
Ageing-Related Changes to H3K4me3, H3K27ac, and H3K27me3 in Purified Mouse Neurons

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Tissue Processing
2.2. Isolation of Nuclei from Mouse Forebrain
2.3. Fluorescence-Activated Nuclei Sorting (FANS) for Neuronal Nuclei
2.4. Chromatin Immunoprecipitation (ChIP)
2.5. ChIP-Seq Library Preparation and Next-Generation Sequencing
2.6. ChIP-Seq Alignment
2.7. Annotation Datasets
2.8. Cell-Identity Gene Expression and ChIP-Seq Coverage
2.9. Sample Quality Control and Filtering
2.10. Genomic Feature Overlap Calculations
2.11. Gene Ontology Enrichment
2.12. Motif Analysis
2.13. Additional Datasets
2.14. Differential Peak Analyses
2.15. ChIP-Seq Profile Plotting
3. Results
3.1. Generation of Neuronal-Specific Epigenome Maps during Mouse Ageing
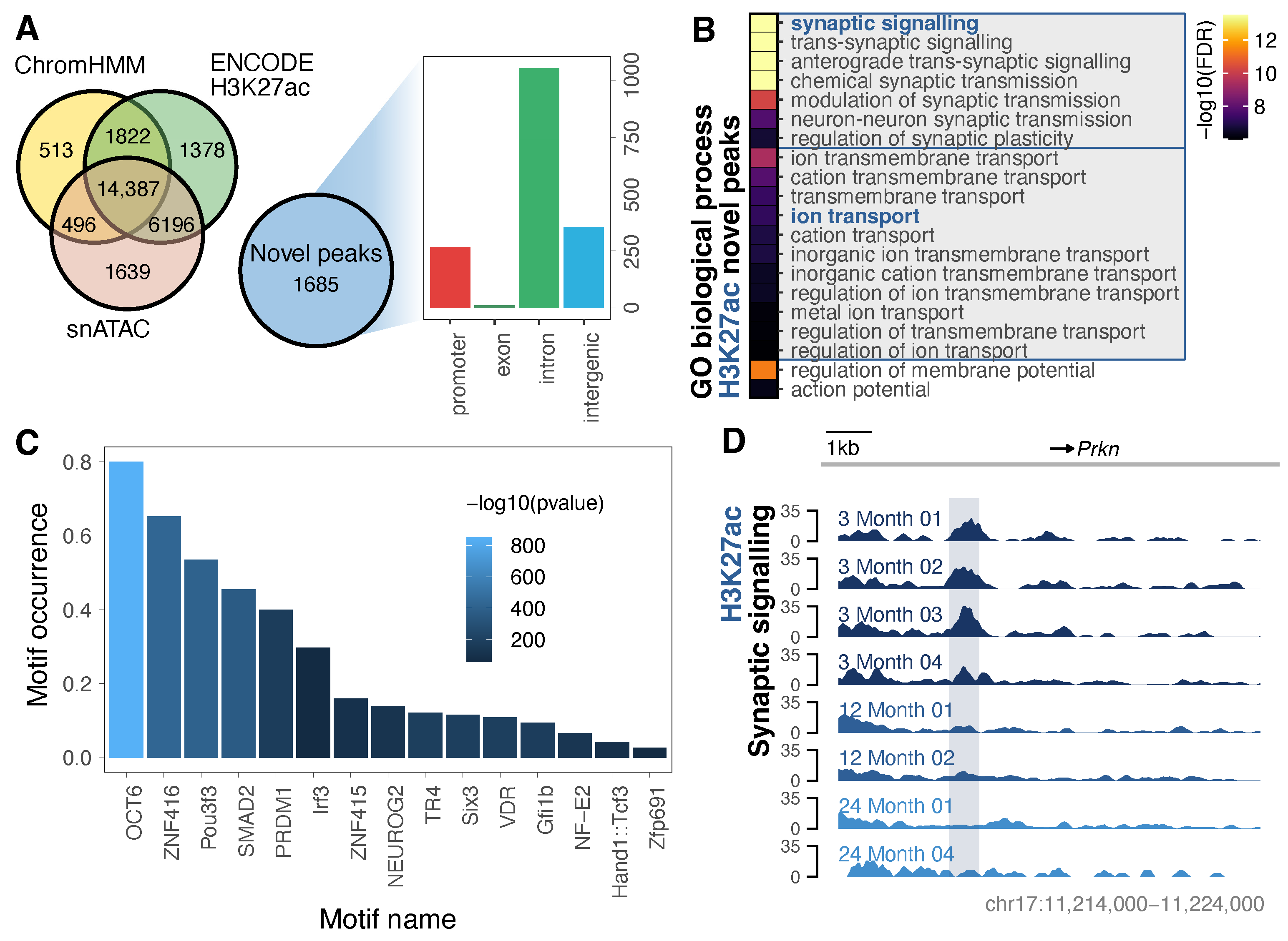
3.2. Purified Neurons Enable Detection of an Expanded Repertoire of H3K27ac-Marked Neuronal Enhancers
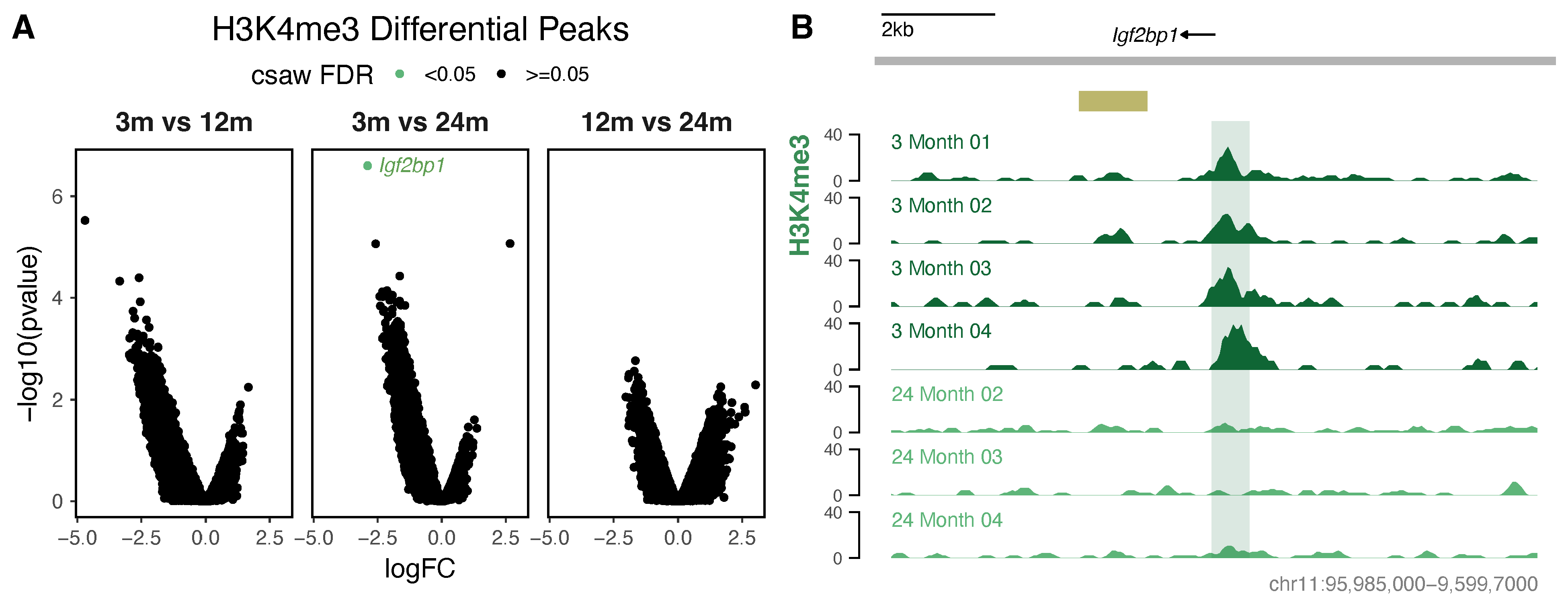
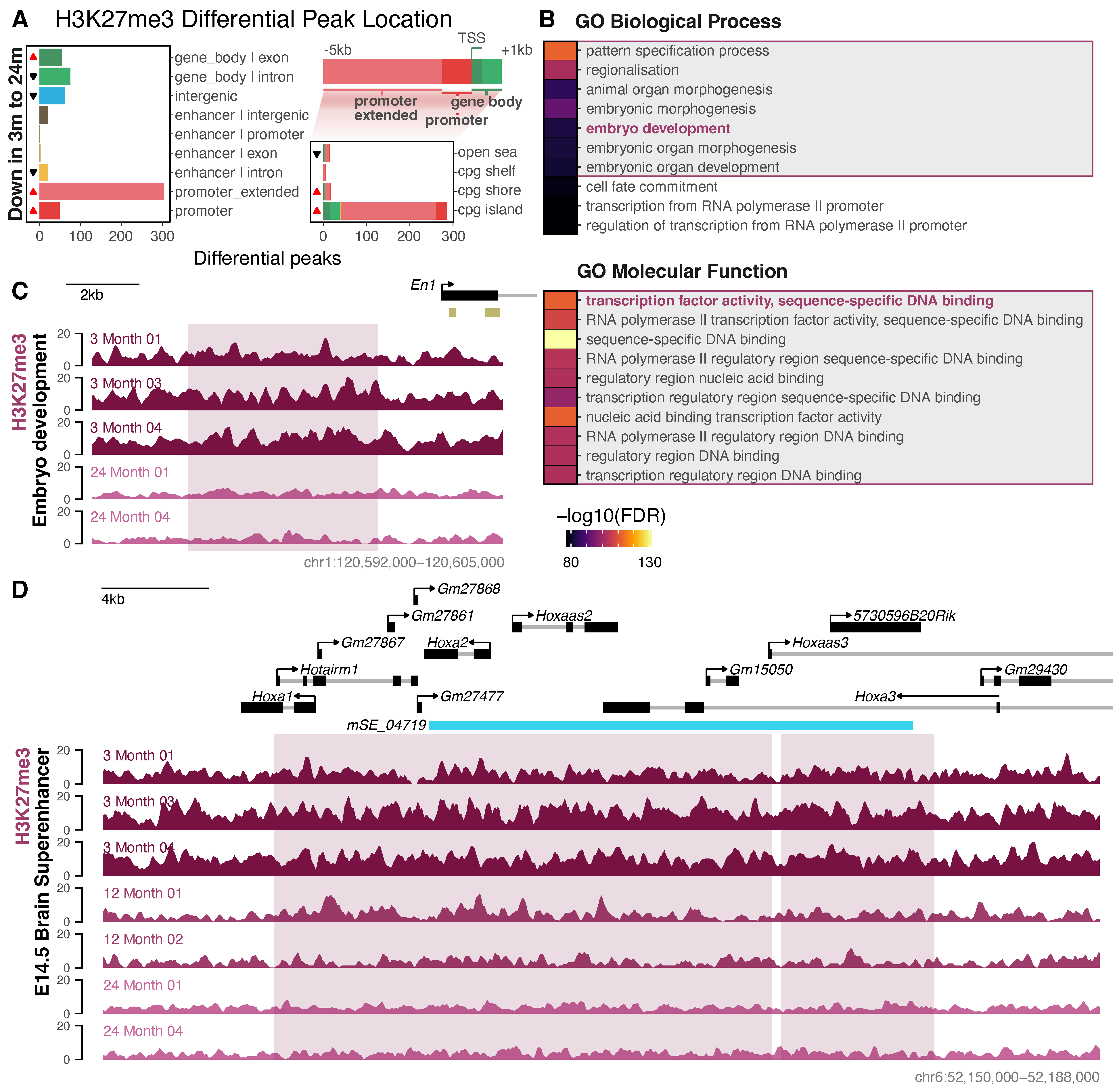
3.3. Differential Histone Modification Reveals a Redistribution of H3K27ac from Intronic Enhancers to Promoters with Neuronal Age, and a Loss of Repressive H3K27me3 from Developmental Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murman, D. The Impact of Age on Cognition. Semin. Hear. 2015, 36, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.M.; Disterhoft, J.F. Learning and aging affect neuronal excitability and learning. Neurobiol. Learn. Mem. 2020, 167, 107133. [Google Scholar] [CrossRef]
- Oliveira, A.M.M.; Hemstedt, T.J.; Bading, H. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 2012, 15, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- McQuown, S.C.; Barrett, R.M.; Matheos, D.P.; Post, R.J.; Rogge, G.A.; Alenghat, T.; Mullican, S.E.; Jones, S.; Rusche, J.R.; Lazar, M.A.; et al. HDAC3 Is a Critical Negative Regulator of Long-Term Memory Formation. J. Neurosci. 2011, 31, 764–774. [Google Scholar] [CrossRef]
- Shu, G.; Kramár, E.A.; López, A.J.; Huynh, G.; Wood, M.A.; Kwapis, J.L. Deleting HDAC3 rescues long-term memory impairments induced by disruption of the neuron-specific chromatin remodeling subunit BAF53b. Learn. Mem. 2018, 25, 109–114. [Google Scholar] [CrossRef]
