
Contribution of AurkA/TPX2 Overexpression to Chromosomal Imbalances and Cancer

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
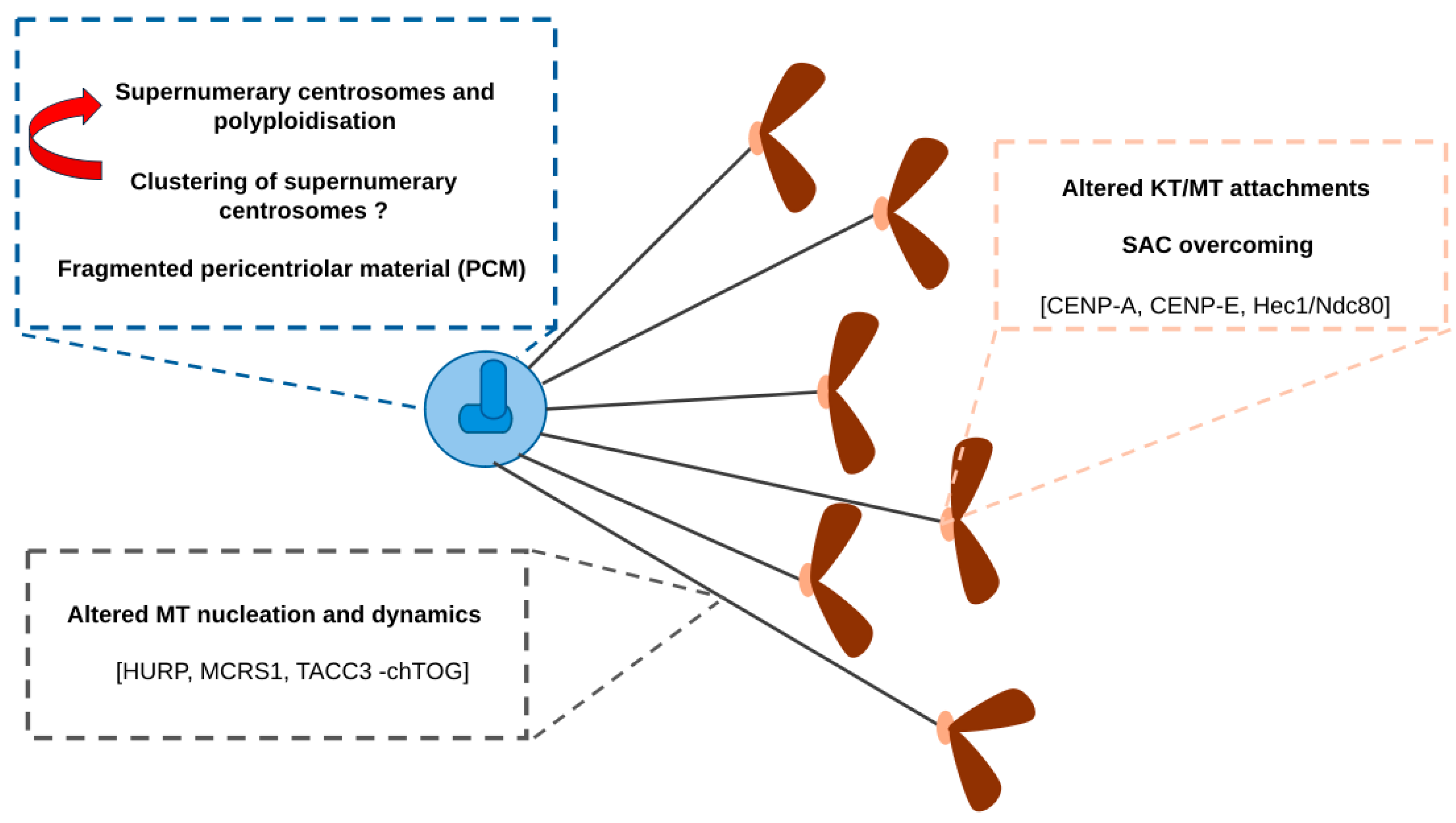
2. Contribution of AurkA Kinase Deregulation to Aneuploidy and CIN
2.1. Effects of AurkA Overexpression on Centrosome Amplification and Fragmentation
2.2. Effects of AurkA Overexpression on MT Dynamics and Chromosome Congression
3. TPX2 and CIN: A Tangled Link
4. AurkA/TPX2 Complex Deregulation Promotes Genome Instability in Cancer
5. The AurkA/TPX2 Complex as a Therapeutic Target
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bakhoum, S.F.; Thompson, S.L.; Manning, A.L.; Compton, D.A. Genome Stability Is Ensured by Temporal Control of Kinetochore–Microtubule Dynamics. Nat. Cell Biol. 2009, 11, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Silkworth, W.T.; Nardi, I.K.; Nicholson, J.M.; Compton, D.A.; Cimini, D. The Mitotic Origin of Chromosomal Instability. Curr. Biol. 2014, 24, 148–149. [Google Scholar] [CrossRef]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic Kinetochore Orientation Is a Major Mechanism of Aneuploidy in Mitotic Mammalian Tissue Cells. J. Cell Biol. 2001, 153, 517. [Google Scholar] [CrossRef] [PubMed]
- van Jaarsveld, R.H.; Kops, G.J.P.L. Difference Makers: Chromosomal Instability versus Aneuploidy in Cancer. Trends Cancer 2016, 2, 561–571. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J.; et al. Chromosomal Instability Accelerates the Evolution of Resistance to Anti-Cancer Therapies. Dev. Cell 2021, 56, 2427–2439. [Google Scholar] [CrossRef]
- Ippolito, M.R.; Martis, V.; Martin, S.; Tijhuis, A.E.; Hong, C.; Wardenaar, R.; Dumont, M.; Zerbib, J.; Spierings, D.C.J.; Fachinetti, D.; et al. Gene Copy-Number Changes and Chromosomal Instability Induced by Aneuploidy Confer Resistance to Chemotherapy. Dev. Cell 2021, 56, 2440–2454. [Google Scholar] [CrossRef]
- Girish, V.; Lakhani, A.A.; Thompson, S.L.; Scaduto, C.M.; Brown, L.M.; Hagenson, R.A.; Sausville, E.L.; Mendelson, B.E.; Kandikuppa, P.K.; Lukow, D.A.; et al. Oncogene-like Addiction to Aneuploidy in Human Cancers. Science 2023, 381, eadg4521. [Google Scholar] [CrossRef]
- Lakhani, A.A.; Thompson, S.L.; Sheltzer, J.M. Aneuploidy in Human Cancer: New Tools and Perspectives. Trends Genet. 2023, 39, 968–980. [Google Scholar] [CrossRef]
- Asteriti, I.A.; Rensen, W.M.; Lindon, C.; Lavia, P.; Guarguaglini, G. The Aurora-A/TPX2 Complex: A Novel Oncogenic Holoenzyme? Biochim. Biophys. Acta Rev. Cancer 2010, 1806, 230–239. [Google Scholar] [CrossRef]
- Szász, A.M.; Li, Q.; Eklund, A.C.; Sztupinszki, Z.; Rowan, A.; Tokés, A.M.; Székely, B.; Kiss, A.; Szendroi, M.; Gyorffy, B.; et al. The CIN4 Chromosomal Instability QPCR Classifier Defines Tumor Aneuploidy and Stratifies Outcome in Grade 2 Breast Cancer. PLoS ONE 2013, 8, e56707. [Google Scholar] [CrossRef]
- Hochegger, H.; Hégarat, N.; Pereira-Leal, J.B. Aurora at the Pole and Equator: Overlapping Functions of Aurora Kinases in the Mitotic Spindle. Open Biol. 2013, 3, 120185. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The Functional Diversity of Aurora Kinases: A Comprehensive Review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef]
- Joukov, V.; De Nicolo, A. Aurora-PLK1 Cascades as Key Signaling Modules in the Regulation of Mitosis. Sci. Signal. 2018, 11, eaar4195. [Google Scholar] [CrossRef] [PubMed]
- Magnaghi-Jaulin, L.; Eot-Houllier, G.; Gallaud, E.; Giet, R. Aurora A Protein Kinase: To the Centrosome and Beyond. Biomolecules 2019, 9, 28. [Google Scholar] [CrossRef]
- van der Waal, M.S.; Hengeveld, R.C.C.; van der Horst, A.; Lens, S.M.A. Cell Division Control by the Chromosomal Passenger Complex. Exp. Cell Res. 2012, 318, 1407–1420. [Google Scholar] [CrossRef]
