
Context-Dependent Distinct Roles of SOX9 in Combined Hepatocellular Carcinoma–Cholangiocarcinoma
 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mouse Husbandry and Breeding
2.2. Patient Data
3. Results
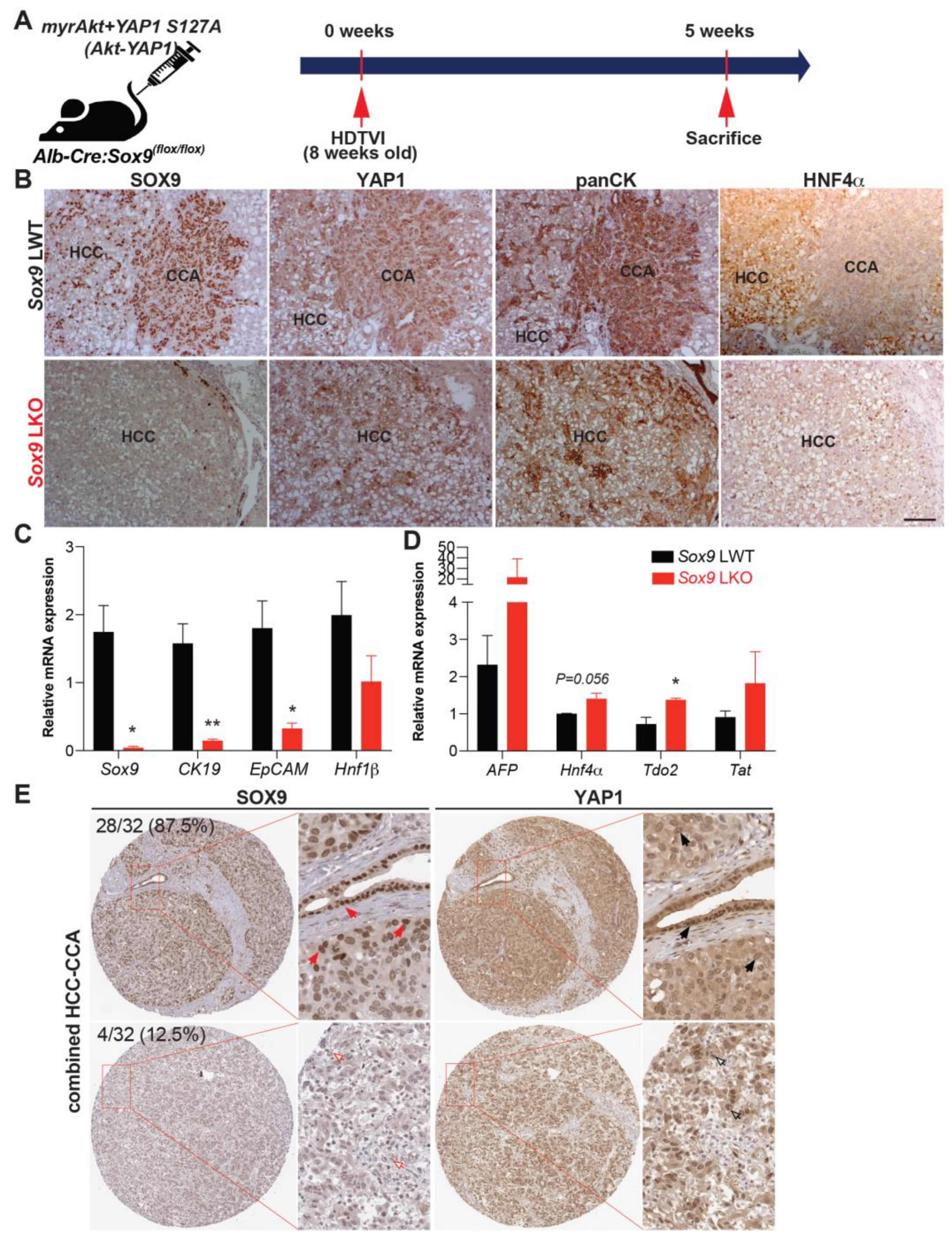
3.1. Forced Expression of Myristoylated Akt and YAP1 in HC Yields cHCC-CCA, While the Chronic Elimination of Sox9 Induces Molecular Phenotype Switch to an Aggressive HCC
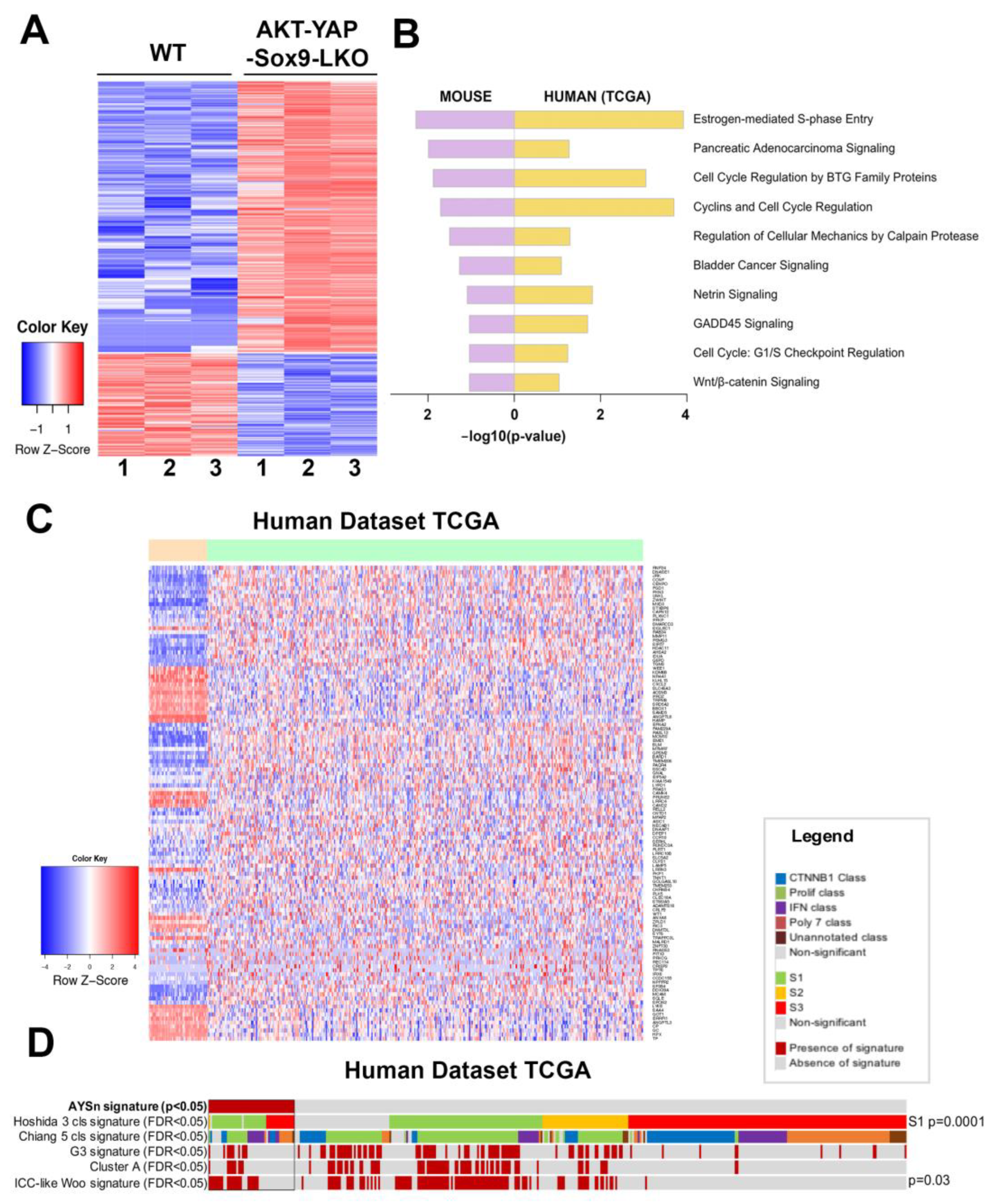
3.2. Transcriptomic Analysis of HCC in Akt-YAP1 Model in Sox9-LKO Reveals Significant Similarity to a Subset of Human HCCs
3.3. SOX9 Is Dispensable for Akt-YAP1-Mediated LPC-like Immature CCA Nodule Formation but Required for Their Transformation into Mature CCA
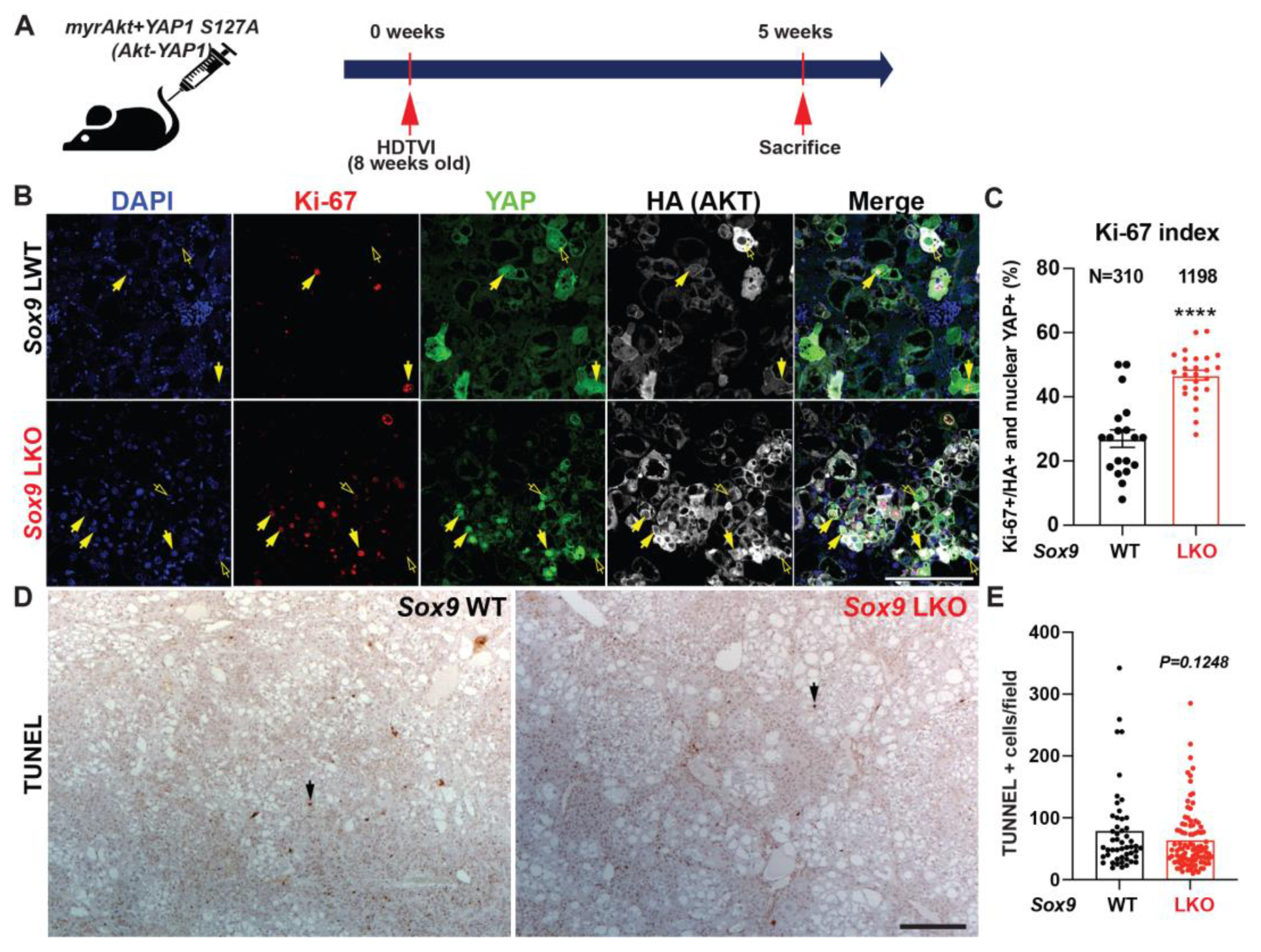
3.4. Distinct Roles SOX9 in Akt-YAP1-Driven HCC Tumor in Regulating Proliferation
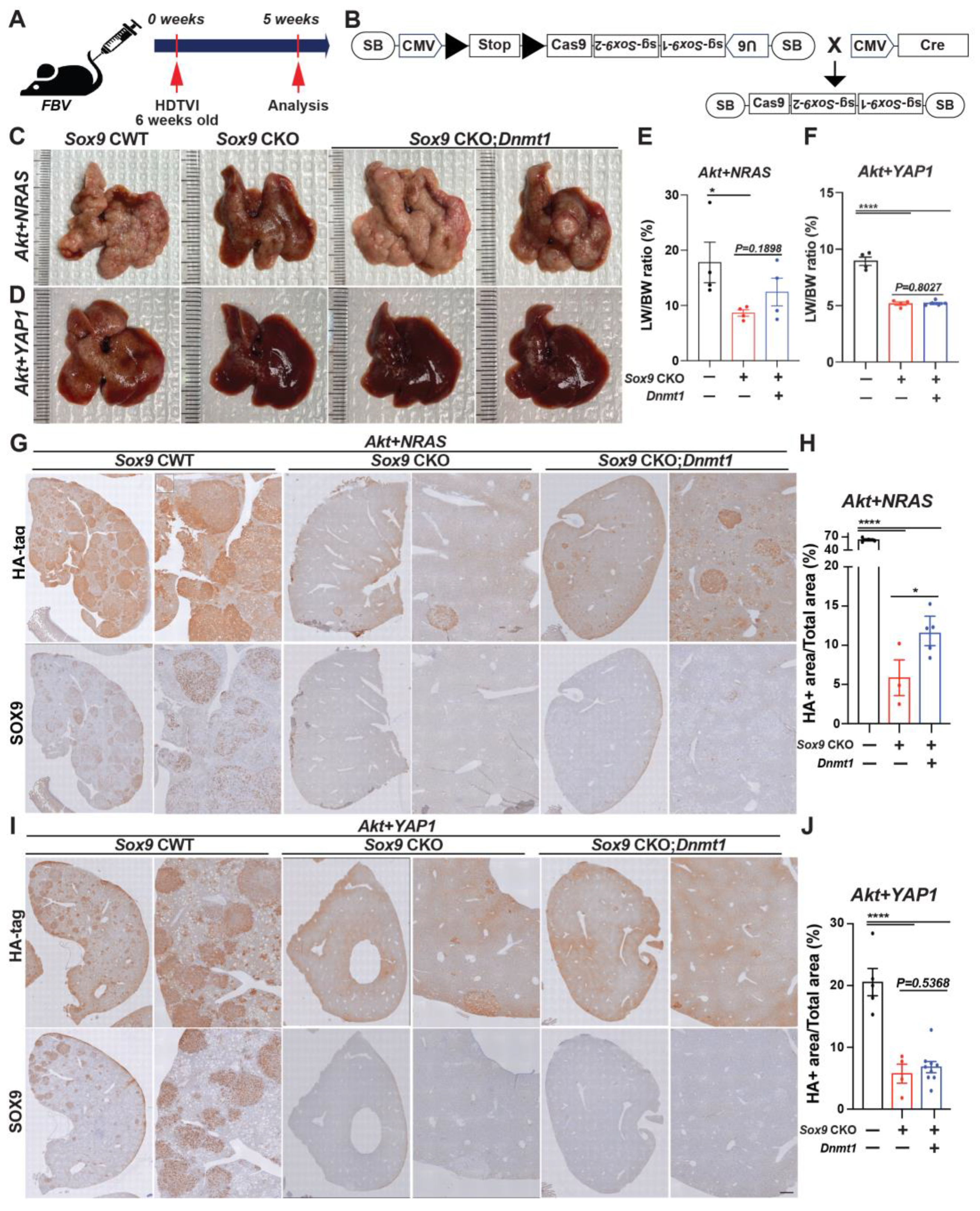
3.5. Tumor-Specific Acute Sox9 Loss Represses YAP1 or NRAS-Dependent cHCC-CCA Development
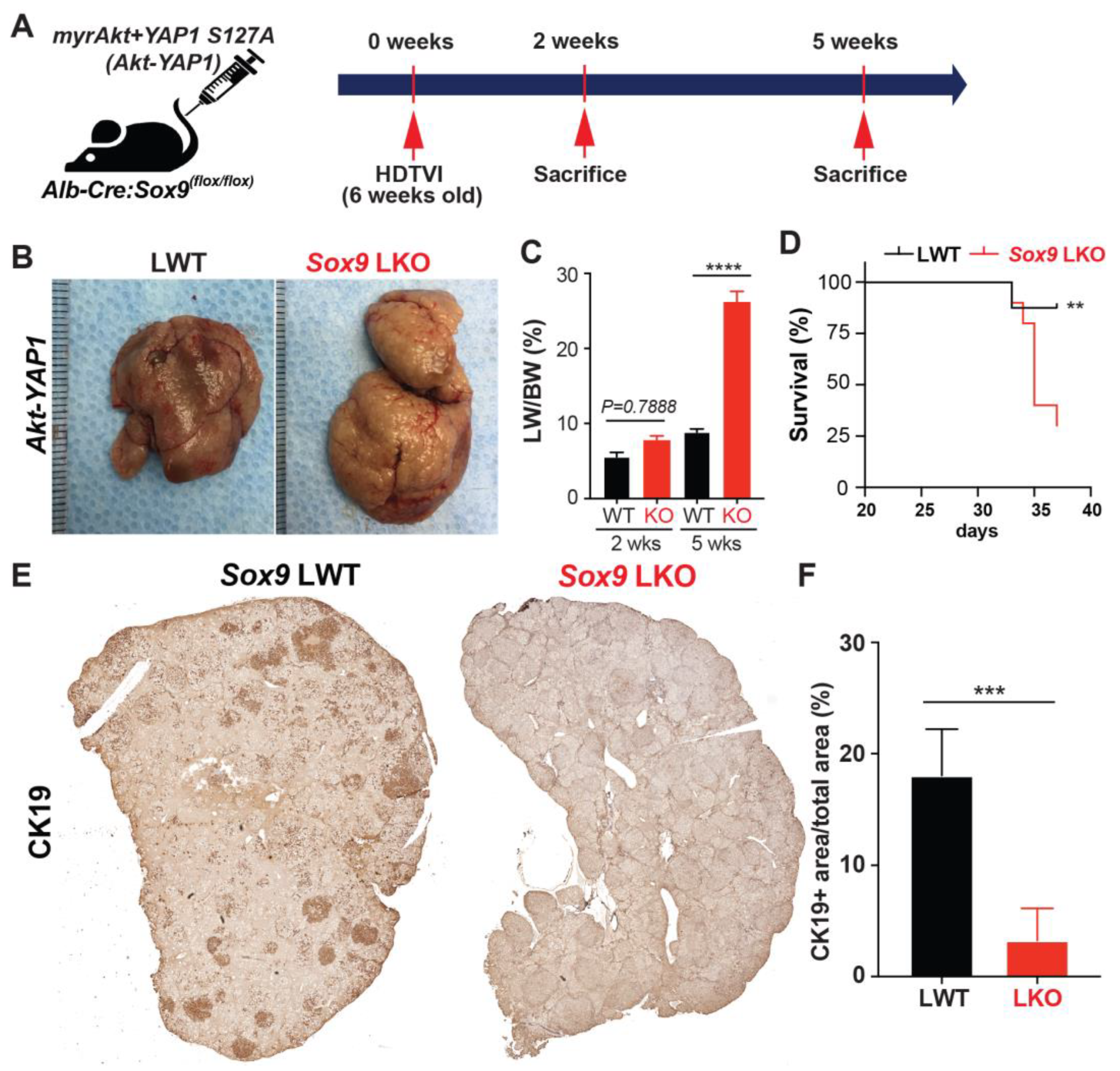
3.6. Therapeutic Deletion of Sox9 Significantly Reduces Advanced Akt-YAP1 Liver Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gigante, E.; Paradis, V.; Ronot, M.; Cauchy, F.; Soubrane, O.; Ganne-Carrie, N.; Nault, J.C. New insights into the pathophysiology and clinical care of rare primary liver cancers. JHEP Rep. 2021, 3, 100174. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Sia, D.; Zhang, Z.; Camprecios, G.; Stueck, A.; Dong, H.; Montal, R.; Torrens, L.; Martinez-Quetglas, I.; Fiel, M.I.; et al. Mixed hepatocellular cholangiocarcinoma tumors: Cholangiolocellular carcinoma is a distinct molecular entity. J. Hepatol. 