
Therapeutic Targets in Innate Immunity to Tackle Alzheimer’s Disease

 and
and 
Abstract
1. Introduction
2. Current Therapies: Limitations of Past and Current Strategies
3. Neuroinflammation as a Central Mechanism in AD
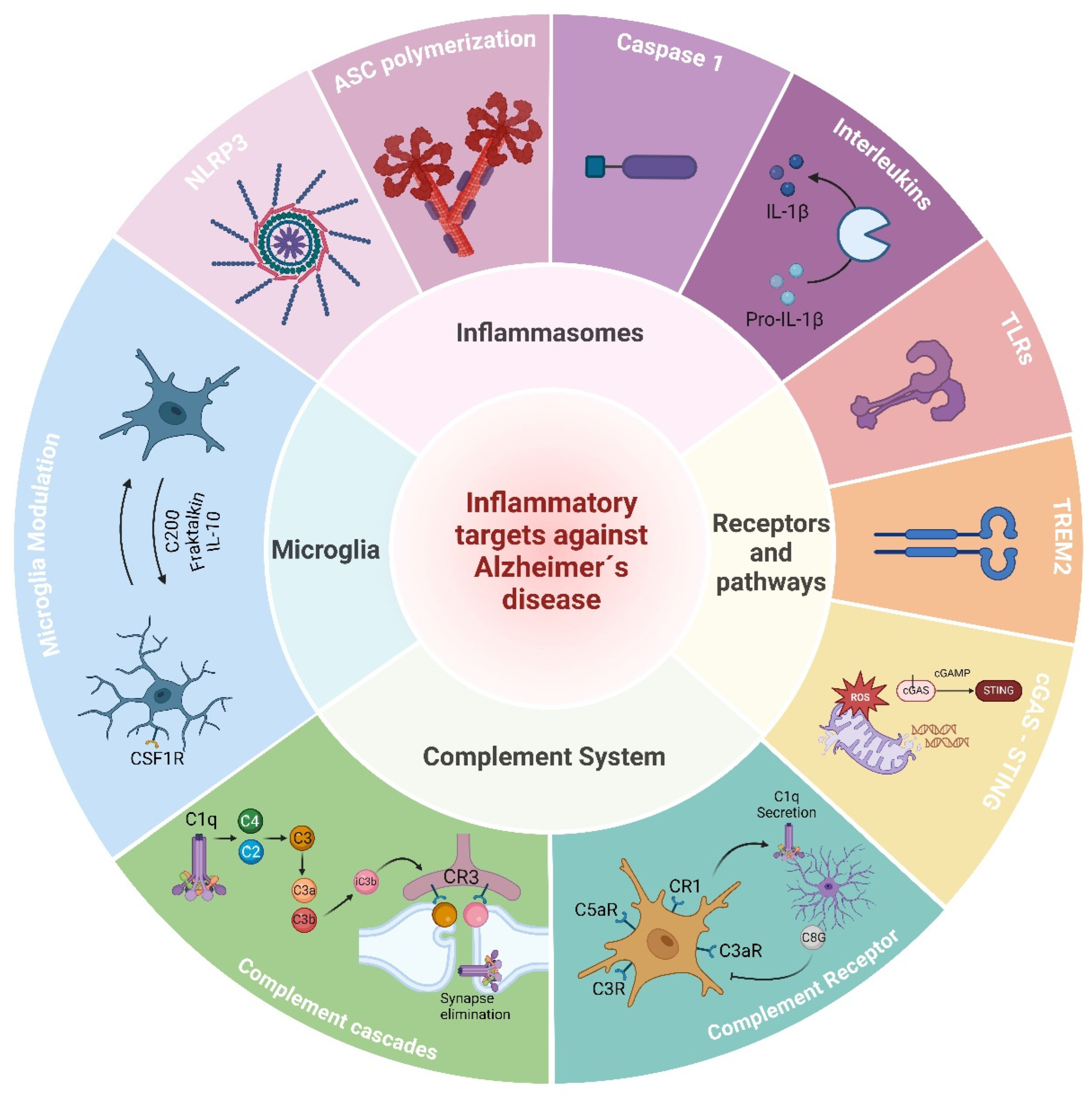
4. Targeting Inflammation and the Immune System in AD
4.1. Microglia
4.1.1. The Role of Microglia in Alzheimer’s Disease
4.1.2. Microglia Modulators against Alzheimer’s Disease
4.2. Triggering Receptor Expressed on Myeloid Cells 2 (TREM2)
4.2.1. Role of TREM2 in Alzheimer’s Disease Pathogenesis and Progression
4.2.2. Therapeutic Targeting of TREM2 against Alzheimer’s Disease
4.3. The Complement System
4.3.1. Complement System in Alzheimer’s Disease
4.3.2. Complement-Targeting Therapy in AD
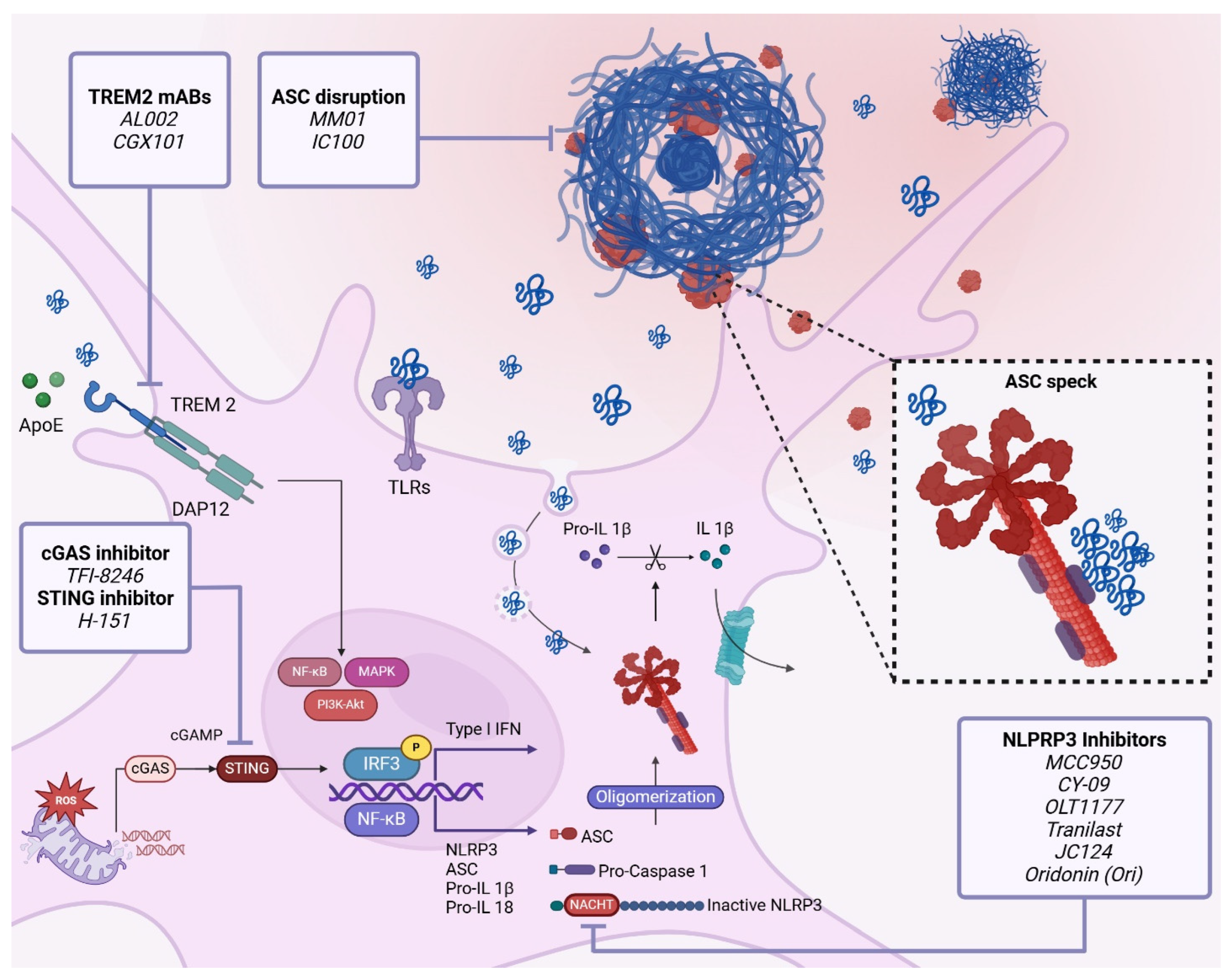
4.4. Inflammasomes
4.4.1. Inflammasome Activation
4.4.2. Relationship between Inflammasomes and Alzheimer’s Disease Pathogenesis and Progression
4.4.3. Therapeutic Targeting of Inflammasomes in AD
NLRP3 Inhibitors
ASC Speck Disruption
Caspase-1 Inhibitors
4.5. Cytosolic DNA Sensors
4.5.1. GAS-STING Pathway in AD
4.5.2. Therapeutic Targeting of the cGAS-STING-IFN Axis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adenovirus-associated virus vectors |
| AD | Alzheimer’s disease |
| ADAM10 | Disintegrin and metalloproteinase domain-containing protein 10 |
| AIM2 | Interferon-inducible protein absent in melanoma 2 |
| ALR | Absent in melanoma 2 protein like receptors |
| ALS | Amyotrophic lateral sclerosis |
| ApoE4 | Apolipoprotein E |
| APP | Amyloid precursor protein |
| ARIA | Amyloid-related imaging abnormalities |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| Aβ | Amyloid β |
| CAA | Cerebral amyloid angiopathy |
| CARD | Caspase recruitment domain |
| CCF | Cytoplasmic chromatin fragments |
| CCL5 | C-C motif chemokine ligand 5 |
| CD200 | Cluster of differentiation 200 |
| CD68 | Cluster of differentiation 68 |
| CDR | Cytosolic DNA receptors |
| cGAMP | Cyclic GMP-AMP |
| cGAS | Cyclic GMP-AMP synthase |
| CLR | C-type lectin-like receptor |
| CNS | Central nervous system |
| CSF1R | Colony stimulating factor 1 receptor |
| CTT | C-terminal tail |
| CX3Cl1 | C-X3-C motif chemokine ligand 1 |
| CX3CR1 | C-X3-C motif chemokine receptor 1 |
| CXCl10 | C-X-C motif chemokine ligand 10 |
| DAI | DNA-dependent activator of IFN-regulatory factors |
| DAMP | Damage-associated molecular pattern |
| DAP12 | DNAX activation protein of 12 kDa |
| DDX41 | DEAD-box helicase 41 |
| ERGIC | ER–Golgi intermediate compartment |
| FDA | Food and Drug Administration |
| FTD | Frontotemporal dementia |
| gDNA | Genomic-DNA |
| GWAS | Genome-wide association studies |
| HT | Huntington’s disease |
| IFI16 | Interferon-gamma-induced protein 16 |
| IFN | Type I interferon |
| IFNAR | Interferon-α receptor |
| IRF3 | interferon regulatory factor 3 |
| ISG | Interferon-stimulated genes |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| LBD | Lligand-binding domain |
| LRR | Leucine-rich repeat |
| MAPK | Mitogen-activated protein kinase |
| MAPT | Microtubule-associated protein tau |
| MCI | Mild cognitive impairment |
| MEF2C | Myocyte enhancer factor 2c |
| mtDNA | Mitochondrial-DNA |
| MyD88 | Myeloid differentiation primary response 88 |
| NFT | Neurofibrillary tangles |
| NF-κB | Nuclear factor kappa B |
| NLR | Nucleotide-binding domain with leucine-rich repeat-containing receptors |
| NLRP | NLR containing a pyrin domain |
| NMDA | N-Methyl-D-aspartic acid |
| NSAID | Nonsteroidal anti-inflammatory drug |
| PAMP | Pathogen-associated molecular pattern |
| PD | Parkinson’s disease |
| PET | Positron emission tomography |
| PQBP1 | Polyglutamine binding protein 1 |
| PRR | Pattern recognition receptor |
| PYD | Pyrin domain |
| PYHIN | Pyrin and hematopoietic interferon-inducible nuclear family |
| RHIM | Receptor-interacting protein homotypic interaction motifs |
| RLR | Retinoic acid-inducible gene-like receptors |
| Stat1 | Signal transducer and activator of transcription 1 |
| STING | Stimulator of interferon genes protein |
| TBI | Traumatic brain injury |
| TBK1 | Tank-binding kinase 1 |
| TLR | Toll-like receptors |
| TREM2 | Triggering receptors expressed on myeloid cells 2 |
| ZBP1 | Z-form DNA binding protein 1 |
References
- Li, X.; Feng, X.; Sun, X.; Hou, N.; Han, F.; Liu, Y. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990–2019. Front. Aging Neurosci. 2022, 14, 937486. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Castro-Gomez, S.; Heneka, M.T. Innate Immune Activation in Neurodegenerative Diseases. Immunity 2024, 57, 790–814. [Google Scholar] [CrossRef]
- Chen, Y.; He, Y.; Han, J.; Wei, W.; Chen, F. Blood-Brain Barrier Dysfunction and Alzheimer’s Disease: Associations, Pathogenic Mechanisms, and Therapeutic Potential. Front. Aging Neurosci. 2023, 15, 1258640. [Google Scholar] [CrossRef]
- Singh, S.; Bhatt, L.K. Targeting Cellular Senescence: A Potential Therapeutic Approach forAlzheimer’s Disease. Curr. Mol. Pharmacol. 2023, 17, e010623217543. [Google Scholar] [CrossRef]
- Yamada, K.; Iwatsubo, T. Involvement of the Glymphatic/Meningeal Lymphatic System in Alzheimer’s Disease: Insights into Proteostasis and Future Directions. Cell. Mol. Life Sci. 2024, 81, 192. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gomez, S.; Binder, J.; Heneka, M.T. [Neuroinflammation as motor of Alzheimer’s disease]. Nervenarzt 2019, 90, 898–906. [Google Scholar] [CrossRef]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An Early and Late Peak in Microglial Activation in Alzheimer’s Disease Trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Boza-Serrano, A.; Yang, Y.; Paulus, A.; Deierborg, T. Innate Immune Alterations Are Elicited in Microglial Cells before Plaque Deposition in the Alzheimer’s Disease Mouse Model 5xFAD. Sci. Rep. 2018, 8, 1550. [Google Scholar] [CrossRef] [PubMed]
- Pizzirusso, G.; Preka, E.; Goikolea, J.; Aguilar-Ruiz, C.; Rodriguez-Rodriguez, P.; Vazquez-Cabrera, G.; Laterza, S.; Latorre-Leal, M.; Eroli, F.; Blomgren, K.; et al. Dynamic Microglia Alterations Associate with Hippocampal Network Impairments: A Turning Point in Amyloid Pathology Progression. Brain Behav. Immun. 2024, 119, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Lopez, O.L.; Becker, J.T.; Wahed, A.S.; Saxton, J.; Sweet, R.A.; Wolk, D.A.; Klunk, W.; Dekosky, S.T. Long-Term Effects of the Concomitant Use of Memantine with Cholinesterase Inhibition in Alzheimer Disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, J.R.M.; Resende, R.; Custódio, J.B.A.; Salvador, J.A.R.; Santos, A.E. BACE1 Inhibitors for Alzheimer’s Disease: Current Challenges and Future Perspectives. J. Alzheimer’s Dis. 2024, 1–26, pre-press. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512. [Google Scholar] [CrossRef]