- Gupta, D.; Kumar, M.; Saifi, S.; Rawat, S.; Ethayathulla, A.S.; Kaur, P. A Comprehensive Review on Role of Aurora Kinase Inhibitors (AKIs) in Cancer Therapeutics. Int. J. Biol. Macromol. 2024, 265, 130913. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Anderson, L.; Zhu, Y.; Mossie, K.; Ng, L.; Souza, B.; Schryver, B.; Flanagan, P.; Clairvoyant, F.; Ginther, C.; et al. A Homologue of Drosophila Aurora Kinase Is Oncogenic and Amplified in Human Colorectal Cancers. EMBO J. 1998, 17, 3052–3065. [Google Scholar] [CrossRef] [PubMed]
- Lakkaniga, N.R.; Wang, Z.; Xiao, Y.; Kharbanda, A.; Lan, L.; Li, H. yu Revisiting Aurora Kinase B: A Promising Therapeutic Target for Cancer Therapy. Med. Res. Rev. 2024, 44, 686–706. [Google Scholar] [CrossRef]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef]
- Lassmann, S.; Shen, Y.; Jütting, U.; Wiehle, P.; Walch, A.; Gitsch, G.; Hasenburg, A.; Werner, M. Predictive Value of Aurora-A/STK15 Expression for Late Stage Epithelial Ovarian Cancer Patients Treated by Adjuvant Chemotherapy. Clin. Cancer Res. 2007, 13, 4083–4091. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Plum, P.S.; Gockel, I.; Thieme, R. Pan-Cancer Analysis and in Vitro Validation of the Oncogenic and Prognostic Roles of AURKA in Human Cancers. Front. Oncol. 2023, 13, 1186101. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Nosho, K.; Shima, K.; Irahara, N.; Kure, S.; Toyoda, S.; Kirkner, G.J.; Goel, A.; Fuchs, C.S.; Ogino, S. Aurora-A Expression Is Independently Associated with Chromosomal Instability in Colorectal Cancer. Neoplasia 2009, 11, 418–425. [Google Scholar] [CrossRef]
- Ertych, N.; Stolz, A.; Stenzinger, A.; Weichert, W.; Kaulfuß, S.; Burfeind, P.; Aigner, A.; Wordeman, L.; Bastians, H. Increased Microtubule Assembly Rates Influence Chromosomal Instability in Colorectal Cancer Cells. Nat. Cell Biol. 2014, 16, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.P.; Wang, J.Y.; Jao, S.W.; Wu, C.C.; Chen, J.H.; Hsiao, K.H.; Lin, C.Y.; Chen, S.H.; Su, S.Y.; Chen, Y.J.; et al. Over-expression of AURKA, SKA3 and DSN1 contributes to colorectal adenoma to carcinoma progression. Oncotarget 2016, 7, 45803–45818. [Google Scholar] [CrossRef]
- Giet, R.; Petretti, C.; Prigent, C. Aurora Kinases, Aneuploidy and Cancer, a Coincidence or a Real Link? Trends Cell Biol. 2005, 15, 241–250. [Google Scholar] [CrossRef]
- Zhu, J.; Abbruzzese, J.L.; Izzo, J.; Hittelman, W.N.; Li, D. AURKA Amplification, Chromosome Instability, and Centrosome Abnormality in Human Pancreatic Carcinoma Cells. Cancer Genet. Cytogenet. 2005, 159, 10–17. [Google Scholar] [CrossRef]
- Bertolin, G.; Tramier, M. Insights into the non-mitotic functions of Aurora kinase A: More than just cell division. Cell Mol. Life Sci. 2020, 77, 1031–1047. [Google Scholar] [CrossRef]
- Naso, F.D.; Boi, D.; Ascanelli, C.; Pamfil, G.; Lindon, C.; Paiardini, A.; Guarguaglini, G. Nuclear Localisation of Aurora-A: Its Regulation and Significance for Aurora-A Functions in Cancer. Oncogene 2021, 40, 3917–3928. [Google Scholar] [CrossRef]
- Mittal, K.; Kaur, J.; Jaczko, M.; Wei, G.; Toss, M.S.; Rakha, E.A.; Janssen, E.A.M.; Søiland, H.; Kucuk, O.; Reid, M.D.; et al. Centrosome Amplification: A Quantifiable Cancer Cell Trait with Prognostic Value in Solid Malignancies. Cancer Metastasis Rev. 2021, 40, 319–339. [Google Scholar] [CrossRef]
- Song, S.; Jung, S.; Kwon, M. Expanding Roles of Centrosome Abnormalities in Cancers. BMB Rep. 2023, 56, 216–224. [Google Scholar] [CrossRef]
- Landen, C.N.; Lin, Y.G.; Immaneni, A.; Deavers, M.T.; Merritt, W.M.; Spannuth, W.A.; Bodurka, D.C.; Gershenson, D.M.; Brinkley, W.R.; Sood, A.K. Overexpression of the Centrosomal Protein Aurora-A Kinase Is Associated with Poor Prognosis in Epithelial Ovarian Cancer Patients. Clin. Cancer Res. 2007, 13, 4098–4104. [Google Scholar] [CrossRef]