2017, 66, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Rossner, F.; Sinn, B.V.; Horst, D. Pathology of Combined Hepatocellular Carcinoma-Cholangiocarcinoma: An Update. Cancers 2023, 15, 494. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Haber, P.K.; Sia, D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep. 2021, 3, 100226. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. Combined Hepatocellular-Cholangiocarcinoma: An Update on Pathology and Diagnostic Approach. Biomedicines 2022, 10, 1826. [Google Scholar] [CrossRef]
- Feng, M.; Pan, Y.; Kong, R.; Shu, S. Therapy of Primary Liver Cancer. Innovation 2020, 1, 100032. [Google Scholar] [CrossRef]
- Brindley, P.J.; Bachini, M.; Ilyas, S.I.; Khan, S.A.; Loukas, A.; Sirica, A.E.; Teh, B.T.; Wongkham, S.; Gores, G.J. Cholangiocarcinoma. Nat. Rev. Dis. Primers 2021, 7, 65. [Google Scholar] [CrossRef]
- Hsu, B.Y.; Driscoll, J.; Tateno, C.; Mattis, A.N.; Kelley, R.K.; Willenbring, H. Human Hepatocytes Can Give Rise to Intrahepatic Cholangiocarcinomas. Gastroenterology 2024, in press. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Zhang, S.; Jia, J.; Liu, X.; Zhang, J.; Wang, P.; Song, X.; Che, L.; Liu, K.; et al. Distinct and Overlapping Roles of Hippo Effectors YAP and TAZ During Human and Mouse Hepatocarcinogenesis. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1095–1117. [Google Scholar] [CrossRef]
- Wang, J.; Dong, M.; Xu, Z.; Song, X.; Zhang, S.; Qiao, Y.; Che, L.; Gordan, J.; Hu, K.; Liu, Y.; et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018, 37, 3229–3242. [Google Scholar] [CrossRef]