- Ninkovic, J.; Mori, T.; Götz, M. Distinct Modes of Neuron Addition in Adult Mouse Neurogenesis. J. Neurosci. 2007, 27, 10906–10911. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Ohtsuka, T.; Takao, K.; Miyakawa, T.; Yamaguchi, M.; Mori, K.; Ikeda, T.; Itohara, S.; Kageyama, R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 2008, 11, 1153–1161. [Google Scholar] [CrossRef]
- Giles, K.A.; Taberlay, P.C. The Role of Nucleosomes in Epigenetic Gene Regulation. In Clinical Epigenetics; Springer Singapore: Singapore, 2019; Volume 87–117, pp. 87–117. [Google Scholar] [CrossRef]
- Collins, B.E.; Greer, C.B.; Coleman, B.C.; Sweatt, J.D. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin 2019, 12, 7. [Google Scholar] [CrossRef]
- Howe, F.S.; Fischl, H.; Murray, S.C.; Mellor, J. Is H3K4me3 instructive for transcription activation? BioEssays 2017, 39, 1–12. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Kang, J.; Kang, Y.; Kim, A. Histone H3K4ac, as a marker of active transcription start sites and enhancers, plays roles in histone eviction and RNA transcription. Biochim. et Biophys. Acta (BBA)–Gene Regul. Mech. 2024, 1867, 195021. [Google Scholar] [CrossRef]
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of Histone Lysine Methylation across Transcribed Mammalian Chromatin. Mol. Cell. Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef]
- Xiao, C.; Fan, T.; Tian, H.; Zheng, Y.; Zhou, Z.; Li, S.; Li, C.; He, J. H3K36 trimethylation-mediated biological functions in cancer. Clin. Epigenetics 2021, 13, 199. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef] [PubMed]
- Benito, E.; Urbanke, H.; Ramachandran, B.; Barth, J.; Halder, R.; Awasthi, A.; Jain, G.; Capece, V.; Burkhardt, S.; Navarro-Sala, M.; et al. HDAC inhibitor–dependent transcriptome and memory reinstatement in cognitive decline models. J. Clin. Investig. 2015, 125, 3572–3584. [Google Scholar] [CrossRef]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef]
- Cheung, I.; Shulha, H.P.; Jiang, Y.; Matevossian, A.; Wang, J.; Weng, Z.; Akbarian, S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 8824–8829. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Singh, P.P.; Mahmoudi, S.; Harel, I.; Casey, K.M.; Dulken, B.W.; Kundaje, A.; Brunet, A. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 2019, 29, 697–709. [Google Scholar] [CrossRef]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered Histone Acetylation Is Associated with Age-Dependent Memory Impairment in Mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.M.; Coleman, P.D.; Rutten, B.P.F.; van den Hove, D.L.A. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef]