- Zhang, D.; Hirota, T.; Marumoto, T.; Shimizu, M.; Kunitoku, N.; Sasayama, T.; Arima, Y.; Feng, L.; Suzuki, M.; Takeya, M.; et al. Cre-LoxP-Controlled Periodic Aurora-A Overexpression Induces Mitotic Abnormalities and Hyperplasia in Mammary Glands of Mouse Models. Oncogene 2004, 23, 8720–8730. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, Y.X.; Qiao, W.; Tominaga, Y.; Ouchi, M.; Ouchi, T.; Deng, C.X. Overexpression of Aurora Kinase A in Mouse Mammary Epithelium Induces Genetic Instability Preceding Mammary Tumor Formation. Oncogene 2006, 25, 7148–7158. [Google Scholar] [CrossRef] [PubMed]
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-A Overexpression Reveals Tetraploidization as a Major Route to Centrosome Amplification in P53-/- Cells. EMBO J. 2002, 21, 483–492. [Google Scholar] [CrossRef]
- Lentini, L.; Amato, A.; Schillaci, T.; Di Leonardo, A. Simultaneous Aurora-A/STK15 Overexpression and Centrosome Amplification Induce Chromosomal Instability in Tumour Cells with a MIN Phenotype. BMC Cancer 2007, 7, 212. [Google Scholar] [CrossRef]
- Torchia, E.C.; Chen, Y.; Sheng, H.; Katayama, H.; Fitzpatrick, J.; Brinkley, W.R.; Caulin, C.; Sen, S.; Roop, D.R. A Genetic Variant of Aurora Kinase A Promotes Genomic Instability Leading to Highly Malignant Skin Tumors. Cancer Res. 2009, 69, 7207–7215. [Google Scholar] [CrossRef]
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.L.; Gray, J.W.; Sahin, A.; Brinkley, B.R.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193. [Google Scholar] [CrossRef]
- Zhang, D.; Shimizu, T.; Araki, N.; Hirota, T.; Yoshie, M.; Ogawa, K.; Nakagata, N.; Takeya, M.; Saya, H. Aurora A Overexpression Induces Cellular Senescence in Mammary Gland Hyperplastic Tumors Developed in P53-Deficient Mice. Oncogene 2008, 27, 4305–4314. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Penrhyn-Lowe, S.; Venkitaraman, A.R. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 2003, 3, 51–62. [Google Scholar] [CrossRef]
- Naso, F.D.; Polverino, F.; Cilluffo, D.; Latini, L.; Stagni, V.; Asteriti, I.A.; Rosa, A.; Soddu, S.; Guarguaglini, G. AurkA/TPX2 Co-Overexpression in Nontransformed Cells Promotes Genome Instability through Induction of Chromosome Mis-Segregation and Attenuation of the P53 Signalling Pathway. Biochim. Biophys. Acta Mol. Basis. Dis. 2024, 1870, 167116. [Google Scholar] [CrossRef]
- Holland, A.J.; Fachinetti, D.; Zhu, Q.; Bauer, M.; Verma, I.M.; Nigg, E.A.; Cleveland, D.W. The Autoregulated Instability of Polo-like Kinase 4 Limits Centrosome Duplication to Once per Cell Cycle. Genes Dev. 2012, 26, 2684–2689. [Google Scholar] [CrossRef]
- Ganem, N.J.; Cornils, H.; Chiu, S.Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Thery, M.; Camargo, F.D.; Pellman, D. Cytokinesis Failure Triggers Hippo Tumor Suppressor Pathway Activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Fava, L.L.; Schuler, F.; Sladky, V.; Haschka, M.D.; Soratroi, C.; Eiterer, L.; Demetz, E.; Weiss, G.; Geley, S.; Nigg, E.A.; et al. The PIDDosome Activates P53 in Response to Supernumerary Centrosomes. Genes Dev. 2017, 31, 34–45. [Google Scholar] [CrossRef]
- Contadini, C.; Monteonofrio, L.; Virdia, I.; Prodosmo, A.; Valente, D.; Chessa, L.; Musio, A.; Fava, L.L.; Rinaldo, C.; Di Rocco, G.; et al. P53 Mitotic Centrosome Localization Preserves Centrosome Integrity and Works as Sensor for the Mitotic Surveillance Pathway. Cell Death Dis. 2019, 10, 850. [Google Scholar] [CrossRef]
- Sasai, K.; Treekitkarnmongkol, W.; Kai, K.; Katayama, H.; Sen, S. Functional Significance of Aurora Kinases-p53 Protein Family Interactions in Cancer. Front. Oncol. 2016, 6, 247. [Google Scholar] [CrossRef]
