
Centrosomal Protein 55 Regulates Chromosomal Instability in Cancer Cells by Controlling Microtubule Dynamics
 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture and Stable Lentiviral Knock-Down of CEP55
2.2. Stable Lentiviral Re-Expression of CEP55WT and CEP5559–428
2.3. Western Blotting
2.4. Real Time PCR
2.5. Immunofluorescence Staining
2.6. EB3 Tracking for Evaluation of MT Velocity
2.7. Cold-Induced Depolymerisation in Cellulo
2.8. Chromosome Isolation
2.9. Next-Generation Sequencing
2.10. Structure Prediction of CEP55
2.11. Recombinant Expression of His-GFP-CEP55 and CEP55 Mutants
2.12. MT-Binding
2.13. MT-Pulldown
2.14. MT-Polymerisation
2.15. Cold-Induced In Vitro MT-Depolymerisation
3. Results
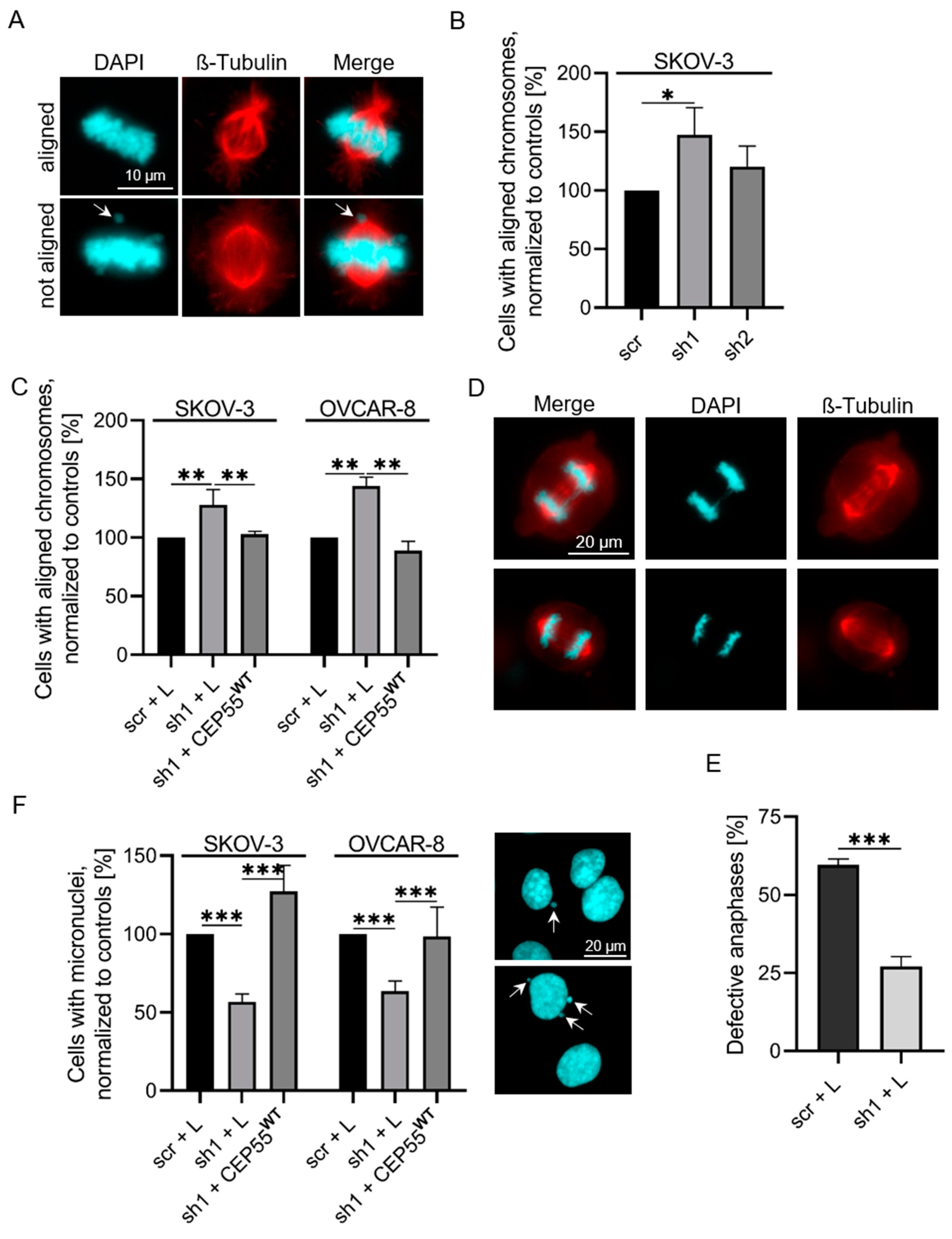
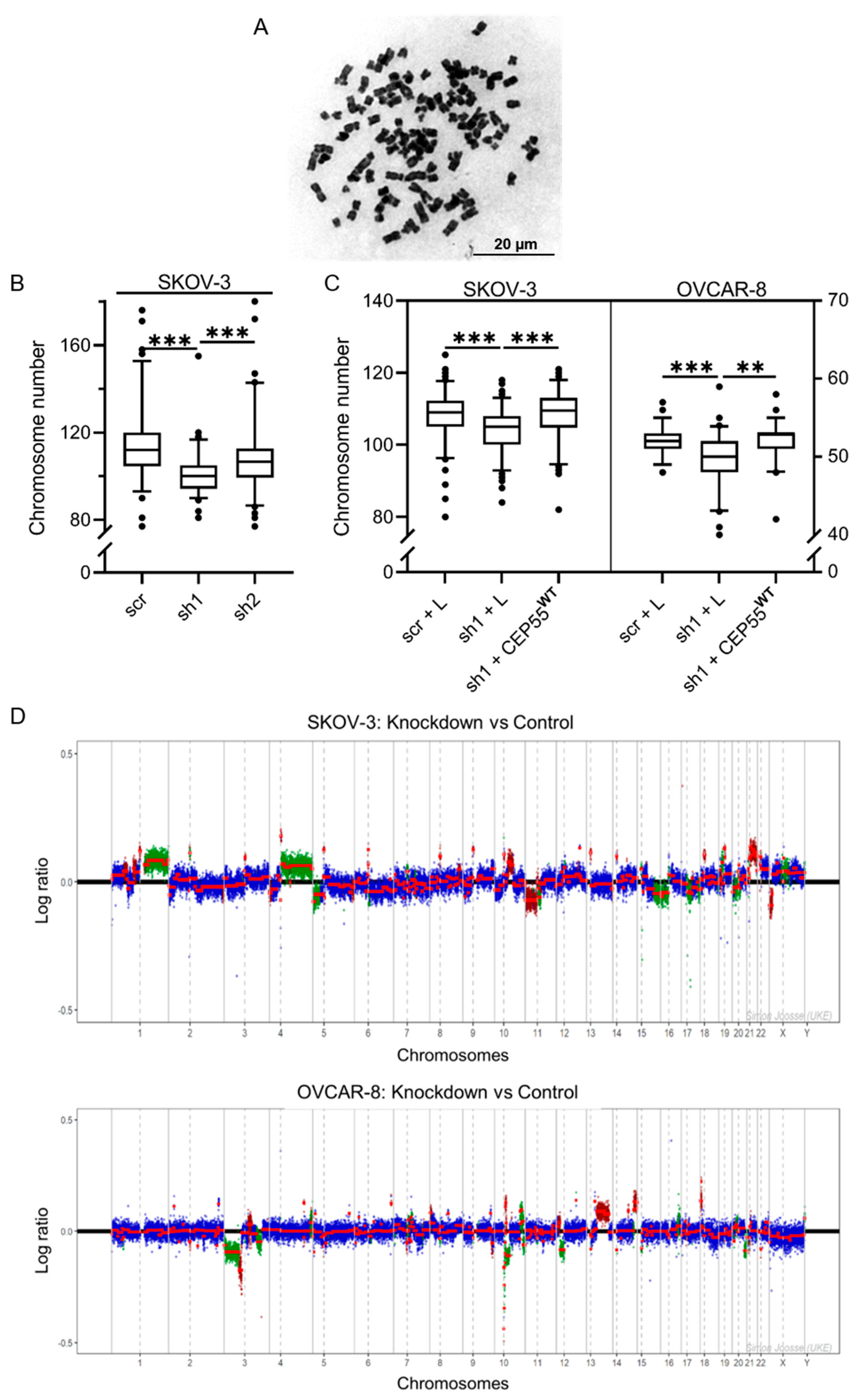
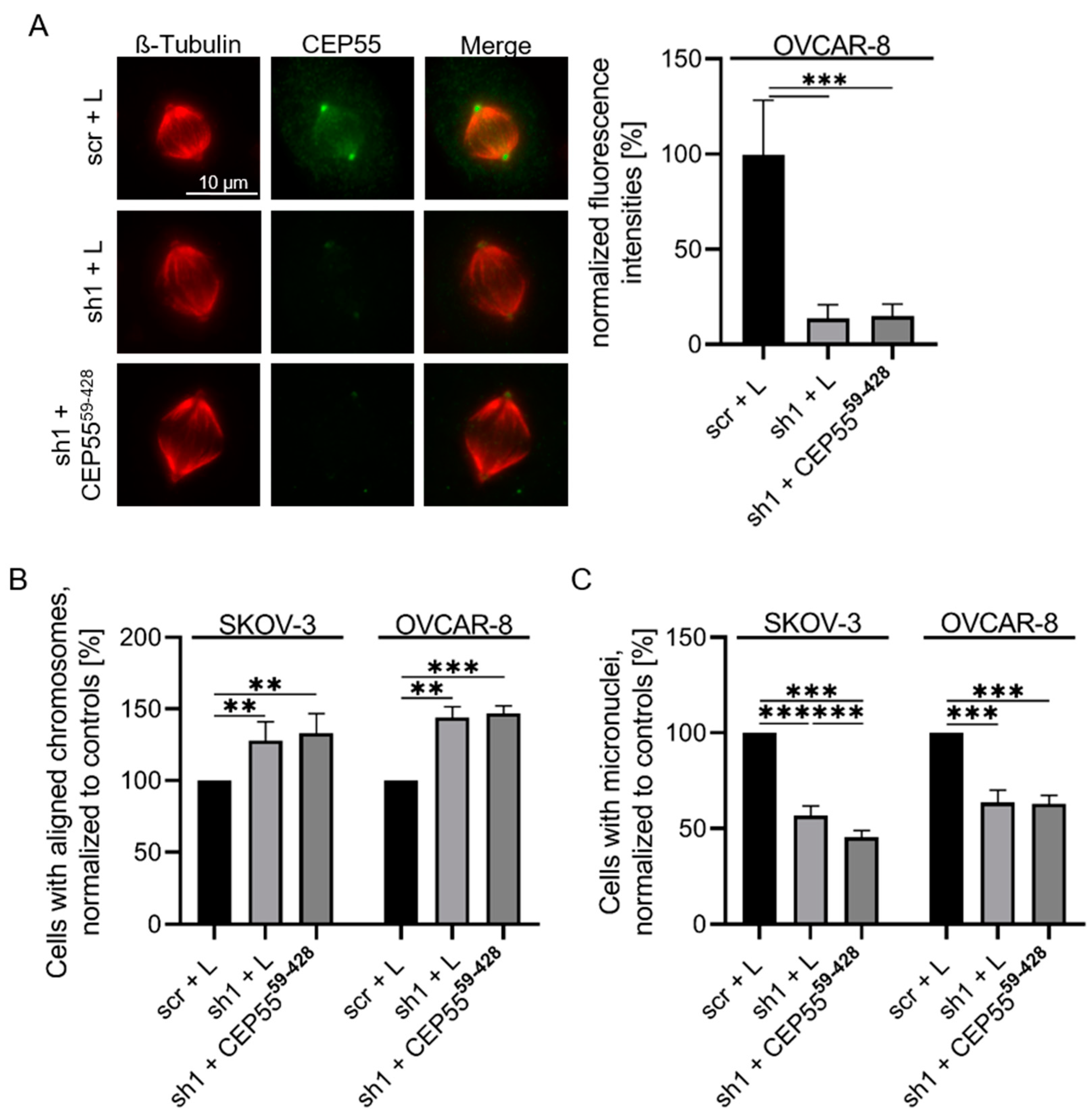
3.1. Depletion of CEP55 Decreases CIN
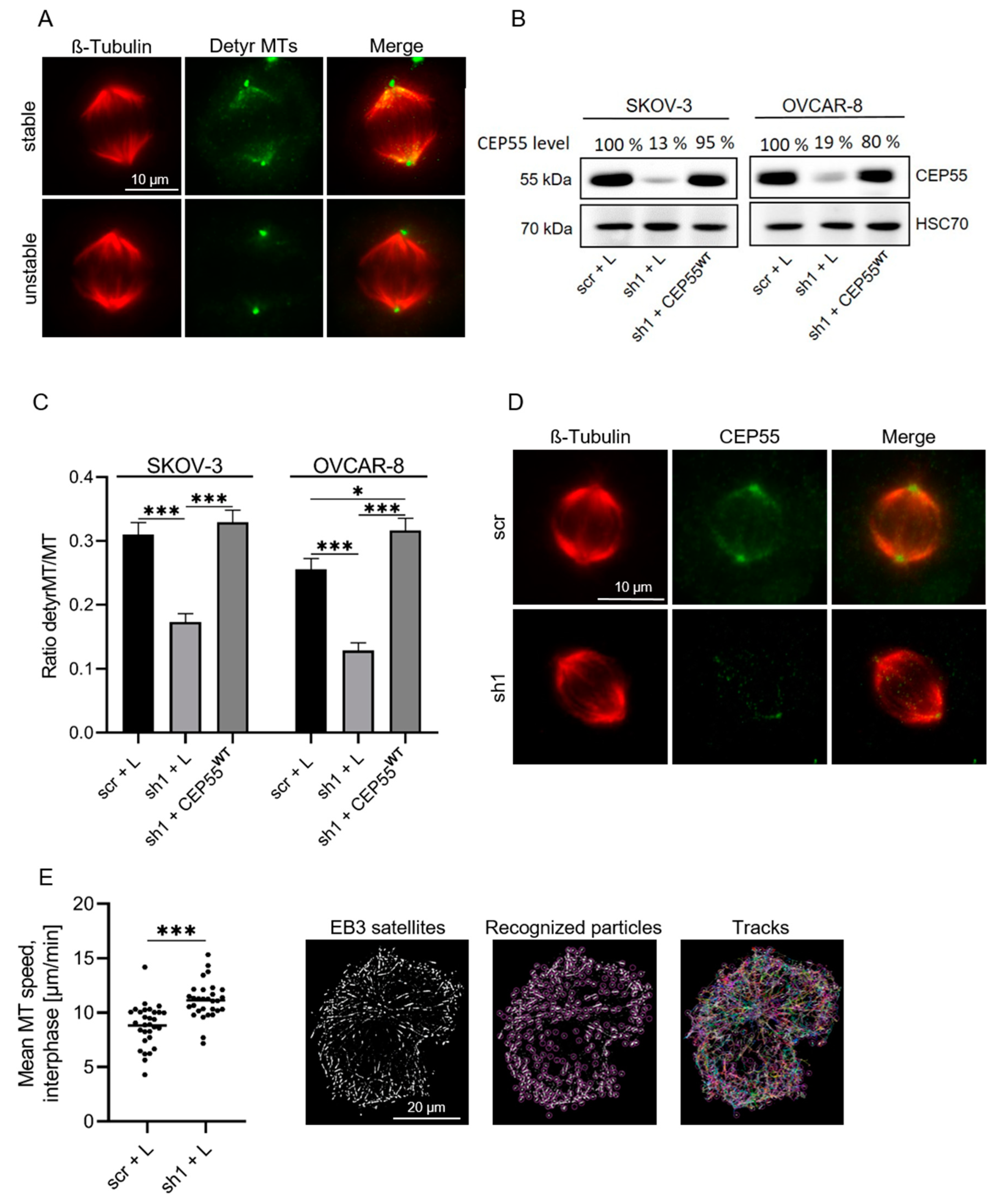
3.2. CEP55 Depletion Reduces the Stability but Increases the Dynamics of MTs
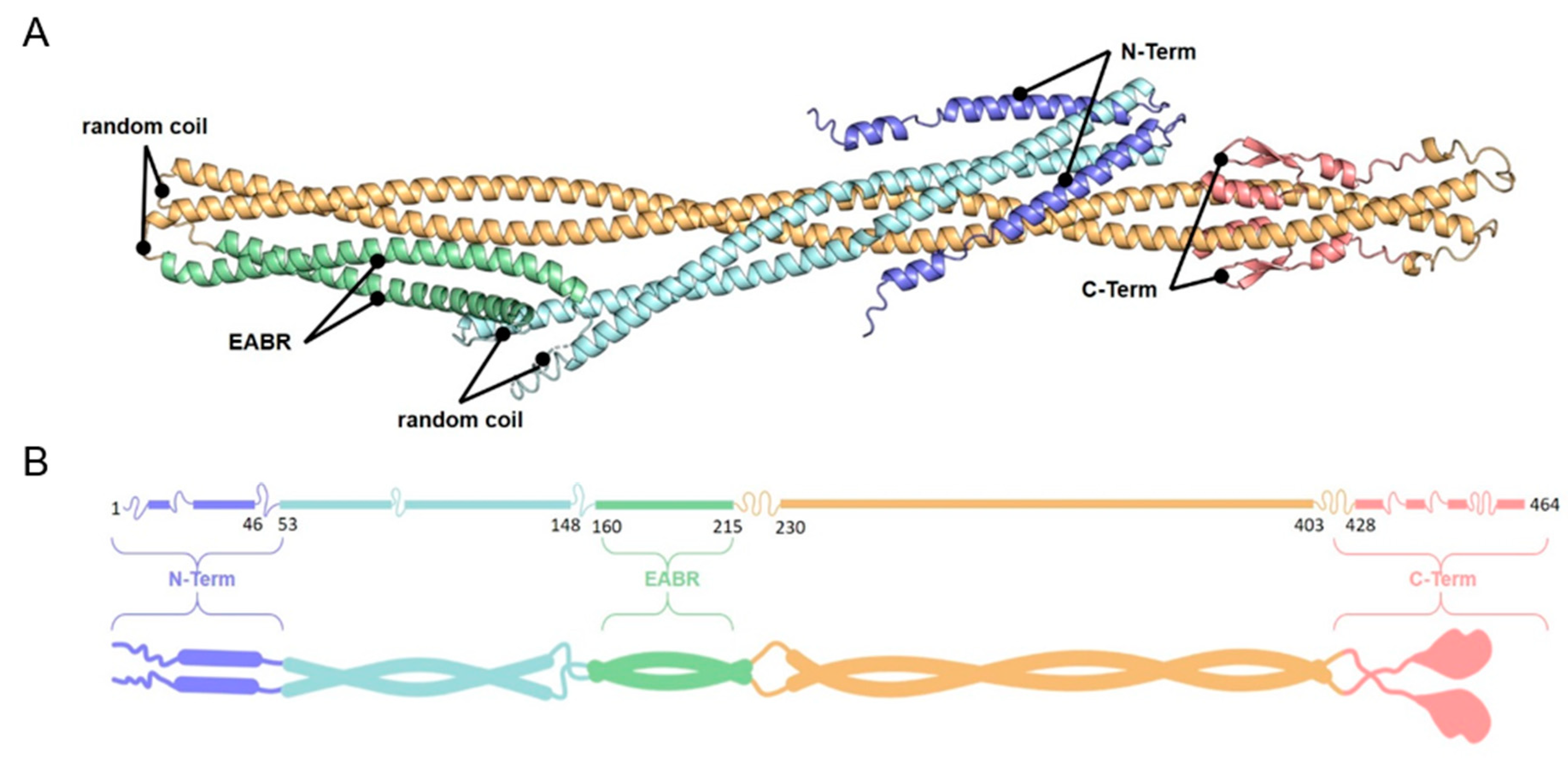
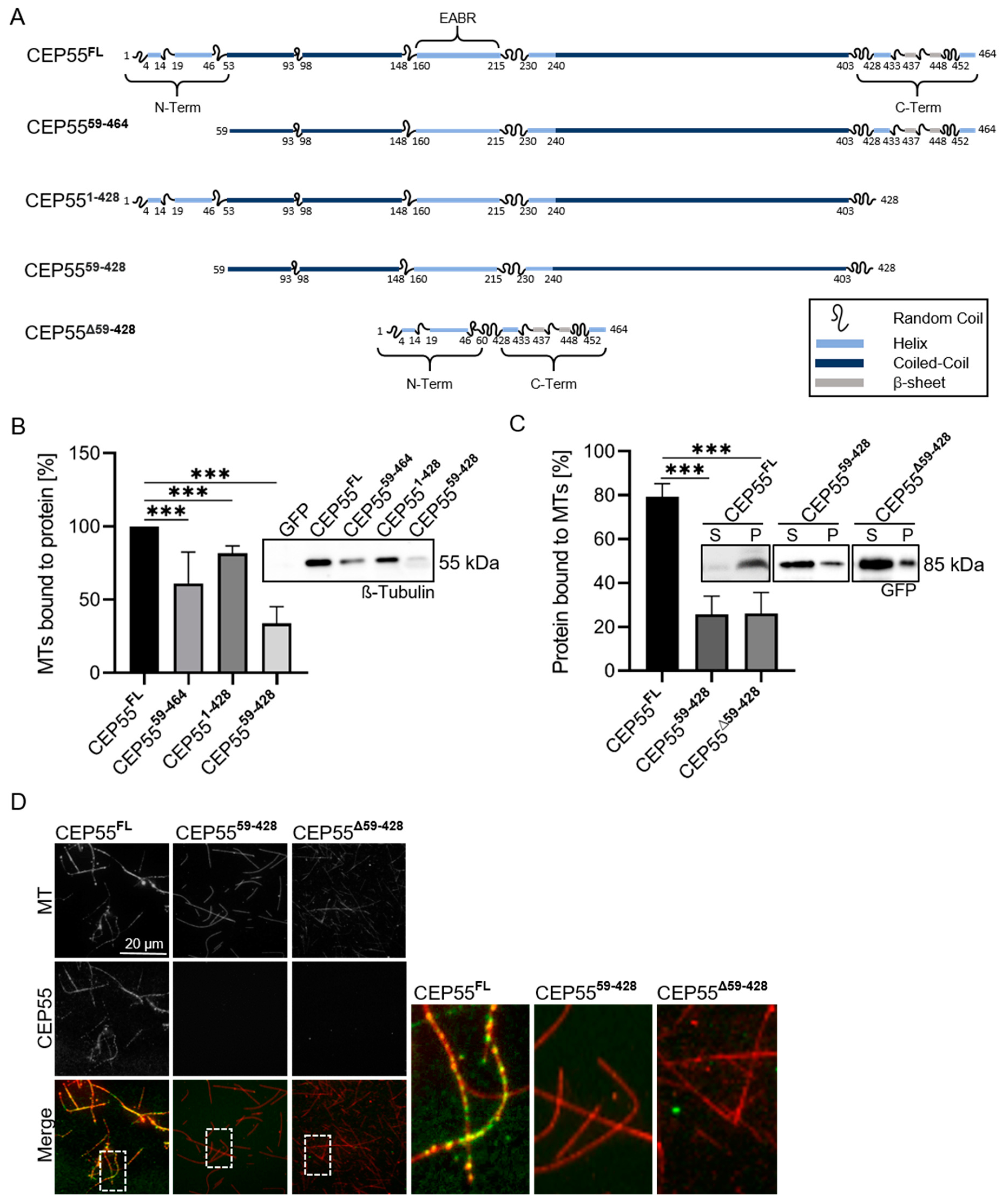
3.3. Identification of CEP55 Peptides Required for MT-Binding
3.4. Re-Expression of a MT-Binding Deficient CEP55 Mutant Does Not Rescue Reduced CIN of CEP55 shRNA Cells
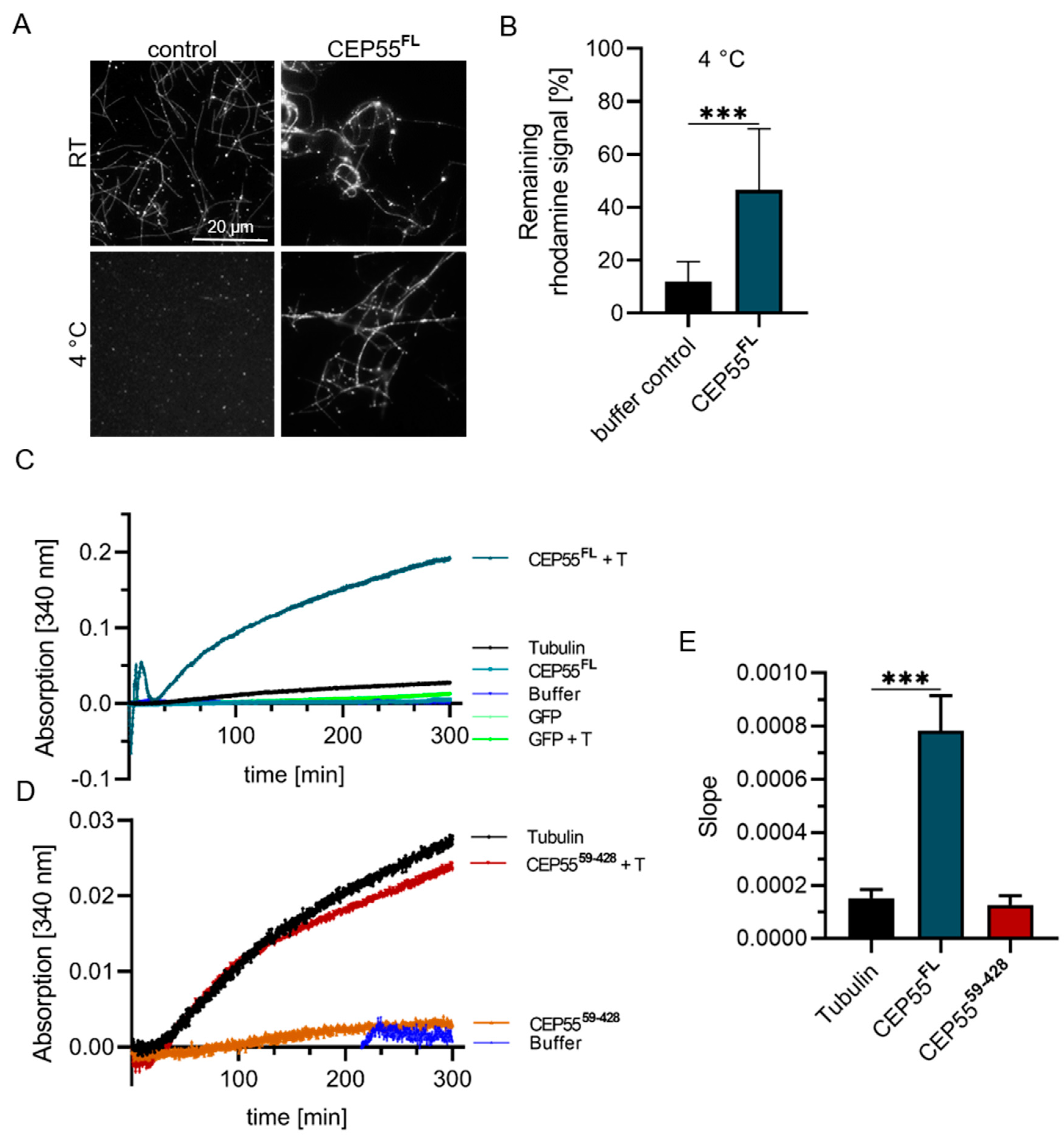
3.5. Impact of CEP55 on MT-Dynamics In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhang, W.; Niu, C.; He, W.; Hou, T.; Sun, X.; Xu, L.; Zhang, Y. Upregulation of centrosomal protein 55 is associated with unfavorable prognosis and tumor invasion in epithelial ovarian carcinoma. Tumour Biol. 2016, 37, 6239–6254. [Google Scholar] [CrossRef]