- Dagnas, M.; Guillou, J.-L.; Prévôt, T.; Mons, N. HDAC Inhibition Facilitates the Switch between Memory Systems in Young But Not Aged Mice. J. Neurosci. 2013, 33, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T.; et al. Histone Deacetylase Inhibitors Enhance Memory and Synaptic Plasticity via CREB: CBP-Dependent Transcriptional Activation. J. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.F.J.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B.; et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 2018, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Keown, C.L.; Kurihara, L.; Zhou, J.; He, Y.; Li, J.; Castanon, R.; Lucero, J.; Nery, J.R.; Sandoval, J.P.; et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 2017, 357, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef]
- Signal, B.; Pérez Suárez, T.G.; Taberlay, P.C.; Woodhouse, A. Cellular specificity is key to deciphering epigenetic changes underlying Alzheimer’s disease. Neurobiol. Dis. 2023, 186, 106284. [Google Scholar] [CrossRef]
- Girdhar, K.; Hoffman, G.E.; Jiang, Y.; Brown, L.; Kundakovic, M.; Hauberg, M.E.; Francoeur, N.J.; Wang, Y.; Shah, H.; Kavanagh, D.H.; et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci. 2018, 21, 1126–1136. [Google Scholar] [CrossRef]
- Ximerakis, M.; Lipnick, S.L.; Innes, B.T.; Simmons, S.K.; Adiconis, X.; Dionne, D.; Mayweather, B.A.; Nguyen, L.; Niziolek, Z.; Ozek, C.; et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019, 22, 1696–1708. [Google Scholar] [CrossRef]
- Shulha, H.P.; Cheung, I.; Guo, Y.; Akbarian, S.; Weng, Z. Coordinated Cell Type–Specific Epigenetic Remodeling in Prefrontal Cortex Begins before Birth and Continues into Early Adulthood. PLoS Genet. 2013, 9, e1003433. [Google Scholar] [CrossRef]