- Vukmir, R.B. Amyloid-related Imaging Abnormalities (ARIA): Diagnosis, Management, and Care in the Setting of Amyloid-modifying Therapy. Ann. Clin. Transl. Neurol. 2024, 11, 1669–1680. [Google Scholar] [CrossRef]
- Nguyen, H.V.; Mital, S.; Knopman, D.S.; Alexander, G.C. Cost-Effectiveness of Lecanemab for Individuals With Early-Stage Alzheimer Disease. Neurology 2024, 102, e209218. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Rogers, J.; Kirby, L.C.; Hempelman, S.R.; Berry, D.L.; McGeer, P.L.; Kaszniak, A.W.; Zalinski, J.; Cofield, M.; Mansukhani, L.; Willson, P.; et al. Clinical Trial of Indomethacin in Alzheimer’s Disease. Neurology 1993, 43, 1609. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Anti-Inflammatory Agents and Alzheimer Disease: The Last 12 Years. J. Alzheimer’s Dis. 2006, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Andrews, S.J.; Renton, A.E.; Fulton-Howard, B.; Podlesny-Drabiniok, A.; Marcora, E.; Goate, A.M. The Complex Genetic Architecture of Alzheimer’s Disease: Novel Insights and Future Directions. EBioMedicine 2023, 90, 104511. [Google Scholar] [CrossRef]
- Mohapatra, L.; Mishra, D.; Shiomurti Tripathi, A.; Kumar Parida, S. Immunosenescence as a Convergence Pathway in Neurodegeneration. Int. Immunopharmacol. 2023, 121, 110521. [Google Scholar] [CrossRef] [PubMed]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Muzambi, R.; Bhaskaran, K.; Brayne, C.; Davidson, J.A.; Smeeth, L.; Warren-Gash, C. Common Bacterial Infections and Risk of Dementia or Cognitive Decline: A Systematic Review. J. Alzheimer’s Dis. 2020, 76, 1609–1626. [Google Scholar] [CrossRef]
- Asby, D.; Boche, D.; Allan, S.; Love, S.; Miners, J.S. Systemic Infection Exacerbates Cerebrovascular Dysfunction in Alzheimer’s Disease. Brain 2021, 144, 1869–1883. [Google Scholar] [CrossRef]
- Zhuang, Q.-S.; Meng, L.; Wang, Z.; Shen, L.; Ji, H.-F. Associations Between Obesity and Alzheimer’s Disease: Multiple Bioinformatic Analyses. J. Alzheimer’s Dis. 2021, 80, 271–281. [Google Scholar] [CrossRef]
- Grubman, A.; Choo, X.Y.; Chew, G.; Ouyang, J.F.; Sun, G.; Croft, N.P.; Rossello, F.J.; Simmons, R.; Buckberry, S.; Landin, D.V.; et al. Transcriptional Signature in Microglia Associated with Aβ Plaque Phagocytosis. Nat. Commun. 2021, 12, 3015. [Google Scholar] [CrossRef]
- Sun, N.; Victor, M.B.; Park, Y.P.; Xiong, X.; Scannail, A.N.; Leary, N.; Prosper, S.; Viswanathan, S.; Luna, X.; Boix, C.A.; et al. Human Microglial State Dynamics in Alzheimer’s Disease Progression. Cell 2023, 186, 4386–4403.e29. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Shmuel-Galia, L.; Klug, Y.; Porat, Z.; Charni, M.; Zarmi, B.; Shai, Y. Intramembrane Attenuation of the TLR4-TLR6 Dimer Impairs Receptor Assembly and Reduces Microglia-Mediated Neurodegeneration. J. Biol. Chem. 2017, 292, 13415–13427. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 Ligands Promote Sterile Inflammation through Assembly of a Toll-like Receptor 4 and 6 Heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Microglial Dysfunction in Brain Aging and Alzheimer’s Disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained Microglial Depletion with CSF1R Inhibitor Impairs Parenchymal Plaque Development in an Alzheimer’s Disease Model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Kiani Shabestari, S.; Morabito, S.; Danhash, E.P.; McQuade, A.; Sanchez, J.R.; Miyoshi, E.; Chadarevian, J.P.; Claes, C.; Coburn, M.A.; Hasselmann, J.; et al. Absence of Microglia Promotes Diverse Pathologies and Early Lethality in Alzheimer’s Disease Mice. Cell Rep. 2022, 39, 110961. [Google Scholar] [CrossRef]
- Munro, D.A.D.; Bestard-Cuche, N.; McQuaid, C.; Chagnot, A.; Shabestari, S.K.; Chadarevian, J.P.; Maheshwari, U.; Szymkowiak, S.; Morris, K.; Mohammad, M.; et al. Microglia Protect against Age-Associated Brain Pathologies. Neuron 2024, 112, 2732–2748.e8. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial Stimulation of Glioblastoma Invasion Involves Epidermal Growth Factor Receptor (EGFR) and Colony Stimulating Factor 1 Receptor (CSF-1R) Signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Dagher, N.N.; Najafi, A.R.; Kayala, K.M.N.; Elmore, M.R.P.; White, T.E.; Medeiros, R.; West, B.L.; Green, K.N. Colony-Stimulating Factor 1 Receptor Inhibition Prevents Microglial Plaque Association and Improves Cognition in 3xTg-AD Mice. J. Neuroinflamm. 2015, 12, 139. [Google Scholar] [CrossRef]
- Krauser, J.A.; Jin, Y.; Walles, M.; Pfaar, U.; Sutton, J.; Wiesmann, M.; Graf, D.; Pflimlin-Fritschy, V.; Wolf, T.; Camenisch, G.; et al. Phenotypic and Metabolic Investigation of a CSF-1R Kinase Receptor Inhibitor (BLZ945) and Its Pharmacologically Active Metabolite. Xenobiotica 2015, 45, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Uemura, Y.; Ohno, H.; Ohzeki, Y.; Takanashi, H.; Murooka, H.; Kubo, K.; Serizawa, I. The Selective M-CSF Receptor Tyrosine Kinase Inhibitor Ki20227 Suppresses Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2008, 195, 73–80. [Google Scholar] [CrossRef]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R Inhibitor JNJ-40346527 Attenuates Microglial Proliferation and Neurodegeneration in P301S Mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Dudek, A.Z.; Sukari, A.; Call, J.; Kunk, P.R.; Lewis, K.; Gainor, J.F.; Sarantopoulos, J.; Lee, P.; Golden, A.; et al. ARRY-382 in Combination with Pembrolizumab in Patients with Advanced Solid Tumors: Results from a Phase 1b/2 Study. Clin. Cancer Res. 2022, 28, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.G.; McDonald, B.; Parham, J.; Keith, B.; Rusnak, D.W.; Shaw, E.; Jansen, M.; Lin, P.; Payne, A.; Crosby, R.M.; et al. Inhibition of Colony-Stimulating-Factor-1 Signaling in Vivo with the Orally Bioavailable cFMS Kinase Inhibitor GW2580. Proc. Natl. Acad. Sci. USA 2005, 102, 16078–16083. [Google Scholar] [CrossRef]
- Sosna, J.; Philipp, S.; Albay, R.; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early Long-Term Administration of the CSF1R Inhibitor PLX3397 Ablates Microglia and Reduces Accumulation of Intraneuronal Amyloid, Neuritic Plaque Deposition and Pre-Fibrillar Oligomers in 5XFAD Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef]
- Son, Y.; Jeong, Y.J.; Shin, N.-R.; Oh, S.J.; Nam, K.R.; Choi, H.-D.; Choi, J.Y.; Lee, H.-J. Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. Int. J. Mol. Sci. 2020, 21, 5553. [Google Scholar] [CrossRef]