- Jeffery, J.; Sinha, D.; Srihari, S.; Kalimutho, M.; Khanna, K.K. Beyond cytokinesis: The emerging roles of cep55 in tumorigenesis. Oncogene 2016, 35, 683–690. [Google Scholar] [CrossRef]
- Tang, W.; Li, J.; Chang, X.; Jia, L.; Tang, Q.; Wang, Y.; Zheng, Y.; Sun, L.; Feng, Z. Construction of a novel prognostic-predicting model correlated to ovarian cancer. Biosci. Rep. 2020, 40, BSR20201261. [Google Scholar] [CrossRef] [PubMed]
- Schiewek, J.; Schumacher, U.; Lange, T.; Joosse, S.A.; Wikman, H.; Pantel, K.; Mikhaylova, M.; Kneussel, M.; Linder, S.; Schmalfeldt, B.; et al. Clinical relevance of cytoskeleton associated proteins for ovarian cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 2195–2205. [Google Scholar] [CrossRef]
- Chen, C.H.; Lai, J.M.; Chou, T.Y.; Chen, C.Y.; Su, L.J.; Lee, Y.C.; Cheng, T.S.; Hong, Y.R.; Chou, C.K.; Whang-Peng, J.; et al. Vegfa upregulates flj10540 and modulates migration and invasion of lung cancer via pi3k/akt pathway. PLoS ONE 2009, 4, e5052. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, P.J.; Chen, Y.C.; Fu, S.L.; Wu, K.J.; Tsou, A.P.; Lee, Y.C.; Lin, T.C.; Hsu, S.L.; Lin, W.J.; et al. Flj10540-elicited cell transformation is through the activation of pi3-kinase/akt pathway. Oncogene 2007, 26, 4272–4283. [Google Scholar] [CrossRef]