- Oakford, P.C.; James, S.R.; Qadi, A.; West, A.C.; Ray, S.N.; Bert, A.G.; Cockerill, P.N.; Holloway, A.F. Transcriptional and epigenetic regulation of the GM-CSF promoter by RUNX1. Leuk. Res. 2010, 34, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Taberlay, P.C.; Kelly, T.K.; Liu, C.-C.; You, J.S.; De Carvalho, D.D.; Miranda, T.B.; Zhou, X.J.; Liang, G.; Jones, P.A. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell 2011, 147, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Taberlay, P.C.; Statham, A.L.; Kelly, T.K.; Clark, S.J.; Jones, P.A. Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res. 2014, 24, 1421–1432. [Google Scholar] [CrossRef]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Wingett, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 28 July 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Xu, S.; Grullon, S.; Ge, K.; Peng, W. Spatial Clustering for Identification of ChIP-Enriched Regions (SICER) to Map Regions of Histone Methylation Patterns in Embryonic Stem Cells. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2014; pp. 97–111. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef]
- Carroll, T.S.; Liang, Z.; Salama, R.; Stark, R.; de Santiago, I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 2014, 5, 75. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Cavalcante, R.G.; Sartor, M.A. annotatr: Genomic regions in context. Bioinformatics 2017, 33, 2381–2383. [Google Scholar] [CrossRef]
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; de Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef] [PubMed]
- van der Velde, A.; Fan, K.; Tsuji, J.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Weng, Z. Annotation of chromatin states in 66 complete mouse epigenomes during development. Commun. Biol. 2021, 4, 239. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Khan, A.; Zhang, X. dbSUPER: A database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016, 44, D164–D171. [Google Scholar] [CrossRef]
- Lawrence, M.; Gentleman, R.; Carey, V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics 2009, 25, 1841–1842. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pagès, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef]
- Welch, R.P.; Lee, C.; Imbriano, P.M.; Patil, S.; Weymouth, T.E.; Smith, R.A.; Scott, L.J.; Sartor, M.A. ChIP-Enrich: Gene set enrichment testing for ChIP-seq data. Nucleic Acids Res. 2014, 42, e105. [Google Scholar] [CrossRef] [PubMed]
- Pagès, H.; Aboyoun, P.; Gentleman, R.; DebRoy, S. Biostrings: Efficient Manipulation of Biological Strings. Available online: https://bioconductor.org/packages/Biostrings (accessed on 28 July 2023).