- Matter, M.S.; Marquardt, J.U.; Andersen, J.B.; Quintavalle, C.; Korokhov, N.; Stauffer, J.K.; Kaji, K.; Decaens, T.; Quagliata, L.; Elloumi, F.; et al. Oncogenic driver genes and the inflammatory microenvironment dictate liver tumor phenotype. Hepatology 2016, 63, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Wang, C.; Mattu, S.; Destefanis, G.; Ladu, S.; Delogu, S.; Armbruster, J.; Fan, L.; Lee, S.A.; Jiang, L.; et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012, 55, 833–845. [Google Scholar] [CrossRef]
- Hu, S.; Molina, L.; Tao, J.; Liu, S.; Hassan, M.; Singh, S.; Poddar, M.; Bell, A.; Sia, D.; Oertel, M.; et al. NOTCH-YAP1/TEAD-DNMT1 Axis Drives Hepatocyte Reprogramming Into Intrahepatic Cholangiocarcinoma. Gastroenterology 2022, 163, 449–465. [Google Scholar] [CrossRef]
- Liu, Y.; Zhuo, S.; Zhou, Y.; Ma, L.; Sun, Z.; Wu, X.; Wang, X.W.; Gao, B.; Yang, Y. Yap-Sox9 signaling determines hepatocyte plasticity and lineage-specific hepatocarcinogenesis. J. Hepatol. 2022, 76, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Poncy, A.; Antoniou, A.; Cordi, S.; Pierreux, C.E.; Jacquemin, P.; Lemaigre, F.P. Transcription factors SOX4 and SOX9 cooperatively control development of bile ducts. Dev. Biol. 2015, 404, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Panikkar, A.; Xu, J.; Antoniou, A.; Raynaud, P.; Lemaigre, F.; Stanger, B.Z. Notch signaling controls liver development by regulating biliary differentiation. Development 2009, 136, 1727–1739. [Google Scholar] [CrossRef]
- Yin, C. Molecular mechanisms of Sox transcription factors during the development of liver, bile duct, and pancreas. Semin. Cell Dev. Biol. 2017, 63, 68–78. [Google Scholar] [CrossRef]