- Casali, B.T.; MacPherson, K.P.; Reed-Geaghan, E.G.; Landreth, G.E. Microglia Depletion Rapidly and Reversibly Alters Amyloid Pathology by Modification of Plaque Compaction and Morphologies. Neurobiol. Dis. 2020, 142, 104956. [Google Scholar] [CrossRef]
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological Targeting of CSF1R Inhibits Microglial Proliferation and Prevents the Progression of Alzheimer’s-like Pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef]
- Boissonneault, V.; Filali, M.; Lessard, M.; Relton, J.; Wong, G.; Rivest, S. Powerful Beneficial Effects of Macrophage Colony-Stimulating Factor on Beta-Amyloid Deposition and Cognitive Impairment in Alzheimer’s Disease. Brain 2009, 132, 1078–1092. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Wu, L.-J. How Microglia Sense and Regulate Neuronal Activity. Glia 2021, 69, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Understanding and Exploiting the Endogenous Interleukin-10/STAT3-Mediated Anti-Inflammatory Response. Curr. Opin. Pharmacol. 2006, 6, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Lobo-Silva, D.; Carriche, G.M.; Castro, A.G.; Roque, S.; Saraiva, M. Balancing the Immune Response in the Brain: IL-10 and Its Regulation. J. Neuroinflamm. 2016, 13, 297. [Google Scholar] [CrossRef]
- Shemer, A.; Scheyltjens, I.; Frumer, G.R.; Kim, J.-S.; Grozovski, J.; Ayanaw, S.; Dassa, B.; Van Hove, H.; Chappell-Maor, L.; Boura-Halfon, S.; et al. Interleukin-10 Prevents Pathological Microglia Hyperactivation Following Peripheral Endotoxin Challenge. Immunity 2020, 53, 1033–1049.e7. [Google Scholar] [CrossRef]
- Weston, L.L.; Jiang, S.; Chisholm, D.; Jantzie, L.L.; Bhaskar, K. Interleukin-10 Deficiency Exacerbates Inflammation-Induced Tau Pathology. J. Neuroinflamm. 2021, 18, 161. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.-V.; Doty, K.R.; Gate, D.; Rodriguez, J.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 Deficiency Rebalances Innate Immunity to Mitigate Alzheimer-like Pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 Alters Immunoproteostasis in APP Mice, Increasing Plaque Burden and Worsening Cognitive Behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Ingraham, K.L.; Swan, R.J.; Jacobsen, M.T.; Andrews, S.J.; Ikezu, T. AAV Serotype 2/1-Mediated Gene Delivery of Anti-Inflammatory Interleukin-10 Enhances Neurogenesis and Cognitive Function in APP+PS1 Mice. Gene Ther. 2012, 19, 724–733. [Google Scholar] [CrossRef]
- Hatherley, D.; Lea, S.M.; Johnson, S.; Barclay, A.N. Structures of CD200/CD200 Receptor Family and Implications for Topology, Regulation, and Evolution. Structure 2013, 21, 820–832. [Google Scholar] [CrossRef]
- Manich, G.; Recasens, M.; Valente, T.; Almolda, B.; González, B.; Castellano, B. Role of the CD200-CD200R Axis During Homeostasis and Neuroinflammation. Neuroscience 2019, 405, 118–136. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-Regulation of the Macrophage Lineage Through Interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Dalsing-Hernandez, J.E.; Campbell, N.A.; Lue, L.-F. Decreased Expression of CD200 and CD200 Receptor in Alzheimer’s Disease: A Potential Mechanism Leading to Chronic Inflammation. Exp. Neurol. 2009, 215, 5–19. [Google Scholar] [CrossRef]
- Feng, D.; Huang, A.; Yan, W.; Chen, D. CD200 Dysfunction in Neuron Contributes to Synaptic Deficits and Cognitive Impairment. Biochem. Biophys. Res. Commun. 2019, 516, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for Neuronally Derived Fractalkine in Mediating Interactions between Neurons and CX3CR1-Expressing Microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Nishiyori, A.; Minami, M.; Ohtani, Y.; Takami, S.; Yamamoto, J.; Kawaguchi, N.; Kume, T.; Akaike, A.; Satoh, M. Localization of Fractalkine and CX 3 CR1 mRNAs in Rat Brain: Does Fractalkine Play a Role in Signaling from Neuron to Microglia? FEBS Lett. 1998, 429, 167–172. [Google Scholar] [CrossRef]
- Iemmolo, M.; Ghersi, G.; Bivona, G. The Cytokine CX3CL1 and ADAMs/MMPs in Concerted Cross-Talk Influencing Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 8026. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V Cortical Neurons Require Microglial Support for Survival during Postnatal Development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Maggi, L. CX3CR1 Deficiency Alters Hippocampal-Dependent Plasticity Phenomena Blunting the Effects of Enriched Environment. Front. Cell. Neurosci. 2011, 5, 22. [Google Scholar] [CrossRef]
- Rogers, J.T.; Morganti, J.M.; Bachstetter, A.D.; Hudson, C.E.; Peters, M.M.; Grimmig, B.A.; Weeber, E.J.; Bickford, P.C.; Gemma, C. CX3CR1 Deficiency Leads to Impairment of Hippocampal Cognitive Function and Synaptic Plasticity. J. Neurosci. 2011, 31, 16241–16250. [Google Scholar] [CrossRef]
- Zujovic, V.; Benavides, J.; Vigé, X.; Carter, C.; Taupin, V. Fractalkine Modulates TNF-Alpha Secretion and Neurotoxicity Induced by Microglial Activation. Glia 2000, 29, 305–315. [Google Scholar] [CrossRef]
- Zujovic, V.; Schussler, N.; Jourdain, D.; Duverger, D.; Taupin, V. In Vivo Neutralization of Endogenous Brain Fractalkine Increases Hippocampal TNFalpha and 8-Isoprostane Production Induced by Intracerebroventricular Injection of LPS. J. Neuroimmunol. 2001, 115, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of Microglial Neurotoxicity by the Fractalkine Receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Llorens-Martín, M.; Perea, J.R.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 Impairs the Internalization of Tau by Microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef]
- Kulczyńska-Przybik, A.; Słowik, A.; Mroczko, P.; Borawski, B.; Groblewska, M.; Borawska, R.; Mroczko, B. Cerebrospinal Fluid and Blood CX3CL1 as a Potential Biomarker in Early Diagnosis and Prognosis of Dementia. Curr. Alzheimer Res. 2020, 17, 709–721. [Google Scholar] [CrossRef]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 Deficiency Alters Microglial Activation and Reduces Beta-Amyloid Deposition in Two Alzheimer’s Disease Mouse Models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; Coleman, U.; Kingery-Gallagher, N.D.; El Khoury, J. Heterozygous CX3CR1 Deficiency in Microglia Restores Neuronal β-Amyloid Clearance Pathways and Slows Progression of Alzheimer’s Like-Disease in PS1-APP Mice. Front. Immunol. 2019, 10, 2780. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive Microglia Drive Tau Pathology and Contribute to the Spreading of Pathological Tau in the Brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Nash, K.R.; Lee, D.C.; Hunt, J.B.; Morganti, J.M.; Selenica, M.-L.; Moran, P.; Reid, P.; Brownlow, M.; Guang-Yu Yang, C.; Savalia, M.; et al. Fractalkine Overexpression Suppresses Tau Pathology in a Mouse Model of Tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.E.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 Knockout Prevents Neuron Loss in a Mouse Model of Alzheimer’s Disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef]