- Jia, Y.; Xiao, Z.; Gongsun, X.; Xin, Z.; Shang, B.; Chen, G.; Wang, Z.; Jiang, W. Cep55 promotes the proliferation, migration and invasion of esophageal squamous cell carcinoma via the pi3k/akt pathway. Onco Targets Ther. 2018, 11, 4221–4232. [Google Scholar] [CrossRef]
- Sinha, D.; Nag, P.; Nanayakkara, D.; Duijf, P.H.G.; Burgess, A.; Raninga, P.; Smits, V.A.J.; Bain, A.L.; Subramanian, G.; Wall, M.; et al. Cep55 overexpression promotes genomic instability and tumorigenesis in mice. Commun. Biol. 2020, 3, 593. [Google Scholar] [CrossRef]
- Lee, H.H.; Elia, N.; Ghirlando, R.; Lippincott-Schwartz, J.; Hurley, J.H. Midbody targeting of the escrt machinery by a noncanonical coiled coil in cep55. Science 2008, 322, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Colf, L.A.; Karren, M.A.; Sandrin, V.; Rodesch, C.K.; Sundquist, W.I. Human escrt-iii and vps4 proteins are required for centrosome and spindle maintenance. Proc. Natl. Acad. Sci. USA 2010, 107, 12889–12894. [Google Scholar] [CrossRef] [PubMed]
- Carlton, J.G.; Agromayor, M.; Martin-Serrano, J. Differential requirements for alix and escrt-iii in cytokinesis and hiv-1 release. Proc. Natl. Acad. Sci. USA 2008, 105, 10541–10546. [Google Scholar] [CrossRef]
- Fabbro, M.; Zhou, B.B.; Takahashi, M.; Sarcevic, B.; Lal, P.; Graham, M.E.; Gabrielli, B.G.; Robinson, P.J.; Nigg, E.A.; Ono, Y.; et al. Cdk1/erk2- and plk1-dependent phosphorylation of a centrosome protein, cep55, is required for its recruitment to midbody and cytokinesis. Dev. Cell 2005, 9, 477–488. [Google Scholar] [CrossRef]