- Weisend, C.M.; Kundert, J.A.; Suvorova, E.S.; Prigge, J.R.; Schmidt, E.E. Cre activity in fetal albCre mouse hepatocytes: Utility for developmental studies. Genesis 2009, 47, 789–792. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E.; Mouse Genome Database Group. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef]
- Hoshida, Y. Nearest template prediction: A single-sample-based flexible class prediction with confidence assessment. PLoS ONE 2010, 5, e15543. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Lee, J.H.; Yoon, J.H.; Kim, C.Y.; Lee, H.S.; Jang, J.J.; Yi, N.J.; Suh, K.S.; Lee, K.U.; Park, E.S.; et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 2010, 70, 3034–3041. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, M.; Simbula, G. Role of the Hippo pathway in liver regeneration and repair: Recent advances. Inflamm. Regen. 2022, 42, 59. [Google Scholar] [CrossRef]
- Hu, S.; Cao, C.; Poddar, M.; Delgado, E.; Singh, S.; Singh-Varma, A.; Stolz, D.B.; Bell, A.; Monga, S.P. Hepatocyte β-catenin loss is compensated by Insulin-mTORC1 activation to promote liver regeneration. Hepatology 2023, 77, 1593–1611. [Google Scholar] [CrossRef]
- Antoniou, A.; Raynaud, P.; Cordi, S.; Zong, Y.; Tronche, F.; Stanger, B.Z.; Jacquemin, P.; Pierreux, C.E.; Clotman, F.; Lemaigre, F.P. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology 2009, 136, 2325–2333. [Google Scholar] [CrossRef]
- Xu, W.P.; Cui, Y.L.; Chen, L.L.; Ding, K.; Ding, C.H.; Chen, F.; Zhang, X.; Xie, W.F. Deletion of Sox9 in the liver leads to hepatic cystogenesis in mice by transcriptionally downregulating Sec63. J. Pathol. 2021, 254, 57–69. [Google Scholar] [CrossRef]