- Karlström, S.; Nordvall, G.; Sohn, D.; Hettman, A.; Turek, D.; Åhlin, K.; Kers, A.; Claesson, M.; Slivo, C.; Lo-Alfredsson, Y.; et al. Substituted 7-Amino-5-Thio-Thiazolo[4,5-d ]Pyrimidines as Potent and Selective Antagonists of the Fractalkine Receptor (CX 3 CR1). J. Med. Chem. 2013, 56, 3177–3190. [Google Scholar] [CrossRef] [PubMed]
- Ridderstad Wollberg, A.; Ericsson-Dahlstrand, A.; Juréus, A.; Ekerot, P.; Simon, S.; Nilsson, M.; Wiklund, S.-J.; Berg, A.-L.; Ferm, M.; Sunnemark, D.; et al. Pharmacological Inhibition of the Chemokine Receptor CX3CR1 Attenuates Disease in a Chronic-Relapsing Rat Model for Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5409–5414. [Google Scholar] [CrossRef]
- Chen, G.; Zhou, Z.; Sha, W.; Wang, L.; Yan, F.; Yang, X.; Qin, X.; Wu, M.; Li, D.; Tian, S.; et al. A Novel CX3CR1 Inhibitor AZD8797 Facilitates Early Recovery of Rat Acute Spinal Cord Injury by Inhibiting Inflammation and Apoptosis. Int. J. Mol. Med. 2020, 45, 1373–1384. [Google Scholar] [CrossRef]
- Gayen, M.; Benoit, M.R.; Fan, Q.; Hudobenko, J.; Yan, R. The CX3CL1 Intracellular Domain Exhibits Neuroprotection via Insulin Receptor/Insulin-like Growth Factor Receptor Signaling. J. Biol. Chem. 2022, 298, 102532. [Google Scholar] [CrossRef]
- Tanaka, Y.; Takeuchi, T.; Yamanaka, H.; Nanki, T.; Umehara, H.; Yasuda, N.; Tago, F.; Kitahara, Y.; Kawakubo, M.; Torii, K.; et al. Long-Term Safety and Efficacy of E6011, an Anti-Fractalkine Monoclonal Antibody, in Patients with Rheumatoid Arthritis Inadequately Responding to Methotrexate. Mod. Rheumatol. 2023, 34, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. The Biology of TREM Receptors. Nat. Rev. Immunol. 2023, 23, 580–594. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New Insights into the Role of TREM2 in Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The Role of TREM2 in Alzheimer’s Disease and Other Neurodegenerative Disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef]
- Shirotani, K.; Hori, Y.; Yoshizaki, R.; Higuchi, E.; Colonna, M.; Saito, T.; Hashimoto, S.; Saito, T.; Saido, T.C.; Iwata, N. Aminophospholipids Are Signal-Transducing TREM2 Ligands on Apoptotic Cells. Sci. Rep. 2019, 9, 7508. [Google Scholar] [CrossRef]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein, E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid That Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef]
- Kulkarni, B.; Kumar, D.; Cruz-Martins, N.; Sellamuthu, S. Role of TREM2 in Alzheimer’s Disease: A Long Road Ahead. Mol. Neurobiol. 2021, 58, 5239–5252. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Xiang, X.; Piers, T.M.; Wefers, B.; Zhu, K.; Mallach, A.; Brunner, B.; Kleinberger, G.; Song, W.; Colonna, M.; Herms, J.; et al. The Trem2 R47H Alzheimer’s Risk Variant Impairs Splicing and Reduces Trem2 mRNA and Protein in Mice but Not in Humans. Mol. Neurodegener. 2018, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, X.; He, P. The Most Prevalent Rare Coding Variants of TREM2 Conferring Risk of Alzheimer’s Disease: A Systematic Review and Meta-analysis. Exp. Ther. Med. 2021, 21, 347. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-Mediated Early Microglial Response Limits Diffusion and Toxicity of Amyloid Plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 Deficiency Eliminates TREM2+ Inflammatory Macrophages and Ameliorates Pathology in Alzheimer’s Disease Mouse Models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Finn, M.B.; Wang, Y.; Shen, A.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Piccio, L.; Colonna, M.; Holtzman, D.M. Altered Microglial Response to Aβ Plaques in APPPS1-21 Mice Heterozygous for TREM2. Mol. Neurodegener. 2014, 9, 20. [Google Scholar] [CrossRef]
- Jiang, T.; Tan, L.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Tan, M.-S.; Gu, L.-Z.; Wang, H.-F.; Ding, Z.-Z.; Zhang, Y.-D.; et al. Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.P.; Neumann, H. Clearance of Apoptotic Neurons without Inflammation by Microglial Triggering Receptor Expressed on Myeloid Cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef]
- Schlepckow, K.; Monroe, K.M.; Kleinberger, G.; Cantuti-Castelvetri, L.; Parhizkar, S.; Xia, D.; Willem, M.; Werner, G.; Pettkus, N.; Brunner, B.; et al. Enhancing Protective Microglial Activities with a Dual Function TREM2 Antibody to the Stalk Region. EMBO Mol. Med. 2020, 12, e11227. [Google Scholar] [CrossRef]
- Schlepckow, K.; Morenas-Rodríguez, E.; Hong, S.; Haass, C. Stimulation of TREM2 with Agonistic Antibodies-an Emerging Therapeutic Option for Alzheimer’s Disease. Lancet Neurol. 2023, 22, 1048–1060. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-Human TREM2 Induces Microglia Proliferation and Reduces Pathology in an Alzheimer’s Disease Model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Fassler, M.; Rappaport, M.S.; Cuño, C.B.; George, J. Engagement of TREM2 by a Novel Monoclonal Antibody Induces Activation of Microglia and Improves Cognitive Function in Alzheimer’s Disease Models. J. Neuroinflamm. 2021, 18, 19. [Google Scholar] [CrossRef]
- West, E.E.; Woodruff, T.; Fremeaux-Bacchi, V.; Kemper, C. Complement in Human Disease: Approved and up-and-Coming Therapeutics. Lancet 2024, 403, 392–405. [Google Scholar] [CrossRef]
- Cavaillon, J.-M.; Sansonetti, P.; Goldman, M. 100th Anniversary of Jules Bordet’s Nobel Prize: Tribute to a Founding Father of Immunology. Front. Immunol. 2019, 10, 2114. [Google Scholar] [CrossRef]
- Ehrlich, P.; Morgenroth, J. Zur Theorie Der Lysinwirkung. Berl. Klin. Wochenschr. 1899, 36, 1–9. [Google Scholar]
- Buchner, H. Zur Nomenklatur Der Schützenden Eiweisskörper. Centr. F Bakteriol. Parasitenk. 1891, 10, 699–701. [Google Scholar]
- Nomenclature of Complement. Bull. World Health Organ. 1968, 39, 935–938.
- Garred, P.; Tenner, A.J.; Mollnes, T.E. Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics. Pharmacol. Rev. 2021, 73, 792–827. [Google Scholar] [CrossRef]