- Little, J.N.; Dwyer, N.D. Cep55: Abscission boss or assistant? Trends Cell Biol. 2021, 31, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Little, J.N.; McNeely, K.C.; Michel, N.; Bott, C.J.; Lettieri, K.S.; Hecht, M.R.; Martin, S.A.; Dwyer, N.D. Loss of coiled-coil protein cep55 impairs neural stem cell abscission and results in p53-dependent apoptosis in developing cortex. J. Neurosci. 2021, 41, 3344–3365. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Swanton, C. The role of aneuploidy in cancer evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a028373. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Thompson, S.L.; Manning, A.L.; Compton, D.A. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol. 2009, 11, 27–35. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, L.; Yi, Q.; Lv, L.; Wang, Z.; Zhao, X.; Zhong, L.; Jiang, H.; Rasool, S.; Hao, Q.; et al. Lagging chromosomes entrapped in micronuclei are not ‘lost’ by cells. Cell Res. 2012, 22, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Krupina, K.; Goginashvili, A.; Cleveland, D.W. Causes and consequences of micronuclei. Curr. Opin. Cell Biol. 2021, 70, 91–99. [Google Scholar] [CrossRef]
- Windhorst, S.; Fliegert, R.; Blechner, C.; Mollmann, K.; Hosseini, Z.; Gunther, T.; Eiben, M.; Chang, L.; Lin, H.Y.; Fanick, W.; et al. Inositol 1,4,5-trisphosphate 3-kinase-a is a new cell motility-promoting protein that increases the metastatic potential of tumor cells by two functional activities. J. Biol. Chem. 2010, 285, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Heinz, L.S.; Muhs, S.; Schiewek, J.; Grub, S.; Nalaskowski, M.; Lin, Y.N.; Wikman, H.; Oliveira-Ferrer, L.; Lange, T.; Wellbrock, J.; et al. Strong fascin expression promotes metastasis independent of its f-actin bundling activity. Oncotarget 2017, 8, 110077–110091. [Google Scholar] [CrossRef] [PubMed]
- Ershov, D.; Phan, M.S.; Pylvanainen, J.W.; Rigaud, S.U.; Le Blanc, L.; Charles-Orszag, A.; Conway, J.R.W.; Laine, R.F.; Roy, N.H.; Bonazzi, D.; et al. Trackmate 7: Integrating state-of-the-art segmentation algorithms into tracking pipelines. Nat. Methods 2022, 19, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Grueb, S.S.; Muhs, S.; Popp, Y.; Schmitt, S.; Geyer, M.; Lin, Y.N.; Windhorst, S. The formin drosophila homologue of diaphanous2 (diaph2) controls microtubule dynamics in colorectal cancer cells independent of its fh2-domain. Sci. Rep. 2019, 9, 5352. [Google Scholar] [CrossRef]
- Riebensahm, C.; Joosse, S.A.; Mohme, M.; Hanssen, A.; Matschke, J.; Goy, Y.; Witzel, I.; Lamszus, K.; Kropidlowski, J.; Petersen, C.; et al. Clonality of circulating tumor cells in breast cancer brain metastasis patients. Breast Cancer Res. 2019, 21, 101. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-freec: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The psipred protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- Delorenzi, M.; Speed, T. An hmm model for coiled-coil domains and a comparison with pssm-based predictions. Bioinformatics 2002, 18, 617–625. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.Z.; Till, S.; Mirdita, M.; Steinegger, M.; Soding, J.; Lupas, A.N.; Alva, V. Protein sequence analysis using the mpi bioinformatics toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sonderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. Netsurfp-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins 2019, 87, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Ludwiczak, J.; Winski, A.; Szczepaniak, K.; Alva, V.; Dunin-Horkawicz, S. Deepcoil-a fast and accurate prediction of coiled-coil domains in protein sequences. Bioinformatics 2019, 35, 2790–2795. [Google Scholar] [CrossRef]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef]
- Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.; Honigschmid, P.; Schafferhans, A.; Roos, M.; Bernhofer, M.; et al. Predictprotein--an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014, 42, W337–W343. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented mpi bioinformatics toolkit with a new hhpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Genovese, G.; Compton, D.A. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr. Biol. 2009, 19, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Gama Braga, L.; Prifti, D.K.; Garand, C.; Saini, P.K.; Elowe, S. A quantitative and semiautomated method for determining misaligned and lagging chromosomes during mitosis. Mol. Biol. Cell 2021, 32, 880–891. [Google Scholar] [CrossRef]