- Lesaffer, B.; Verboven, E.; Van Huffel, L.; Moya, I.M.; van Grunsven, L.A.; Leclercq, I.A.; Lemaigre, F.P.; Halder, G. Comparison of the Opn-CreER and Ck19-CreER Drivers in Bile Ducts of Normal and Injured Mouse Livers. Cells 2019, 8, 380. [Google Scholar] [CrossRef]
- Ko, S.; Russell, J.O.; Molina, L.M.; Monga, S.P. Liver Progenitors and Adult Cell Plasticity in Hepatic Injury and Repair: Knowns and Unknowns. Annu. Rev. Pathol. 2020, 15, 23–50. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef]
- Jiang, M.; Ren, J.; Belmonte, J.C.I.; Liu, G.H. Hepatocyte reprogramming in liver regeneration: Biological mechanisms and applications. FEBS J. 2023, 290, 5674–5688. [Google Scholar] [CrossRef] [PubMed]
- Romanazzo, S.; Lin, K.; Srivastava, P.; Kilian, K.A. Targeting cell plasticity for regeneration: From in vitro to in vivo reprogramming. Adv. Drug. Deliv. Rev. 2020, 161-162, 124–144. [Google Scholar] [CrossRef]
- Gadd, V.L.; Aleksieva, N.; Forbes, S.J. Epithelial Plasticity during Liver Injury and Regeneration. Cell Stem Cell 2020, 27, 557–573. [Google Scholar] [CrossRef]
- Li, W.; Li, L.; Hui, L. Cell Plasticity in Liver Regeneration. Trends Cell Biol. 2020, 30, 329–338. [Google Scholar] [CrossRef]
- Cigliano, A.; Zhang, S.; Ribback, S.; Steinmann, S.; Sini, M.; Ament, C.E.; Utpatel, K.; Song, X.; Wang, J.; Pilo, M.G.; et al. The Hippo pathway effector TAZ induces intrahepatic cholangiocarcinoma in mice and is ubiquitously activated in the human disease. J. Exp. Clin. Cancer Res. 2022, 41, 192. [Google Scholar] [CrossRef]