- Navarro-Serer, B.; Childers, E.P.; Hermance, N.M.; Mercadante, D.; Manning, A.L. Aurora A Inhibition Limits Centrosome Clustering and Promotes Mitotic Catastrophe in Cells with Supernumerary Centrosomes. Oncotarget 2019, 10, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Sasai, K.; Kloc, M.; Brinkley, B.R.; Sen, S. Aurora Kinase-A Regulates Kinetochore/Chromatin Associated Microtubule Assembly in Human Cells. Cell Cycle 2008, 7, 2691–2704. [Google Scholar] [CrossRef]
- Meunier, S.; Timón, K.; Vernos, I. Aurora-A Regulates MCRS1 Function during Mitosis. Cell Cycle 2016, 15, 1779–1786. [Google Scholar] [CrossRef]
- Wu, J.M.; Chen, C.T.; Coumar, M.S.; Lin, W.H.; Chen, Z.J.; Hsu, J.T.A.; Peng, Y.H.; Shiao, H.Y.; Lin, W.H.; Chu, C.Y.; et al. Aurora Kinase Inhibitors Reveal Mechanisms of HURP in Nucleation of Centrosomal and Kinetochore Microtubules. Proc. Natl. Acad. Sci. USA 2013, 110, E1779–E1787. [Google Scholar] [CrossRef]
- Polverino, F.; Naso, F.D.; Asteriti, I.A.; Palmerini, V.; Singh, D.; Valente, D.; Bird, A.W.; Rosa, A.; Mapelli, M.; Guarguaglini, G. The Aurora-A/TPX2 Axis Directs Spindle Orientation in Adherent Human Cells by Regulating NuMA and Microtubule Stability. Curr. Biol. 2021, 31, 658–667. [Google Scholar] [CrossRef]
- Kunitoku, N.; Sasayama, T.; Marumoto, T.; Zhang, D.; Honda, S.; Kobayashi, O.; Hatakeyama, K.; Ushio, Y.; Saya, H.; Hirota, T. CENP-A Phosphorylation by Aurora-A in Prophase Is Required for Enrichment of Aurora-B at Inner Centromeres and for Kinetochore Function. Dev. Cell 2003, 5, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Eot-Houllier, G.; Magnaghi-Jaulin, L.; Fulcrand, G.; Moyroud, F.X.; Monier, S.; Jaulin, C. Aurora A-Dependent CENP-A Phosphorylation at Inner Centromeres Protects Bioriented Chromosomes against Cohesion Fatigue. Nat. Commun. 2018, 9, 1888. [Google Scholar] [CrossRef] [PubMed]
- Ye, A.A.; Deretic, J.; Hoel, C.M.; Hinman, A.W.; Cimini, D.; Welburn, J.P.; Maresca, T.J.; Aurora, A. Kinase Contributes to a Pole-Based Error Correction Pathway. Curr. Biol. 2015, 25, 1842–1851. [Google Scholar] [CrossRef]
- Zhang, X.; Ling, Y.; Guo, Y.; Bai, Y.; Shi, X.; Gong, F.; Tan, P.; Zhang, Y.; Wei, C.; He, X.; et al. Mps1 Kinase Regulates Tumor Cell Viability via Its Novel Role in Mitochondria. Cell Death Dis. 2016, 7, e2292. [Google Scholar] [CrossRef]
- Leça, N.; Barbosa, F.; Rodriguez-Calado, S.; Moura, M.; Pedroso, P.D.; Pinto, I.; Verza, A.E.; Bange, T.; Sunkel, C.E.; Barisic, M.; et al. Proximity-Based Activation of AURORA A by MPS1 Potentiates Error Correction. bioRxiv 2024, 11, 598300. [Google Scholar] [CrossRef]
- Kim, Y.; Holland, A.J.; Lan, W.; Cleveland, D.W. Aurora Kinases and Protein Phosphatase 1 Mediate Chromosome Congression through Regulation of CENP-E. Cell 2010, 142, 444–455. [Google Scholar] [CrossRef]
- Eibes, S.; Rajendraprasad, G.; Guasch-Boldu, C.; Kubat, M.; Steblyanko, Y.; Barisic, M. CENP-E Activation by Aurora A and B Controls Kinetochore Fibrous Corona Disassembly. Nat. Commun. 2023, 14, 5317. [Google Scholar] [CrossRef]
- De Luca, M.; Brunetto, L.; Asteriti, I.A.; Giubettini, M.; Lavia, P.; Guarguaglini, G. Aurora-A and Ch-TOG Act in a Common Pathway in Control of Spindle Pole Integrity. Oncogene 2008, 27, 6539–6549. [Google Scholar] [CrossRef]
- Iemura, K.; Natsume, T.; Maehara, K.; Kanemaki, M.T.; Tanaka, K. Chromosome Oscillation Promotes Aurora a–Dependent Hec1 Phosphorylation and Mitotic Fidelity. J. Cell Biol. 2021, 220, e202006116. [Google Scholar] [CrossRef] [PubMed]
