. Increased expression of mature cathepsin B in aging rat liver. Cell Tissue Res. 2000, 302, 181–188. [Google Scholar] [CrossRef]
- Folick, A.; Oakley, H.D.; Yu, Y.; Armstrong, E.H.; Kumari, M.; Sanor, L.; Moore, D.D.; Ortlund, E.A.; Zechner, R.; Wang, M.C. Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 2015, 347, 83–86. [Google Scholar] [CrossRef]
- Lapierre, L.R.; Gelino, S.; Melendez, A.; Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol. 2011, 21, 1507–1514. [Google Scholar] [CrossRef]
- Settembre, C.; Perera, R.M. Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology. Nat. Rev. Mol. Cell Biol. 2024, 25, 223–245. [Google Scholar] [CrossRef]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagne, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell Proteom. 2013, 12, 1572–1588. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Recent progress in autophagic lysosome reformation. Traffic 2017, 18, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Alsaqati, M.; Thomas, R.S.; Kidd, E.J. Upregulation of endocytic protein expression in the Alzheimer’s disease male human brain. Aging Brain 2023, 4, 100084. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Dai, Y.; Zhu, X.; Chen, Q.; Zhu, H.; Zhou, B.; Tang, H.; Pang, S. Saturated very long chain fatty acid configures glycosphingolipid for lysosome homeostasis in long-lived C. elegans. Nat. Commun. 2021, 12, 5073. [Google Scholar] [CrossRef]
- Villalobos, T.V.; Ghosh, B.; DeLeo, K.R.; Alam, S.; Ricaurte-Perez, C.; Wang, A.; Mercola, B.M.; Butsch, T.J.; Ramos, C.D.; Das, S.; et al. Tubular lysosome induction couples animal starvation to healthy aging. Nat. Aging 2023, 3, 1091–1106. [Google Scholar] [CrossRef]
- Chapin, H.C.; Okada, M.; Merz, A.J.; Miller, D.L. Tissue-specific autophagy responses to aging and stress in C. elegans. Aging 2015, 7, 419–434. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Step | Individual Step | Molecular Mechanism | Model System | Reference |
|---|---|---|---|---|
| 1 | Autophagosome formation and elongation | Decline in Atg2, Atg5, and Atg7 transcriptional levels | Human, Drosophila | [6,9] |
| Decreased ULK1 Ser555 phosphorylation | Mouse | [16] | ||
| VPS34 acetylation by increased p300 | Human | [17] | ||
| BECN-1 inhibition by RUBCN upregulation | Mouse | [18] | ||
| Reduced WIPI2 protein levels and WIPI2 phosphorylation | Mouse | [14] | ||
| Oxidation of ATG3 and ATG7 leading to intermolecular disulfide formation between ATG3 and ATG7 | Rat, Mouse | [19] | ||
| Dysregulation of PKA activity resulting in aberrant LC3B/ATG8 S12 phosphorylation | Human | [20] | ||
| 2 | Cargo sequestration | Reduction in p62/SQSTM1/Ref(2)P transcription | Mouse, Drosophila | [21,22] |
| Cholesterol accumulation disrupts mitochondrial OPTN translocation to prevent mitochondrial clearance | Human, Mouse | [23] | ||
| 3 | Autophagosome closure and autophagosome–lysosome fusion | Decreased translation or increased turnover of Vps4 | Yeast | [24] |
| Reduced RILP expression to prevent perinuclear localization of autophagosomes | Human | [25] | ||
| Decrease protein levels of Rab2/RAB2A, Arl8/ARL8B, and Rab7/RAB7A | Drosophila | [26] | ||
| Decreased interaction between dynein and dynactin | Monkey | [27,28] | ||
| 4 | Autophagic cargo degradation and autophagic lysosome reformation | Lysosomal pH increased with age | Yeast, C. elegans | [4,29,30] |
| Cathepsin activity decreased via declining lysosomal acidity | Rat, C. elegans | [4,31] | ||
| Transcriptional decrease in V-ATPase components | Yeast | [24] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.H.Y.; Hansen, M.; Kumsta, C. Molecular Mechanisms of Autophagy Decline during Aging. Cells 2024, 13, 1364. https://doi.org/10.3390/cells13161364
Lim SHY, Hansen M, Kumsta C. Molecular Mechanisms of Autophagy Decline during Aging. Cells. 2024; 13(16):1364. https://doi.org/10.3390/cells13161364
Chicago/Turabian StyleLim, Shaun H. Y., Malene Hansen, and Caroline Kumsta. 2024. "Molecular Mechanisms of Autophagy Decline during Aging" Cells 13, no. 16: 1364. https://doi.org/10.3390/cells13161364
APA StyleLim, S. H. Y., Hansen, M., & Kumsta, C. (2024). Molecular Mechanisms of Autophagy Decline during Aging. Cells, 13(16), 1364. https://doi.org/10.3390/cells13161364