- Kim, M.; Delgado, E.; Ko, S. DNA methylation in cell plasticity and malignant transformation in liver diseases. Pharmacol. Ther. 2023, 241, 108334. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.; Kim, M.; Molina, L.; Sirica, A.E.; Monga, S.P. YAP1 activation and Hippo pathway signaling in the pathogenesis and treatment of intrahepatic cholangiocarcinoma. Adv. Cancer Res. 2022, 156, 283–317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H.; Cui, G.; Liang, B.; Chen, X.; Ko, S.; Affo, S.; Song, X.; Liao, Y.; Feng, J.; et al. beta-Catenin Sustains and Is Required for YES-associated Protein Oncogenic Activity in Cholangiocarcinoma. Gastroenterology 2022, 163, 481–494. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Driskill, J.H.; Pan, D. The Hippo Pathway in Liver Homeostasis and Pathophysiology. Annu. Rev. Pathol. 2021, 16, 299–322. [Google Scholar] [CrossRef]
- Yamamoto, M.; Xin, B.; Watanabe, K.; Ooshio, T.; Fujii, K.; Chen, X.; Okada, Y.; Abe, H.; Taguchi, Y.; Miyokawa, N.; et al. Oncogenic Determination of a Broad Spectrum of Phenotypes of Hepatocyte-Derived Mouse Liver Tumors. Am. J. Pathol. 2017, 187, 2711–2725. [Google Scholar] [CrossRef]
- Russell, J.O.; Camargo, F.D. Hippo signalling in the liver: Role in development, regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 297–312. [Google Scholar] [CrossRef]
- Zhang, S.; Song, X.; Cao, D.; Xu, Z.; Fan, B.; Che, L.; Hu, J.; Chen, B.; Dong, M.; Pilo, M.G.; et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef]
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu-Ebinger, S.; et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014, 147, 690–701. [Google Scholar] [CrossRef]