- Sobajima, T.; Kowalczyk, K.M.; Skylakakis, S.; Hayward, D.; Fulcher, L.J.; Neary, C. PP6 Regulation of Aurora A—TPX2 Limits NDC80 Phosphorylation and Mitotic Spindle Size. J. Cell Biol. 2023, 222, e202205117. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, Y.; Lees, E.; Seghezzi, W. AuroraA Overexpression Overrides the Mitotic Spindle Checkpoint Triggered by Nocodazole, a Microtubule Destabilizer. Oncogene 2003, 22, 8293–8301. [Google Scholar] [CrossRef]
- de Castro, I.P.; Malumbres, M. Mitotic Stress and Chromosomal Instability in Cancer: The Case for TPX2. Genes Cancer 2012, 3, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Neumayer, G.; Belzil, C.; Gruss, O.J.; Nguyen, M.D. TPX2: Of Spindle Assembly, DNA Damage Response, and Cancer. Cell. Mol. Life Sci. 2014, 71, 3027–3047. [Google Scholar] [CrossRef]
- Wittmann, T.; Boleti, H.; Antony, C.; Karsenti, E.; Vernos, I. Localization of the Kinesin-like Protein Xklp2 to Spindle Poles Requires a Leucine Zipper, a Microtubule-Associated Protein, and Dynein. J. Cell Biol. 1998, 143, 673–685. [Google Scholar] [CrossRef]
- Gruss, O.J.; Carazo-Salas, R.E.; Schatz, C.A.; Guarguaglini, G.; Kast, J.; Wilm, M.; Le Bot, N.; Vernos, I.; Karsenti, E.; Mattaj, I.W. Ran induces spindle assembly by reversing the inhibitory effect of importin alpha on TPX2 activity. Cell 2001, 104, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gruss, O.J.; Wittmann, M.; Yokoyama, H.; Pepperkok, R.; Kufer, T.; Silljé, H.; Karsenti, E.; Mattaj, I.W.; Vernos, I. Chromosome-Induced Microtubule Assembly Mediated by TPX2 Is Required for Spindle Formation in HeLa Cells. Nat. Cell Biol. 2002, 4, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Gruss, O.J.; Vernos, I. The Mechanism of Spindle Assembly: Functions of Ran and Its Target TPX2. J. Cell Biol. 2004, 166, 949. [Google Scholar] [CrossRef]
- Schatz, C.A.; Santarella, R.; Hoenger, A.; Karsenti, E.; Mattaj, I.W.; Gruss, O.J.; Carazo-Salas, R.E. Importin Alpha-Regulated Nucleation of Microtubules by TPX2. EMBO J. 2003, 22, 2060–2070. [Google Scholar] [CrossRef]
- Alfaro-Aco, R.; Thawani, A.; Petry, S. Structural Analysis of the Role of TPX2 in Branching Microtubule Nucleation. J. Cell Biol. 2017, 216, 983–997. [Google Scholar] [CrossRef]
- Kraus, J.; Alfaro-Aco, R.; Gouveia, B.; Petry, S. Microtubule Nucleation for Spindle Assembly: One Molecule at a Time. Trends Biochem. Sci. 2023, 48, 761–775. [Google Scholar] [CrossRef]
- Mann, B.J.; Balchand, S.K.; Wadsworth, P. Regulation of Kif15 Localization and Motility by the C-Terminus of TPX2 and Microtubule Dynamics. Mol. Biol. Cell 2017, 28, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Balchand, S.K.; Mann, B.J.; Titus, J.; Ross, J.L.; Wadsworth, P. TPX2 Inhibits Eg5 by Interactions with Both Motor and Microtubule. J. Biol. Chem. 2015, 290, 17367–17379. [Google Scholar] [CrossRef]
- Kufer, T.A.; Silljé, H.H.W.; Körner, R.; Gruss, O.J.; Meraldi, P.; Nigg, E.A. Human TPX2 Is Required for Targeting Aurora-A Kinase to the Spindle. J. Cell Biol. 2002, 158, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, R.; Sardon, T.; Vernos, I.; Conti, E. Structural Basis of Aurora-A Activation by TPX2 at the Mitotic Spindle. Mol. Cell 2003, 12, 851–862. [Google Scholar] [CrossRef]
- Giubettini, M.; Asteriti, I.A.; Scrofani, J.; De Luca, M.; Lindon, C.; Lavia, P.; Guarguaglini, G. Control of Aurora-A Stability through Interaction with TPX2. J. Cell Sci. 2011, 124, 113–122. [Google Scholar] [CrossRef]
- Levinson, N.M. The Multifaceted Allosteric Regulation of Aurora Kinase A. Biochem. J. 2018, 475, 2025–2042. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.; Lavia, P.; Guarguaglini, G. A Functional Interplay between Aurora-A, Plk1 and TPX2 at Spindle Poles: Plk1 Controls Centrosomal Localization of Aurora-A and TPX2 Spindle Association. Cell Cycle 2006, 5, 296–303. [Google Scholar] [CrossRef]