- Nesargikar, P.; Spiller, B.; Chavez, R. The Complement System: History, Pathways, Cascade and Inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel Mechanisms and Functions of Complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Freeley, S.; Kemper, C.; Le Friec, G. The “Ins and Outs” of Complement-Driven Immune Responses. Immunol. Rev. 2016, 274, 16–32. [Google Scholar] [CrossRef]
- Kareem, S.; Jacob, A.; Mathew, J.; Quigg, R.J.; Alexander, J.J. Complement: Functions, Location and Implications. Immunology 2023, 170, 180–192. [Google Scholar] [CrossRef]
- Parker, S.E.; Bellingham, M.C.; Woodruff, T.M. Complement Drives Circuit Modulation in the Adult Brain. Prog. Neurobiol. 2022, 214, 102282. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Hawksworth, O.A.; Li, R.; Balachandran, A.; Lee, J.D.; Sepehrband, F.; Kurniawan, N.; Jeanes, A.; Simmons, D.G.; Wolvetang, E.; et al. Complement C5aR1 Signaling Promotes Polarization and Proliferation of Embryonic Neural Progenitor Cells through PKCζ. J. Neurosci. 2017, 37, 5395–5407. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, A.; Sapir, T.; Haffner-Krausz, R.; Olender, T.; Woodruff, T.M.; Reiner, O. Developmental Activities of the Complement Pathway in Migrating Neurons. Nat. Commun. 2017, 8, 15096. [Google Scholar] [CrossRef]
- Warwick, C.A.; Keyes, A.L.; Woodruff, T.M.; Usachev, Y.M. The Complement Cascade in the Regulation of Neuroinflammation, Nociceptive Sensitization, and Pain. J. Biol. Chem. 2021, 297, 101085. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.-D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia Mediate Forgetting via Complement-Dependent Synaptic Elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Zelek, W.M.; Morgan, B.P. Targeting Complement in Neurodegeneration: Challenges, Risks, and Strategies. Trends Pharmacol. Sci. 2022, 43, 615–628. [Google Scholar] [CrossRef]
- Pouw, R.B.; Ricklin, D. Tipping the Balance: Intricate Roles of the Complement System in Disease and Therapy. Semin. Immunopathol. 2021, 43, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Bohlson, S.S.; Tenner, A.J. Complement in the Brain: Contributions to Neuroprotection, Neuronal Plasticity, and Neuroinflammation. Annu. Rev. Immunol. 2023, 41, 431–452. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-C.; Tang, B.-F.; Zhu, M.-Z.; Lu, J.; Lin, H.-X.; Tang, J.-M.; Li, R.; Ma, T. Analysis of Complement System and Its Related Factors in Alzheimer’s Disease. BMC Neurol. 2023, 23, 446. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning during Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef]
- Wen, L.; Bi, D.; Shen, Y. Complement-Mediated Synapse Loss in Alzheimer’s Disease: Mechanisms and Involvement of Risk Factors. Trends Neurosci. 2024, 47, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Goetzl, E.J.; Schwartz, J.B.; Elahi, F.M.; Rissman, R.A. Complement Protein Levels in Plasma Astrocyte-Derived Exosomes Are Abnormal in Conversion from Mild Cognitive Impairment to Alzheimer’s Disease Dementia. Alzheimers Dement. (Amst.) 2019, 11, 61–66. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High Complement Levels in Astrocyte-Derived Exosomes of Alzheimer Disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Kovács, R.Á.; Vadászi, H.; Bulyáki, É.; Török, G.; Tóth, V.; Mátyás, D.; Kun, J.; Hunyadi-Gulyás, É.; Fedor, F.Z.; Csincsi, Á.; et al. Identification of Neuronal Pentraxins as Synaptic Binding Partners of C1q and the Involvement of NP1 in Synaptic Pruning in Adult Mice. Front. Immunol. 2020, 11, 599771. [Google Scholar] [CrossRef]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.-A.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.-C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Gasque, P.; Vaudry, D.; Gonzalez, B.; Fontaine, M. Expression of a Complete and Functional Complement System by Human Neuronal Cells in Vitro. Int. Immunol. 2000, 12, 1015–1023. [Google Scholar] [CrossRef]
- Lv, Z.; Chen, L.; Chen, P.; Peng, H.; Rong, Y.; Hong, W.; Zhou, Q.; Li, N.; Li, B.; Paolicelli, R.C.; et al. Clearance of β-Amyloid and Synapses by the Optogenetic Depolarization of Microglia Is Complement Selective. Neuron 2024, 112, 740–754.e7. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Stam, F.C. Immunoglobulins and Complement Factors in Senile Plaques. An Immunoperoxidase Study. Acta Neuropathol. 1982, 57, 239–242. [Google Scholar] [CrossRef]
- Spurrier, J.; Nicholson, L.; Fang, X.T.; Stoner, A.J.; Toyonaga, T.; Holden, D.; Siegert, T.R.; Laird, W.; Allnutt, M.A.; Chiasseu, M.; et al. Reversal of Synapse Loss in Alzheimer Mouse Models by Targeting mGluR5 to Prevent Synaptic Tagging by C1Q. Sci. Transl. Med. 2022, 14, eabi8593. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Fraser, D.A.; Arora, M.; Bohlson, S.S.; Lozano, E.; Tenner, A.J. Generation of Inhibitory NFkappaB Complexes and Phosphorylated cAMP Response Element-Binding Protein Correlates with the Anti-Inflammatory Activity of Complement Protein C1q in Human Monocytes. J. Biol. Chem. 2007, 282, 7360–7367. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.E.; Hernandez, M.X.; Dinh, M.L.; Benavente, F.; Vasquez, O.; Tenner, A.J. C1q-Induced LRP1B and GPR6 Proteins Expressed Early in Alzheimer Disease Mouse Models, Are Essential for the C1q-Mediated Protection against Amyloid-β Neurotoxicity. J. Biol. Chem. 2013, 288, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.A.; Pisalyaput, K.; Tenner, A.J. C1q Enhances Microglial Clearance of Apoptotic Neurons and Neuronal Blebs, and Modulates Subsequent Inflammatory Cytokine Production. J. Neurochem. 2010, 112, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lue, L.; Yang, L.; Roher, A.; Kuo, Y.; Strohmeyer, R.; Goux, W.J.; Lee, V.; Johnson, G.V.; Webster, S.D.; et al. Complement Activation by Neurofibrillary Tangles in Alzheimer’s Disease. Neurosci. Lett. 2001, 305, 165–168. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.-M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef]
- Stokowska, A.; Atkins, A.L.; Morán, J.; Pekny, T.; Bulmer, L.; Pascoe, M.C.; Barnum, S.R.; Wetsel, R.A.; Nilsson, J.A.; Dragunow, M.; et al. Complement Peptide C3a Stimulates Neural Plasticity after Experimental Brain Ischaemia. Brain 2017, 140, 353–369. [Google Scholar] [CrossRef]
- Hernandez, M.X.; Namiranian, P.; Nguyen, E.; Fonseca, M.I.; Tenner, A.J. C5a Increases the Injury to Primary Neurons Elicited by Fibrillar Amyloid Beta. ASN Neuro 2017, 9, 1759091416687871. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Ager, R.R.; Chu, S.-H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR Antagonist Decreases Pathology and Enhances Behavioral Performance in Murine Models of Alzheimer’s Disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Carvalho, K.; Balderrama-Gutierrez, G.; Chu, S.-H.; Liang, H.Y.; Schartz, N.D.; Selvan, P.; Petrisko, T.J.; Pan, M.A.; Mortazavi, A.; et al. C5aR1 Antagonism Alters Microglial Polarization and Mitigates Disease Progression in a Mouse Model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2022, 10, 116. [Google Scholar] [CrossRef]