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell. 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Abascal, F.; Acosta, R.; Addleman, N.J.; Adrian, J.; Afzal, V.; Ai, R.; Aken, B.; Akiyama, J.A.; Jammal, O.A.; Amrhein, H.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Li, Y.E.; Preissl, S.; Hou, X.; Zhang, Z.; Zhang, K.; Qiu, Y.; Poirion, O.B.; Li, B.; Chiou, J.; Liu, H.; et al. An atlas of gene regulatory elements in adult mouse cerebrum. Nature 2021, 598, 129–136. [Google Scholar] [CrossRef]
- Lun, A.T.L.; Smyth, G. csaw: A Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016, 44, e45. [Google Scholar] [CrossRef]
- Lund, S.P.; Nettleton, D.; McCarthy, D.J.; Smyth, G.K. Detecting Differential Expression in RNA-sequence Data Using Quasi-likelihood with Shrunken Dispersion Estimates. Stat. Appl. Genet. Mol. Biol. 2012, 11, 8. [Google Scholar] [CrossRef]
- Courtney, N.A.; Bao, H.; Briguglio, J.S.; Chapman, E.R. Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin. Nat. Commun. 2019, 10, 4076. [Google Scholar] [CrossRef]
- White, D.N.; Stowell, M.H.B. Room for Two: The Synaptophysin/Synaptobrevin Complex. Front. Synaptic Neurosci. 2021, 13, 740318. [Google Scholar] [CrossRef]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular Anatomy of a Trafficking Organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Beacon, T.H.; Delcuve, G.P.; López, C.; Nardocci, G.; Kovalchuk, I.; van Wijnen, A.J.; Davie, J.R. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin. Epigenetics 2021, 13, 138. [Google Scholar] [CrossRef]
- Macrae, T.A.; Fothergill-Robinson, J.; Ramalho-Santos, M. Regulation, functions and transmission of bivalent chromatin during mammalian development. Nat. Rev. Mol. Cell. Biol. 2023, 24, 6–26. [Google Scholar] [CrossRef]
- Dominguez, M.H.; Ayoub, A.E.; Rakic, P. POU-III Transcription Factors (Brn1, Brn2, and Oct6) Influence Neurogenesis, Molecular Identity, and Migratory Destination of Upper-Layer Cells of the Cerebral Cortex. Cereb. Cortex 2013, 23, 2632–2643. [Google Scholar] [CrossRef] [PubMed]
- Gradari, S.; Herrera, A.; Tezanos, P.; Fontán-Lozano, Á.; Pons, S.; Trejo, J.L. The Role of Smad2 in Adult Neuroplasticity as Seen through Hippocampal-Dependent Spatial Learning/Memory and Neurogenesis. J. Neurosci. 2021, 41, 6836–6849. [Google Scholar] [CrossRef]
- Prajapati, R.S.; Hintze, M.; Streit, A. PRDM1 controls the sequential activation of neural, neural crest and sensory progenitor determinants. Development 2019, 146, dev181107. [Google Scholar] [CrossRef] [PubMed]
- Noack, F.; Vangelisti, S.; Raffl, G.; Carido, M.; Diwakar, J.; Chong, F.; Bonev, B. Multimodal profiling of the transcriptional regulatory landscape of the developing mouse cortex identifies Neurog2 as a key epigenome remodeler. Nat. Neurosci. 2022, 25, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Zhang, Y.; Liu, N.-C.; Yang, D.-R.; Li, G.; Chang, C. Minireview: Pathophysiological Roles of the TR4 Nuclear Receptor: Lessons Learned from Mice Lacking TR4. Mol. Endocrinol. 2014, 28, 805–821. [Google Scholar] [CrossRef]
- Appolloni, I.; Calzolari, F.; Corte, G.; Perris, R.; Malatesta, P. Six3 Controls the Neural Progenitor Status in the Murine CNS. Cereb. Cortex 2008, 18, 553–562. [Google Scholar] [CrossRef]
- Barnes, R.M.; Firulli, B.A.; Conway, S.J.; Vincentz, J.W.; Firulli, A.B. Analysis of the Hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Dev. Dyn. 2010, 239, 3086–3097. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Xie, L.; Duan, C.; Dai, S.; Ren, J.; Yan, Y.; Shen, J.; Lu, H.; Ge, J. Up-Regulation of Interferon Regulatory Factor 3 Involves in Neuronal Apoptosis After Intracerebral Hemorrhage in Adult Rats. Neurochem. Res. 2016, 41, 2937–2947. [Google Scholar] [CrossRef]