- Heidebrecht, H.J.; Buck, F.; Steinmann, J.; Sprenger, R.; Wacker, H.H.; Parwaresch, R. P100: A Novel Proliferation-Associated Nuclear Protein Specifically Restricted to Cell Cycle Phases S, G2, and M. Blood 1997, 90, 226–233. [Google Scholar] [CrossRef]
- He, R.; Zuo, S. A Robust 8-Gene Prognostic Signature for Early-Stage Non-Small Cell Lung Cancer. Front. Oncol. 2019, 9, 693. [Google Scholar] [CrossRef]
- Ouyang, G.; Yi, B.; Pan, G.; Chen, X. A Robust Twelve-Gene Signature for Prognosis Prediction of Hepatocellular Carcinoma. Cancer Cell Int. 2020, 20, 207. [Google Scholar] [CrossRef] [PubMed]
- Mirza, Z.; Ansari, M.S.; Iqbal, M.S.; Ahmad, N.; Alganmi, N.; Banjar, H.; Al-Qahtani, M.H.; Karim, S. Identification of Novel Diagnostic and Prognostic Gene Signature Biomarkers for Breast Cancer Using Artificial Intelligence and Machine Learning Assisted Transcriptomics Analysis. Cancers 2023, 15, 3237. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A Signature of Chromosomal Instability Inferred from Gene Expression Profiles Predicts Clinical Outcome in Multiple Human Cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Matson, D.R.; Denu, R.A.; Zasadil, L.M.; Burkard, M.E.; Weaver, B.A.; Flynn, C.; Stukenberg, P.T. High Nuclear TPX2 Expression Correlates with TP53 Mutation and Poor Clinical Behavior in a Large Breast Cancer Cohort, but Is Not an Independent Predictor of Chromosomal Instability. BMC Cancer 2021, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Ciciarello, M.; Mangiacasale, R.; Thibier, C.; Guarguaglini, G.; Marchetti, E.; Di Fiore, B.; Lavia, P. Importin beta is transported to spindle poles during mitosis and regulates Ran-dependent spindle assembly factors in mammalian cells. J. Cell Sci. 2004, 117 Pt 26, 6511–6522. [Google Scholar] [CrossRef]
- van Gijn, S.E.; Wierenga, E.; van den Tempel, N.; Kok, Y.P.; Heijink, A.M.; Spierings, D.C.J.; Foijer, F.; van Vugt, M.A.T.M.; Fehrmann, R.S.N. TPX2/Aurora Kinase A Signaling as a Potential Therapeutic Target in Genomically Unstable Cancer Cells. Oncogene 2019, 38, 852–867. [Google Scholar] [CrossRef]
- Aguirre-Portolés, C.; Bird, A.W.; Hyman, A.; Cañamero, M.; Pérez De Castro, I.; Malumbres, M. Tpx2 Controls Spindle Integrity, Genome Stability, and Tumor Development. Cancer Res. 2012, 72, 1518–1528. [Google Scholar] [CrossRef]
- Naso, F.D.; Sterbini, V.; Crecca, E.; Asteriti, I.A.; Russo, A.D.; Giubettini, M.; Cundari, E.; Lindon, C.; Rosa, A.; Guarguaglini, G. Excess TPX2 Interferes with Microtubule Disassembly and Nuclei Reformation at Mitotic Exit. Cells 2020, 9, 374. [Google Scholar] [CrossRef]
- Verstraeten, V.L.R.M.; Renes, J.; Ramaekers, F.C.S.; Kamps, M.; Kuijpers, H.J.; Verheyen, F.; Wabitsch, M.; Steijlen, P.M.; Van Steensel, M.A.M.; Broers, J.L.V. Reorganization of the Nuclear Lamina and Cytoskeleton in Adipogenesis. Histochem. Cell Biol. 2011, 135, 251–261. [Google Scholar] [CrossRef]
- Pons, C.; Almacellas, E.; Tauler, A.; Mauvezin, C. Detection of Nuclear Biomarkers for Chromosomal Instability. Methods Mol. Biol. 2022, 2445, 117–125. [Google Scholar] [CrossRef]
- Rohrberg, J.; Van de Mark, D.; Amouzgar, M.; Lee, J.V.; Taileb, M.; Corella, A.; Kilinc, S.; Williams, J.; Jokisch, M.L.; Camarda, R.; et al. MYC Dysregulates Mitosis, Revealing Cancer Vulnerabilities. Cell Rep. 2020, 30, 3368–3382. [Google Scholar] [CrossRef]
- Sillars-Hardebol, A.H.; Carvalho, B.; Tijssen, M.; Beliën, J.A.M.; De Wit, M.; Delis-van Diemen, P.M.; Pontén, F.; Van De Wiel, M.A.; Fijneman, R.J.A.; Meijer, G.A. TPX2 and AURKA Promote 20q Amplicon-Driven Colorectal Adenoma to Carcinoma Progression. Gut 2012, 61, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sheridan, P.; Niida, A.; Sawada, G.; Uchi, R.; Mizuno, H.; Kurashige, J.; Sugimachi, K.; Sasaki, S.; Shimada, Y.; et al. The AURKA/TPX2 Axis Drives Colon Tumorigenesis Cooperatively with MYC. Ann. Oncol. 2015, 26, 935–942. [Google Scholar] [CrossRef]