- Song, S.; Ajani, J.A.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.R.; Xiao, L.; Hofstetter, W.L.; Weston, B.; et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef]
- Goto, H.; Nishio, M.; To, Y.; Oishi, T.; Miyachi, Y.; Maehama, T.; Nishina, H.; Akiyama, H.; Mak, T.W.; Makii, Y.; et al. Loss of Mob1a/b in mice results in chondrodysplasia due to YAP1/TAZ-TEAD-dependent repression of SOX9. Development 2018, 145, dev159244. [Google Scholar] [CrossRef]
- Aguilar-Medina, M.; Avendano-Felix, M.; Lizarraga-Verdugo, E.; Bermudez, M.; Romero-Quintana, J.G.; Ramos-Payan, R.; Ruiz-Garcia, E.; Lopez-Camarillo, C. SOX9 Stem-Cell Factor: Clinical and Functional Relevance in Cancer. J. Oncol. 2019, 2019, 6754040. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Sahoo, R.K.; Biswal, B.K. SOX9 as an emerging target for anticancer drugs and a prognostic biomarker for cancer drug resistance. Drug Discov. Today 2022, 27, 2541–2550. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, R.; Singh, S.; Poddar, M.; Xu, E.; Oertel, M.; Chen, X.; Ganesh, S.; Abrams, M.; Monga, S.P. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology 2017, 65, 1581–1599. [Google Scholar] [CrossRef]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Yu, Y.P.; Tao, J.; Liu, S.; Tseng, G.; Nalesnik, M.; Hamilton, R.; Bhargava, R.; Nelson, J.B.; Pennathur, A.; et al. MAN2A1-FER Fusion Gene Is Expressed by Human Liver and Other Tumor Types and Has Oncogenic Activity in Mice. Gastroenterology 2017, 153, 1120–1132.e15. [Google Scholar] [CrossRef]