- Lasoń, W.; Jantas, D.; Leśkiewicz, M.; Regulska, M.; Basta-Kaim, A. The Vitamin D Receptor as a Potential Target for the Treatment of Age-Related Neurodegenerative Diseases Such as Alzheimer’s and Parkinson’s Diseases: A Narrative Review. Cells 2023, 12, 660. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Lee, C.; Hu, T.; Nguyen, J.M.; Zhang, J.; Martin, M.V.; Vawter, M.P.; Huang, E.J.; Chan, J.Y. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 8408–8413. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Borsari, B.; Villegas-Mirón, P.; Pérez-Lluch, S.; Turpin, I.; Laayouni, H.; Segarra-Casas, A.; Bertranpetit, J.; Guigó, R.; Acosta, S. Enhancers with tissue-specific activity are enriched in intronic regions. Genome Res. 2021, 31, 1325–1336. [Google Scholar] [CrossRef]
- Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [CrossRef]
- Tischfield, D.J.; Saraswat, D.K.; Furash, A.; Fowler, S.C.; Fuccillo, M.V.; Anderson, S.A. Loss of the neurodevelopmental gene Zswim6 alters striatal morphology and motor regulation. Neurobiol. Dis. 2017, 103, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Núñez, L.; Buxbaum, A.R.; Katz, Z.B.; Lopez-Jones, M.; Nwokafor, C.; Czaplinski, K.; Pan, F.; Rosenberg, J.; Monday, H.R.; Singer, R.H. Tagged actin mRNA dysregulation in IGF2BP1−/− mice. Proc. Natl. Acad. Sci. USA 2022, 119, e2208465119. [Google Scholar] [CrossRef]
- Welshhans, K.; Bassell, G.J. Netrin-1-Induced Local -Actin Synthesis and Growth Cone Guidance Requires Zipcode Binding Protein 1. J. Neurosci. 2011, 31, 9800–9813. [Google Scholar] [CrossRef]
- Tiruchinapalli, D.M.; Oleynikov, Y.; Kelič, S.; Shenoy, S.M.; Hartley, A.; Stanton, P.K.; Singer, R.H.; Bassell, G.J. Activity-Dependent Trafficking and Dynamic Localization of Zipcode Binding Protein 1 and β-Actin mRNA in Dendrites and Spines of Hippocampal Neurons. J. Neurosci. 2003, 23, 3251–3261. [Google Scholar] [CrossRef]
- Perycz, M.; Urbanska, A.S.; Krawczyk, P.S.; Parobczak, K.; Jaworski, J. Zipcode Binding Protein 1 Regulates the Development of Dendritic Arbors in Hippocampal Neurons. J. Neurosci. 2011, 31, 5271–5285. [Google Scholar] [CrossRef]
- Zhang, H.L.; Eom, T.; Oleynikov, Y.; Shenoy, S.M.; Liebelt, D.A.; Dictenberg, J.B.; Singer, R.H.; Bassell, G.J. Neurotrophin-Induced Transport of a β-Actin mRNP Complex Increases β-Actin Levels and Stimulates Growth Cone Motility. Neuron 2001, 31, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Eom, T.; Antar, L.N.; Singer, R.H.; Bassell, G.J. Localization of a β-Actin Messenger Ribonucleoprotein Complex with Zipcode-Binding Protein Modulates the Density of Dendritic Filopodia and Filopodial Synapses. J. Neurosci. 2003, 23, 10433–10444. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Jankauskas, R.; Mazari, A.M.A.; Drou, N.; Percipalle, P. β-actin regulates a heterochromatin landscape essential for optimal induction of neuronal programs during direct reprograming. PLoS Genet. 2018, 14, e1007846. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef]
- Ponjavic, J.; Oliver, P.L.; Lunter, G.; Ponting, C.P. Genomic and Transcriptional Co-Localization of Protein-Coding and Long Non-Coding RNA Pairs in the Developing Brain. PLoS Genet. 2009, 5, e1000617. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Chau, Y.-Y.; Martínez-Estrada, O.M. Editorial: WT1 in Development and Disease. Front. Cell Dev. Biol. 2022, 10, 969100. [Google Scholar] [CrossRef]
- Liu, N.; Barbosa, A.C.; Chapman, S.L.; Bezprozvannaya, S.; Qi, X.; Richardson, J.A.; Yanagisawa, H.; Olson, E.N. DNA binding-dependent and -independent functions of the Hand2 transcription factor during mouse embryogenesis. Development 2009, 136, 933–942. [Google Scholar] [CrossRef]
- Liu, Y.; Labosky, P.A. Regulation of Embryonic Stem Cell Self-Renewal and Pluripotency by Foxd3. Stem. Cells 2008, 26, 2475–2484. [Google Scholar] [CrossRef]
- Sun, Z.; Xu, X.; He, J.; Murray, A.; Sun, M.; Wei, X.; Wang, X.; McCoig, E.; Xie, E.; Jiang, X.; et al. EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity. Nat. Commun. 2019, 10, 3892. [Google Scholar] [CrossRef]