- Zeng, K.; Bastos, R.N.; Barr, F.A.; Gruneberg, U. Protein Phosphatase 6 Regulates Mitotic Spindle Formation by Controlling the T-Loop Phosphorylation State of Aurora A Bound to Its Activator TPX2. J. Cell Biol. 2010, 191, 1315–1332. [Google Scholar] [CrossRef]
- Hammond, D.; Zeng, K.; Espert, A.; Bastos, R.N.; Baron, R.D.; Gruneberg, U.; Barr, F.A. Melanoma-Associated Mutations in Protein Phosphatase 6 Cause Chromosome Instability and DNA Damage Owing to Dysregulated Aurora-A. J. Cell Sci. 2013, 126, 3429–3440. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.N. Aurora Kinase A Drives the Evolution of Resistance to Third Generation EGFR Inhibitors in Lung Cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef]
- Yang, N.; Wang, C.; Wang, J.; Wang, Z.; Huang, D.; Yan, M.; Kamran, M.; Liu, Q.; Xu, B.L. Aurora Kinase A Stabilizes FOXM1 to Enhance Paclitaxel Resistance in Triple-Negative Breast Cancer. J. Cell Mol. Med. 2019, 23, 6442–6453. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Unger, K.; Schoetz, U.; Belka, C.; Lauber, K. Taxane-Mediated Radiosensitization Derives from Chromosomal Missegregation on Tripolar Mitotic Spindles Orchestrated by AURKA and TPX2. Oncogene 2018, 37, 52–62. [Google Scholar] [CrossRef]
- Eyers, P.A.; Churchill, M.E.A.; Maller, J.L. The Aurora A and Aurora B Protein Kinases: A Single Amino Acid Difference Controls Intrinsic Activity and Activation by TPX2. Cell Cycle 2005, 4, 784–789. [Google Scholar] [CrossRef]
- Pascreau, G.; Eckerdt, F.; Lewellyn, A.L.; Prigent, C.; Maller, J.L. Phosphorylation of P53 Is Regulated by TPX2-Aurora A in Xenopus Oocytes. J. Biol. Chem. 2009, 284, 5497–5505. [Google Scholar] [CrossRef]
- Byrum, A.K.; Carvajal-Maldonado, D.; Mudge, M.C.; Valle-Garcia, D.; Majid, M.C.; Patel, R.; Sowa, M.E.; Gygi, S.P.; Harper, J.W.; Shi, Y.; et al. Mitotic Regulators TPX2 and Aurora A Protect DNA Forks during Replication Stress by Counteracting 53BP1 Function. J. Cell Biol. 2019, 218, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Mosler, T.; Baymaz, H.I.; Gräf, J.F.; Mikicic, I.; Blattner, G.; Bartlett, E.; Ostermaier, M.; Piccinno, R.; Yang, J.; Voigt, A.; et al. PARP1 Proximity Proteomics Reveals Interaction Partners at Stressed Replication Forks. Nucleic Acids Res. 2022, 50, 11600–11618. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz-Sanabria, A.; Nieto-Jimenez, C.; Corrales-Sanchez, V.; Serrano-Oviedo, L.; Andres-Pretel, F.; Montero, J.C.; Burgos, M.; Llopis, J.; Galan-Moya, E.M.; Pandiella, A.; et al. Synthetic Lethality Interaction between Aurora Kinases and CHEK1 Inhibitors in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 2552–2562. [Google Scholar] [CrossRef]
- Do, T.V.; Hirst, J.; Hyter, S.; Roby, K.F.; Godwin, A.K. Aurora A Kinase Regulates Non-Homologous End-Joining and Poly(ADP-Ribose) Polymerase Function in Ovarian Carcinoma Cells. Oncotarget 2017, 8, 50376–50392. [Google Scholar] [CrossRef]
- Lin, C.I.; Chen, Z.C.; Chen, C.H.; Chang, Y.H.; Lee, T.C.; Tang, T.T.; Yu, T.W.; Yang, C.M.; Tsai, M.C.; Huang, C.C.; et al. Co-Inhibition of Aurora A and Haspin Kinases Enhances Survivin Blockage and P53 Induction for Mitotic Catastrophe and Apoptosis in Human Colorectal Cancer. Biochem. Pharmacol. 2022, 206, 115289. [Google Scholar] [CrossRef]