- Chen, Z.H.; Yu, Y.P.; Zuo, Z.H.; Nelson, J.B.; Michalopoulos, G.K.; Monga, S.; Liu, S.; Tseng, G.; Luo, J.H. Targeting genomic rearrangements in tumor cells through Cas9-mediated insertion of a suicide gene. Nat. Biotechnol. 2017, 35, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC—A Quality Control Tool for High throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 26 June 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Villanueva, A.; Alsinet, C.; Yanger, K.; Hoshida, Y.; Zong, Y.; Toffanin, S.; Rodriguez-Carunchio, L.; Sole, M.; Thung, S.; Stanger, B.Z.; et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012, 143, 1660–1669.e1667. [Google Scholar] [CrossRef]
- Oishi, N.; Kumar, M.R.; Roessler, S.; Ji, J.; Forgues, M.; Budhu, A.; Zhao, X.; Andersen, J.B.; Ye, Q.H.; Jia, H.L.; et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012, 56, 1792–1803. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.; Hu, S.; Kim, M.; Oertel, M.; Singhi, A.; Monga, S.P.; Liu, S.; Ko, S. Context-Dependent Distinct Roles of SOX9 in Combined Hepatocellular Carcinoma–Cholangiocarcinoma. Cells 2024, 13, 1451. https://doi.org/10.3390/cells13171451
Park Y, Hu S, Kim M, Oertel M, Singhi A, Monga SP, Liu S, Ko S. Context-Dependent Distinct Roles of SOX9 in Combined Hepatocellular Carcinoma–Cholangiocarcinoma. Cells. 2024; 13(17):1451. https://doi.org/10.3390/cells13171451
Chicago/Turabian StylePark, Yoojeong, Shikai Hu, Minwook Kim, Michael Oertel, Aatur Singhi, Satdarshan P. Monga, Silvia Liu, and Sungjin Ko. 2024. "Context-Dependent Distinct Roles of SOX9 in Combined Hepatocellular Carcinoma–Cholangiocarcinoma" Cells 13, no. 17: 1451. https://doi.org/10.3390/cells13171451