- Scobie, K.N.; Hall, B.J.; Wilke, S.A.; Klemenhagen, K.C.; Fujii-Kuriyama, Y.; Ghosh, A.; Hen, R.; Sahay, A. Krüppel-Like Factor 9 Is Necessary for Late-Phase Neuronal Maturation in the Developing Dentate Gyrus and during Adult Hippocampal Neurogenesis. J. Neurosci. 2009, 29, 9875–9887. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, S.; Yin, M.; Yin, Y.; Zhou, G.; Zhou, J. Ebf2 is required for development of dopamine neurons in the midbrain periaqueductal gray matter of mouse. Dev. Neurobiol. 2015, 75, 1282–1294. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Fu, Y.; Rusznák, Z.; Herculano-Houzel, S.; Watson, C.; Paxinos, G. Cellular composition characterizing postnatal development and maturation of the mouse brain and spinal cord. Brain Struct. Funct. 2013, 218, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Synapse Dysfunction of Layer V Pyramidal Neurons Precedes Neurodegeneration in a Mouse Model of TDP-43 Proteinopathies. Cereb. Cortex 2016, 27, 3630–3647. [Google Scholar] [CrossRef]
- Ding, L.; Wu, H.; Wang, Y.; Li, Y.; Liang, Z.; Xia, X.; Zheng, J.C. m6A Reader Igf2bp1 Regulates the Inflammatory Responses of Microglia by Stabilizing Gbp11 and Cp mRNAs. Front. Immunol. 2022, 13, 872252. [Google Scholar] [CrossRef]
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef]
- Yoon, K.-J.; Ringeling, F.R.; Vissers, C.; Jacob, F.; Pokrass, M.; Jimenez-Cyrus, D.; Su, Y.; Kim, N.-S.; Zhu, Y.; Zheng, L.; et al. Temporal Control of Mammalian Cortical Neurogenesis by m6A Methylation. Cell 2017, 171, 877–889.e17. [Google Scholar] [CrossRef]
- Cheng, H.; Xuan, H.; Green, C.D.; Han, Y.; Sun, N.; Shen, H.; McDermott, J.; Bennett, D.A.; Lan, F.; Han, J.-D.J. Repression of human and mouse brain inflammaging transcriptome by broad gene-body histone hyperacetylation. Proc. Natl. Acad. Sci. USA 2018, 115, 7611–7616. [Google Scholar] [CrossRef]
- Dönertaş, H.M.; İzgi, H.; Kamacıoğlu, A.; He, Z.; Khaitovich, P.; Somel, M. Gene expression reversal toward pre-adult levels in the aging human brain and age-related loss of cellular identity. Sci. Rep. 2017, 7, 5894. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, R.; Wang, G.; Zhang, Y. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef]
- McIntyre, R.L.; Daniels, E.G.; Molenaars, M.; Houtkooper, R.H.; Janssens, G.E. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol. Med. 2019, 11, e9854. [Google Scholar] [CrossRef]
- Malik, A.N.; Vierbuchen, T.; Hemberg, M.; Rubin, A.A.; Ling, E.; Couch, C.H.; Stroud, H.; Spiegel, I.; Farh, K.K.-H.; Harmin, D.A.; et al. Genome-wide identification and characterization of functional neuronal activity–dependent enhancers. Nat. Neurosci. 2014, 17, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, E.W.; Croteau, D.L.; Bohr, V.A. Heterochromatin: An epigenetic point of view in aging. Exp. Mol. Med. 2020, 52, 1466–1474. [Google Scholar] [CrossRef]
- Villeponteau, B. The heterochromatin loss model of aging. Exp. Gerontol. 1997, 32, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Amaral, M.L.; Zhu, C.; Grieco, S.F.; Hou, X.; Lin, L.; Buchanan, J.; Tong, L.; Preissl, S.; Xu, X.; et al. Single-cell epigenome analysis reveals age-associated decay of heterochromatin domains in excitatory neurons in the mouse brain. Cell Res. 2022, 32, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Patrick, R.; Naval-Sanchez, M.; Deshpande, N.; Huang, Y.; Zhang, J.; Chen, X.; Yang, Y.; Tiwari, K.; Esmaeili, M.; Tran, M.; et al. The activity of early-life gene regulatory elements is hijacked in aging through pervasive AP-1-linked chromatin opening. Cell Metab. 2024, 36, 1858–1881.e23. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.F.; Tezanos, P.; Peñarroya, A.; González-Ramón, A.; Urdinguio, R.G.; Gancedo-Verdejo, J.; Tejedor, J.R.; Santamarina-Ojeda, P.; Alba-Linares, J.J.; Sainz-Ledo, L.; et al. A multiomic atlas of the aging hippocampus reveals molecular changes in response to environmental enrichment. Nat. Commun. 2024, 15, 5829. [Google Scholar] [CrossRef]