- Li, C.; Liao, J.; Wang, X.; Chen, F.X.; Guo, X.; Chen, X. Combined Aurora Kinase A and CHK1 Inhibition Enhances Radiosensitivity of Triple-Negative Breast Cancer Through Induction of Apoptosis and Mitotic Catastrophe Associated With Excessive DNA Damage. Int. J. Radiat. Oncol. Biol. Phys. 2023, 117, 1241–1254. [Google Scholar] [CrossRef]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Adhikari, B.; Bozilovic, J.; Diebold, M.; Schwarz, J.D.; Hofstetter, J.; Schröder, M.; Wanior, M.; Narain, A.; Vogt, M.; Dudvarski Stankovic, N.; et al. PROTAC-Mediated Degradation Reveals a Non-Catalytic Function of AURORA-A Kinase. Nat. Chem. Biol. 2020, 16, 1179–1188. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.; Duan, J.; Hou, Z.; Wu, Z.; Liu, L.; Lei, H.; Huang, D.; Ren, Y.; Wang, Y.; et al. A Temporal PROTAC Cocktail-Mediated Sequential Degradation of AURKA Abrogates Acute Myeloid Leukemia Stem Cells. Adv. Sci. 2022, 9, 2104823. [Google Scholar] [CrossRef]
- Wang, R.; Ascanelli, C.; Abdelbaki, A.; Fung, A.; Rasmusson, T.; Michaelides, I.; Roberts, K.; Lindon, C. Selective Targeting of Non-Centrosomal AURKA Functions through Use of a Targeted Protein Degradation Tool. Commun. Biol. 2021, 4, 640. [Google Scholar] [CrossRef]
- Asteriti, I.A.; Polverino, F.; Stagni, V.; Sterbini, V.; Ascanelli, C.; Naso, F.D.; Mastrangelo, A.; Rosa, A.; Paiardini, A.; Lindon, C.; et al. AurkA Nuclear Localization Is Promoted by TPX2 and Counteracted by Protein Degradation. Life Sci. Alliance 2023, 6, e202201726. [Google Scholar] [CrossRef]
- Grover, A.; Singh, R.; Shandilya, A.; Priyandoko, D.; Agrawal, V.; Bisaria, V.S.; Wadhwa, R.; Kaul, S.C.; Sundar, D. Ashwagandha Derived Withanone Targets TPX2-Aurora a Complex: Computational and Experimental Evidence to Its Anticancer Activity. PLoS ONE 2012, 7, e30890. [Google Scholar] [CrossRef]
- Burgess, S.G.; Oleksy, A.; Cavazza, T.; Richards, M.W.; Vernos, I.; Matthews, D.; Bayliss, R. Allosteric Inhibition of Aurora-A Kinase by a Synthetic VNAR Domain. Open Biol. 2016, 6, 160089. [Google Scholar] [CrossRef]
- McIntyre, P.J.; Collins, P.M.; Vrzal, L.; Birchall, K.; Arnold, L.H.; Mpamhanga, C.; Coombs, P.J.; Burgess, S.G.; Richards, M.W.; Winter, A.; et al. Characterization of Three Druggable Hot-Spots in the Aurora-A/TPX2 Interaction Using Biochemical, Biophysical, and Fragment-Based Approaches. ACS Chem. Biol. 2017, 12, 2906–2914. [Google Scholar] [CrossRef]
- Janeček, M.; Rossmann, M.; Sharma, P.; Emery, A.; Huggins, D.J.; Stockwell, S.R.; Stokes, J.E.; Tan, Y.S.; Almeida, E.G.; Hardwick, B.; et al. Allosteric Modulation of AURKA Kinase Activity by a Small-Molecule Inhibitor of Its Protein-Protein Interaction with TPX2. Sci. Rep. 2016, 6, 28528. [Google Scholar] [CrossRef]
- Asteriti, I.A.; Daidone, F.; Colotti, G.; Rinaldo, S.; Lavia, P.; Guarguaglini, G.; Paiardini, A. Identification of Small Molecule Inhibitors of the Aurora-A/TPX2 Complex. Oncotarget 2017, 8, 32117–32133. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, E.; Hwang, N.; Yoo, J.; Nam, Y.; Hwang, I.; Park, J.G.; Park, S.E.; Chung, K.S.; Won Chung, H.; et al. Discovery of N-Benzylbenzamide-Based Allosteric Inhibitors of Aurora Kinase A. Bioorg. Med. Chem. 2024, 102, 117658. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polverino, F.; Mastrangelo, A.; Guarguaglini, G. Contribution of AurkA/TPX2 Overexpression to Chromosomal Imbalances and Cancer. Cells 2024, 13, 1397. https://doi.org/10.3390/cells13161397
Polverino F, Mastrangelo A, Guarguaglini G. Contribution of AurkA/TPX2 Overexpression to Chromosomal Imbalances and Cancer. Cells. 2024; 13(16):1397. https://doi.org/10.3390/cells13161397
Chicago/Turabian StylePolverino, Federica, Anna Mastrangelo, and Giulia Guarguaglini. 2024. "Contribution of AurkA/TPX2 Overexpression to Chromosomal Imbalances and Cancer" Cells 13, no. 16: 1397. https://doi.org/10.3390/cells13161397