- Kuniyasu, K.; Iemura, K.; Tanaka, K. Delayed chromosome alignment to the spindle equator increases the rate of chromosome missegregation in cancer cell lines. Biomolecules 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.A.; Zhu, J.; Li, R. Aneuploidy and chromosomal instability: A vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013, 32, 377–389. [Google Scholar] [CrossRef]
- Zhao, W.M.; Seki, A.; Fang, G. Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol. Biol. Cell 2006, 17, 3881–3896. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Magiera, M.M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Wood, L.; Toramoto, I.; Yagyu, K.; Fukagawa, T.; Earnshaw, W.C. Cenp-32 is required to maintain centrosomal dominance in bipolar spindle assembly. Mol. Biol. Cell 2015, 26, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Manka, S.W.; Moores, C.A. Microtubule structure by cryo-em: Snapshots of dynamic instability. Essays Biochem. 2018, 62, 737–751. [Google Scholar] [PubMed]
- Martinez-Garay, I.; Rustom, A.; Gerdes, H.H.; Kutsche, K. The novel centrosomal associated protein cep55 is present in the spindle midzone and the midbody. Genomics 2006, 87, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Sinha, D.; Jeffery, J.; Nones, K.; Srihari, S.; Fernando, W.C.; Duijf, P.H.; Vennin, C.; Raninga, P.; Nanayakkara, D.; et al. Cep55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol. Med. 2018, 10, e8566. [Google Scholar] [CrossRef]