- Yang, N.; Occean, J.R.; Melters, D.P.; Shi, C.; Wang, L.; Stransky, S.; Doyle, M.E.; Cui, C.-Y.; Delannoy, M.; Fan, J.; et al. A hyper-quiescent chromatin state formed during aging is reversed by regeneration. Mol. Cell 2023, 83, 1659–1676.e11. [Google Scholar] [CrossRef]
- Hagarman, J.A.; Motley, M.P.; Kristjansdottir, K.; Soloway, P.D. Coordinate Regulation of DNA Methylation and H3K27me3 in Mouse Embryonic Stem Cells. PLoS ONE 2013, 8, e53880. [Google Scholar] [CrossRef]
- Murphy, P.J.; Cipriany, B.R.; Wallin, C.B.; Ju, C.Y.; Szeto, K.; Hagarman, J.A.; Benitez, J.J.; Craighead, H.G.; Soloway, P.D. Single-molecule analysis of combinatorial epigenomic states in normal and tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7772–7777. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Gu, H.; Bartels, S.J.J.; Zhang, Y.; Matarese, F.; Simmer, F.; Marks, H.; Bock, C.; Gnirke, A.; Meissner, A.; et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012, 22, 1128–1138. [Google Scholar] [CrossRef]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef]
- Magalhães, J.P. Programmatic features of aging originating in development: Aging mechanisms beyond molecular damage? FASEB J. 2012, 26, 4821–4826. [Google Scholar] [CrossRef]
- Somel, M.; Guo, S.; Fu, N.; Yan, Z.; Hu, H.Y.; Xu, Y.; Yuan, Y.; Ning, Z.; Hu, Y.; Menzel, C.; et al. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res. 2010, 20, 1207–1218. [Google Scholar] [CrossRef]
- Colantuoni, C.; Lipska, B.K.; Ye, T.; Hyde, T.M.; Tao, R.; Leek, J.T.; Colantuoni, E.A.; Elkahloun, A.G.; Herman, M.M.; Weinberger, D.R.; et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 2011, 478, 519–523. [Google Scholar] [CrossRef]
- Yang, J.-H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e27. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.; Ebrahimi, S.; Wang, S.-C.; Cortese, R.; Kaminsky, Z.A.; Gottesman, I.I.; Burke, J.R.; Plassman, B.L.; Petronis, A. Epigenetic assimilation in the aging human brain. Genome Biol. 2016, 17, 76. [Google Scholar] [CrossRef]
- Gonçalves, C.S.; Le Boiteux, E.; Arnaud, P.; Costa, B.M. HOX gene cluster (de)regulation in brain: From neurodevelopment to malignant glial tumours. Cell. Mol. Life Sci. 2020, 77, 3797–3821. [Google Scholar] [CrossRef] [PubMed]
- Gofflot, F.; Lizen, B. Emerging roles for HOX proteins in synaptogenesis. Int. J. Dev. Biol. 2018, 62, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Declerck, K.; Vanden Berghe, W. Back to the future: Epigenetic clock plasticity towards healthy aging. Mech. Ageing Dev. 2018, 174, 18–29. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signal, B.; Phipps, A.J.; Giles, K.A.; Huskins, S.N.; Mercer, T.R.; Robinson, M.D.; Woodhouse, A.; Taberlay, P.C. Ageing-Related Changes to H3K4me3, H3K27ac, and H3K27me3 in Purified Mouse Neurons. Cells 2024, 13, 1393. https://doi.org/10.3390/cells13161393
Signal B, Phipps AJ, Giles KA, Huskins SN, Mercer TR, Robinson MD, Woodhouse A, Taberlay PC. Ageing-Related Changes to H3K4me3, H3K27ac, and H3K27me3 in Purified Mouse Neurons. Cells. 2024; 13(16):1393. https://doi.org/10.3390/cells13161393
Chicago/Turabian StyleSignal, Brandon, Andrew J. Phipps, Katherine A. Giles, Shannon N. Huskins, Timothy R. Mercer, Mark D. Robinson, Adele Woodhouse, and Phillippa C. Taberlay. 2024. "Ageing-Related Changes to H3K4me3, H3K27ac, and H3K27me3 in Purified Mouse Neurons" Cells 13, no. 16: 1393. https://doi.org/10.3390/cells13161393
APA StyleSignal, B., Phipps, A. J., Giles, K. A., Huskins, S. N., Mercer, T. R., Robinson, M. D., Woodhouse, A., & Taberlay, P. C. (2024). Ageing-Related Changes to H3K4me3, H3K27ac, and H3K27me3 in Purified Mouse Neurons. Cells, 13(16), 1393. https://doi.org/10.3390/cells13161393