- Panayiotou, E.; Fella, E.; Andreou, S.; Papacharalambous, R.; Gerasimou, P.; Costeas, P.; Angeli, S.; Kousiappa, I.; Papacostas, S.; Kyriakides, T. C5aR Agonist Enhances Phagocytosis of Fibrillar and Non-Fibrillar Aβ Amyloid and Preserves Memory in a Mouse Model of Familial Alzheimer’s Disease. PLoS ONE 2019, 14, e0225417. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Afridi, R.; Han, J.; Jung, H.-G.; Kim, S.-C.; Hwang, E.M.; Shim, H.S.; Ryu, H.; Choe, Y.; Hoe, H.-S.; et al. Gamma Subunit of Complement Component 8 Is a Neuroinflammation Inhibitor. Brain 2021, 144, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome Signalling in Brain Function and Neurodegenerative Disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Latz, E.; Duewell, P. NLRP3 Inflammasome Activation in Inflammaging. Semin. Immunol. 2018, 40, 61–73. [Google Scholar] [CrossRef]
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a Novel 22-kDa Protein, Aggregates during Apoptosis of Human Promyelocytic Leukemia HL-60 Cells. J. Biol. Chem. 1999, 274, 33835–33838. [Google Scholar] [CrossRef]
- Stutz, A.; Horvath, G.L.; Monks, B.G.; Latz, E. ASC Speck Formation as a Readout for Inflammasome Activation. In The Inflammasome; De Nardo, C.M., Latz, E., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1040, pp. 91–101. ISBN 978-1-62703-522-4. [Google Scholar]
- Hulse, J.; Bhaskar, K. Crosstalk Between the NLRP3 Inflammasome/ASC Speck and Amyloid Protein Aggregates Drives Disease Progression in Alzheimer’s and Parkinson’s Disease. Front. Mol. Neurosci. 2022, 15, 805169. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- McManus, R.M.; Latz, E. NLRP3 Inflammasome Signalling in Alzheimer’s Disease. Neuropharmacology 2024, 252, 109941. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 Inflammasome Activation Drives Tau Pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau Activates NLRP3–ASC Inflammasome Exacerbating Exogenously Seeded and Non-Exogenously Seeded Tau Pathology in Vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-Derived ASC Specks Cross-Seed Amyloid-β in Alzheimer’s Disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The Role of NLRP3 Inflammasome in Alzheimer’s Disease and Potential Therapeutic Targets. Front. Pharmacol. 2022, 13, 845185. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ Oligomers and Protofibrils Induce NLRP3 Inflammasome Activation in Microglia. J. Neurochem. 2020, 155, 650–661. [Google Scholar] [CrossRef]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The Adaptor ASC Has Extracellular and “prionoid” Activities That Propagate Inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef]
- Sahillioğlu, A.C.; Özören, N. Artificial Loading of ASC Specks with Cytosolic Antigens. PLoS ONE 2015, 10, e0134912. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Kancheva, D.; De Ren, S.; Saito, T.; Jans, M.; Boone, F.; Vandendriessche, C.; Paesmans, I.; Maurin, H.; Vandenbroucke, R.E.; et al. Inflammasome Signaling Is Dispensable for SS-Amyloid-Induced Neuropathology in Preclinical Models of Alzheimer’s Disease. Front. Immunol. 2024, 15, 1323409. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 Inflammasome with MCC950 Promotes Non-Phlogistic Clearance of Amyloid-β and Cognitive Function in APP/PS1 Mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a Selective and Direct NLRP3 Inhibitor to Treat Inflammatory Disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-Sulfonyl Nitrile Compound, Safe in Humans, Inhibits the NLRP3 Inflammasome and Reverses the Metabolic Cost of Inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; De Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 Closes the Active Conformation of NLRP3 to an Inactive State. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Fekete, C.; Vastagh, C.; Dénes, Á.; Hrabovszky, E.; Nyiri, G.; Kalló, I.; Liposits, Z.; Sárvári, M. Chronic Amyloid β Oligomer Infusion Evokes Sustained Inflammation and Microglial Changes in the Rat Hippocampus via NLRP3. Neuroscience 2019, 405, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, K.; Zhou, Y.; Wang, H.; Zhou, Y.; Liu, S.; Nie, Y.; Li, Y. NLRP3 Inflammasome Inhibitor CY-09 Reduces Hepatic Steatosis in Experimental NAFLD Mice. Biochem. Biophys. Res. Commun. 2021, 534, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 Inflammasome Inhibitor OLT1177 Rescues Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast Directly Targets NLRP 3 to Treat Inflammasome-driven Diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef] [PubMed]
- Fulp, J.; He, L.; Toldo, S.; Jiang, Y.; Boice, A.; Guo, C.; Li, X.; Rolfe, A.; Sun, D.; Abbate, A.; et al. Structural Insights of Benzenesulfonamide Analogues as NLRP3 Inflammasome Inhibitors: Design, Synthesis, and Biological Characterization. J. Med. Chem. 2018, 61, 5412–5423. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin Is a Covalent NLRP3 Inhibitor with Strong Anti-Inflammasome Activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-Inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Thapak, P.; Bishnoi, M.; Sharma, S.S. Tranilast, a Transient Receptor Potential Vanilloid 2 Channel (TRPV2) Inhibitor Attenuates Amyloid β-Induced Cognitive Impairment: Possible Mechanisms. Neuromol. Med. 2022, 24, 183–194. [Google Scholar] [CrossRef]
- Wang, S.; Yang, H.; Yu, L.; Jin, J.; Qian, L.; Zhao, H.; Xu, Y.; Zhu, X. Oridonin Attenuates Aβ1–42-Induced Neuroinflammation and Inhibits NF-κB Pathway. PLoS ONE 2014, 9, e104745. [Google Scholar] [CrossRef]
- Wang, S.; Yu, L.; Yang, H.; Li, C.; Hui, Z.; Xu, Y.; Zhu, X. Oridonin Attenuates Synaptic Loss and Cognitive Deficits in an Aβ1–42-Induced Mouse Model of Alzheimer’s Disease. PLoS ONE 2016, 11, e0151397. [Google Scholar] [CrossRef]
- Soriano-Teruel, P.M.; García-Laínez, G.; Marco-Salvador, M.; Pardo, J.; Arias, M.; DeFord, C.; Merfort, I.; Vicent, M.J.; Pelegrín, P.; Sancho, M.; et al. Identification of an ASC Oligomerization Inhibitor for the Treatment of Inflammatory Diseases. Cell Death Dis. 2021, 12, 1155. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Wanderley, C.W.; Schneider, A.H.; Schiffelers, L.D.; Wuerth, J.D.; Tödtmann, J.M.; Maasewerd, S.; Hawwari, I.; Duthie, F.; Rohland, C.; et al. Nanobodies Dismantle Post-pyroptotic ASC Specks and Counteract Inflammation in Vivo. EMBO Mol. Med. 2022, 14, e15415. [Google Scholar] [CrossRef]