- Li, C.; Hong, Z.; Ou, M.; Zhu, X.; Zhang, L.; Yang, X. Integrated mirna-mrna expression profiles revealing key molecules in ovarian cancer based on bioinformatics analysis. BioMed Res. Int. 2021, 2021, 6673655. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Effect of CEP55 | Extent |
|---|---|---|
| Cold-induced MT-depolymerization | protection | 80% |
| MT-polymerization rate | slowly increasing slope | 4-fold |
| conclusion | CEP55 attenuates MT depolymerization |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhs, S.; Paraschiakos, T.; Schäfer, P.; Joosse, S.A.; Windhorst, S. Centrosomal Protein 55 Regulates Chromosomal Instability in Cancer Cells by Controlling Microtubule Dynamics. Cells 2024, 13, 1382. https://doi.org/10.3390/cells13161382
Muhs S, Paraschiakos T, Schäfer P, Joosse SA, Windhorst S. Centrosomal Protein 55 Regulates Chromosomal Instability in Cancer Cells by Controlling Microtubule Dynamics. Cells. 2024; 13(16):1382. https://doi.org/10.3390/cells13161382
Chicago/Turabian StyleMuhs, Stefanie, Themistoklis Paraschiakos, Paula Schäfer, Simon A. Joosse, and Sabine Windhorst. 2024. "Centrosomal Protein 55 Regulates Chromosomal Instability in Cancer Cells by Controlling Microtubule Dynamics" Cells 13, no. 16: 1382. https://doi.org/10.3390/cells13161382