- Adriouch, S.; Pelegrin, P. ASC Nanobodies to Counteract the Consequences of Inflammasome Activation. EMBO Mol. Med. 2022, 14, e16087. [Google Scholar] [CrossRef]
- Desu, H.L.; Plastini, M.; Illiano, P.; Bramlett, H.M.; Dietrich, W.D.; De Rivero Vaccari, J.P.; Brambilla, R.; Keane, R.W. IC100: A Novel Anti-ASC Monoclonal Antibody Improves Functional Outcomes in an Animal Model of Multiple Sclerosis. J. Neuroinflamm. 2020, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a Medium-Chain Triglyceride-Based Ketogenic Formula on Cognitive Function in Patients with Mild-to-Moderate Alzheimer’s Disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate Inhibits Inflammasome Activation to Attenuate Alzheimer’s Disease Pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 Inhibition Alleviates Cognitive Impairment and Neuropathology in an Alzheimer’s Disease Mouse Model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.-Q.; Branton, W.G.; Power, C. Caspase-1 Inhibition Prevents Glial Inflammasome Activation and Pyroptosis in Models of Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Beauchet, O.; LeBlanc, A.C. Pre-Symptomatic Caspase-1 Inhibitor Delays Cognitive Decline in a Mouse Model of Alzheimer Disease and Aging. Nat. Commun. 2020, 11, 4571. [Google Scholar] [CrossRef]
- Liang, H.; Sun, Y.; Gao, A.; Zhang, N.; Jia, Y.; Yang, S.; Na, M.; Liu, H.; Cheng, X.; Fang, X.; et al. Ac-YVAD-Cmk Improves Neurological Function by Inhibiting Caspase-1-Mediated Inflammatory Response in the Intracerebral Hemorrhage of Rats. Int. Immunopharmacol. 2019, 75, 105771. [Google Scholar] [CrossRef]
- Lin, X.; Ye, H.; Siaw-Debrah, F.; Pan, S.; He, Z.; Ni, H.; Xu, Z.; Jin, K.; Zhuge, Q.; Huang, L. AC-YVAD-CMK Inhibits Pyroptosis and Improves Functional Outcome after Intracerebral Hemorrhage. BioMed Res. Int. 2018, 2018, 3706047. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wu, H.; Xie, Y.; Xu, L.; Liu, X.; Wang, W. Caspase-1/IL-1β Represses Membrane Transport of GluA1 by Inhibiting the Interaction between Stargazin and GluA1 in Alzheimer’s Disease. Mol. Med. 2021, 27, 8. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.H.; Schipper, J.L.; Clark, A.C. The Potential for Caspases in Drug Discovery. Curr. Opin. Drug Discov. Dev. 2010, 13, 568–576. [Google Scholar]
- Rotem, Z.; Cox, R.A.; Isaacs, A. Inhibition of Virus Multiplication by Foreign Nucleic Acid. Nature 1963, 197, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.G.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and Function of the cGAS–STING Pathway of Cytosolic DNA Sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, C.R.R.; Maurmann, R.M.; Guma, F.T.; Bauer, M.E.; = Barbe-Tuana, F.M. cGAS-STING Pathway as a Potential Trigger of Immunosenescence and Inflammaging. Front. Immunol. 2023, 14, 1132653. [Google Scholar] [CrossRef]
- Guey, B.; Ablasser, A. Emerging Dimensions of Cellular cGAS-STING Signaling. Curr. Opin. Immunol. 2022, 74, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Dvorkin, S.; Cambier, S.; Volkman, H.E.; Stetson, D.B. New Frontiers in the cGAS-STING Intracellular DNA-Sensing Pathway. Immunity 2024, 57, 718–730. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in Action: Expanding Roles in Immunity and Inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Cheng, J.; Ko, H.; Tang, Y. Cytosolic DNA Sensors in Neurodegenerative Diseases: From Physiological Defenders to Pathological Culprits. EMBO Mol. Med. 2024, 16, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS Produces a 2’-5’-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural Mechanism of Cytosolic DNA Sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Wischnewski, M.; Ablasser, A. Interplay of cGAS with Chromatin. Trends Biochem. Sci. 2021, 46, 822–831. [Google Scholar] [CrossRef]
- Schumann, T.; Ramon, S.C.; Schubert, N.; Mayo, M.A.; Hega, M.; Maser, K.I.; Ada, S.-R.; Sydow, L.; Hajikazemi, M.; Badstübner, M.; et al. Deficiency for SAMHD1 Activates MDA5 in a cGAS/STING-Dependent Manner. J. Exp. Med. 2022, 220, e20220829. [Google Scholar] [CrossRef]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef]
- Mukai, K.; Ogawa, E.; Uematsu, R.; Kuchitsu, Y.; Kiku, F.; Uemura, T.; Waguri, S.; Suzuki, T.; Dohmae, N.; Arai, H.; et al. Homeostatic Regulation of STING by Retrograde Membrane Traffic to the ER. Nat. Commun. 2021, 12, 61. [Google Scholar] [CrossRef]
- Jeltema, D.; Abbott, K.; Yan, N. STING Trafficking as a New Dimension of Immune Signaling. J. Exp. Med. 2023, 220, e20220990. [Google Scholar] [CrossRef]
- Luo, Y.; Chang, L.; Ji, Y.; Liang, T. ER: A Critical Hub for STING Signaling Regulation. Trends Cell Biol. 2024. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy Induction via STING Trafficking Is a Primordial Function of the cGAS Pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Mann, C.C.; Orzalli, M.H.; King, D.S.; Kagan, J.C.; Lee, A.S.Y.; Kranzusch, P.J. Modular Architecture of the STING C-Terminal Tail Allows Interferon and NF-κB Signaling Adaptation. Cell Rep. 2019, 27, 1165–1175.e5. [Google Scholar] [CrossRef] [PubMed]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Ouchi, M.; Ouchi, T. Role of IFI16 in DNA Damage and Checkpoint. Front. Biosci. Landmark 2008, 13, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Ponomareva, L.; Veeranki, S.; Panchanathan, R.; Dickerson, E.; Choubey, D. Differential Roles for the Interferon-Inducible IFI16 and AIM2 Innate Immune Sensors for Cytosolic DNA in Cellular Senescence of Human Fibroblasts. Mol. Cancer Res. 2011, 9, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Bosso, M.; Kirchhoff, F. Emerging Role of PYHIN Proteins as Antiviral Restriction Factors. Viruses 2020, 12, 1464. [Google Scholar] [CrossRef]
- Jønsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 Is Required for DNA Sensing in Human Macrophages by Promoting Production and Function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS Cooperate in the Activation of STING during DNA Sensing in Human Keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Zheng, W.; Zhou, R.; Li, S.; He, S.; Luo, J.; Zhu, M.; Chen, N.; Chen, H.; Meurens, F.; Zhu, J. Porcine IFI16 Negatively Regulates cGAS Signaling Through the Restriction of DNA Binding and Stimulation. Front. Immunol. 2020, 11, 1669. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 Mitigate STING-Induced Inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The Helicase DDX41 Senses Intracellular DNA Mediated by the Adaptor STING in Dendritic Cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Rajendrarao, S.; Shahani, N.; Ramírez-Jarquín, U.N.; Subramaniam, S. Cyclic GMP-AMP Synthase Promotes the Inflammatory and Autophagy Responses in Huntington Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 15989–15999. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau Activates Microglia via the PQBP1-cGAS-STING Pathway to Promote Brain Inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Udeochu, J.C.; Amin, S.; Huang, Y.; Fan, L.; Torres, E.R.S.; Carling, G.; Liu, B.; McGurran, H.; Coronas-Samano, G.; Kauwe, G.; et al. Tau Activation of Microglial cGAS–IFN Reduces MEF2C-Mediated Cognitive Resilience. Nat. Neurosci. 2023, 26, 737–750. [Google Scholar] [CrossRef]
- McCauley, M.E.; O’Rourke, J.G.; Yáñez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.K.M.G.; et al. C9orf72 in Myeloid Cells Suppresses STING-Induced Inflammation. Nature 2020, 585, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Ma, G.; Li, X.; Zhao, J.; Zhao, Z.; Zeng, J. Activation of Innate Immune cGAS-STING Pathway Contributes to Alzheimer’s Pathogenesis in 5×FAD Mice. Nat. Aging 2023, 3, 202–212. [Google Scholar] [CrossRef]
- Barrett, J.P.; Knoblach, S.M.; Bhattacharya, S.; Gordish-Dressman, H.; Stoica, B.A.; Loane, D.J. Traumatic Brain Injury Induces cGAS Activation and Type I Interferon Signaling in Aged Mice. Front. Immunol. 2021, 12, 710608. [Google Scholar] [CrossRef]
- Hinkle, J.T.; Patel, J.; Panicker, N.; Karuppagounder, S.S.; Biswas, D.; Belingon, B.; Chen, R.; Brahmachari, S.; Pletnikova, O.; Troncoso, J.C.; et al. STING Mediates Neurodegeneration and Neuroinflammation in Nigrostriatal α-Synucleinopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2118819119. [Google Scholar] [CrossRef]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.-H.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin Inhibits Cytosolic Mitochondrial DNA–Induced Neuroinflammatory Signaling in Accelerated Aging and Neurodegeneration. J. Clin. Investig. 2020, 131, 3124–3136. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, B.; Sinha, S.C.; Amin, S.; Gan, L. Mechanism and Therapeutic Potential of Targeting cGAS-STING Signaling in Neurological Disorders. Mol. Neurodegener. 2023, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Roy, E.; Wang, B.; Wan, Y.-W.; Chiu, G.S.-H.; Cole, A.L.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I Interferon Response Drives Neuroinflammation and Synapse Loss in Alzheimer Disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Chen, J.; Lo, C.-Y.Z.; Guo, Q.; Feng, J.; Zhao, X.-M. Impaired Type I Interferon Signaling Activity Implicated in the Peripheral Blood Transcriptome of Preclinical Alzheimer’s Disease. eBioMedicine 2022, 82, 104175. [Google Scholar] [CrossRef]
- Roy, E.; Cao, W. Glial Interference: Impact of Type I Interferon in Neurodegenerative Diseases. Mol. Neurodegener. 2022, 17, 78. [Google Scholar] [CrossRef]
- Govindarajulu, M.; Ramesh, S.; Beasley, M.; Lynn, G.; Wallace, C.; Labeau, S.; Pathak, S.; Nadar, R.; Moore, T.; Dhanasekaran, M. Role of cGAS–Sting Signaling in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8151. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ Supplementation Reduces Neuroinflammation and Cell Senescence in a Transgenic Mouse Model of Alzheimer’s Disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- McWhirter, S.M.; Jefferies, C.A. Nucleic Acid Sensors as Therapeutic Targets for Human Disease. Immunity 2020, 53, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS–STING Drives Ageing-Related Inflammation and Neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Target | Endogenous Ligands | Physiological Effects | Potential Compounds for AD | Ref. |
|---|---|---|---|---|
| CSF1R-1 | CSF-1, IL-34 | Survival and proliferation of microglia | Inhibitors: PLX3397 (PEXIDARTINIb); PLX5622, BLZ945, Ki20227, JNJ-527, ARRY-382, and GW2580. Agonists: CSF-1 | [39,40,41,42,43,44,45,50] |
| IL-10RB | IL-10 | Decreases proinflammatory activity of microglia | Agonists: IL-10, AAV–IL-10 | [53,58] |
| CD200R | CD200 | Inhibitory effect on microglia activity | CD200, AAV-CD200 | [63] |
| CX3CR1 | Fractalkine (CX3Cl1), sCX3Cl1 | Synaptic pruning during development, control of neuronal network stability, synaptic plasticity, anti-inflammatory | Agonists: AAV-sCX3Cl1, Tet34 Inhibitors: AZD8797, MAB E6011 | [80,82,86] |
| TREM2 | PS, PE, LDL, S1P, ApoE, or Aβ | Regulation of microglia survival, activation, and phagocytosis. | Antagonist: 4D9, AL002, CGX101 | [106,107,108,109] |
| Complement system | Antibodies | Synaptic pruning, neuronal migration, polarization and proliferation, synaptic elimination, forgetting of remote memories | Inhibitors: PMX205, EP67, Avacopan (C5aR1 antagonists), recombinant C8G | [150,151,152,153] |
| NLRP3 | Aβ, MSU, cholesterol crystals, ATP, cation currents | Neuroinflammatory responses | Inhibitors: MCC950, OLT1177, CY-09, Tranilast, JC124, Oridonin, BAY 11-7082, DFV890 (IFM-2427), Selnoflast, Inzomelid (IZD174), NT-0796, RRX-001, VTX2735, and ZYIL1 | [165,176,177,178,179,183,187,188] |
| ASC | Inflammasome activators | Unique adaptor protein for inflammasomes | MM01, IC100 | [192,195] |
| Caspase-1 | ASC and other inflammasome activators | Cleavage of IL-1β, IL-18, and Gasdemin D | VX-765, Ac-YVAD-cmk | [199,200] |
| cGAS- STING | Cytoplasmic DNA, p-tau | Induction of type I IFN responses | cGAS inhibitor: TFI-8246 STING inhibitor: H-151 | [240,253] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serradas, M.L.; Ding, Y.; Martorell, P.V.; Kulińska, I.; Castro-Gomez, S. Therapeutic Targets in Innate Immunity to Tackle Alzheimer’s Disease. Cells 2024, 13, 1426. https://doi.org/10.3390/cells13171426
Serradas ML, Ding Y, Martorell PV, Kulińska I, Castro-Gomez S. Therapeutic Targets in Innate Immunity to Tackle Alzheimer’s Disease. Cells. 2024; 13(17):1426. https://doi.org/10.3390/cells13171426
Chicago/Turabian StyleSerradas, Maria L., Yingying Ding, Paula V. Martorell, Ida Kulińska, and Sergio Castro-Gomez. 2024. "Therapeutic Targets in Innate Immunity to Tackle Alzheimer’s Disease" Cells 13, no. 17: 1426. https://doi.org/10.3390/cells13171426
APA StyleSerradas, M. L., Ding, Y., Martorell, P. V., Kulińska, I., & Castro-Gomez, S. (2024). Therapeutic Targets in Innate Immunity to Tackle Alzheimer’s Disease. Cells, 13(17), 1426. https://doi.org/10.3390/cells13171426