
Synergistic Roles of Non-Homologous End Joining and Homologous Recombination in Repair of Ionizing Radiation-Induced DNA Double Strand Breaks in Mouse Embryonic Stem Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. mES Cell Culture
2.2. Generation of mES DNA-PKcs−/− Rad54−/− and DNA-PKcs−/− Rad54GFP-Knockin Cell Lines
2.3. Western Blotting
2.4. Irradiation
2.5. Clonogenic Survival
2.6. Immunofluorescence Staining
2.7. Microscopy
2.8. Live-Cell Imaging of Rad54-GFP
2.9. Cell Cycle Analysis
2.10. Statistics
3. Results
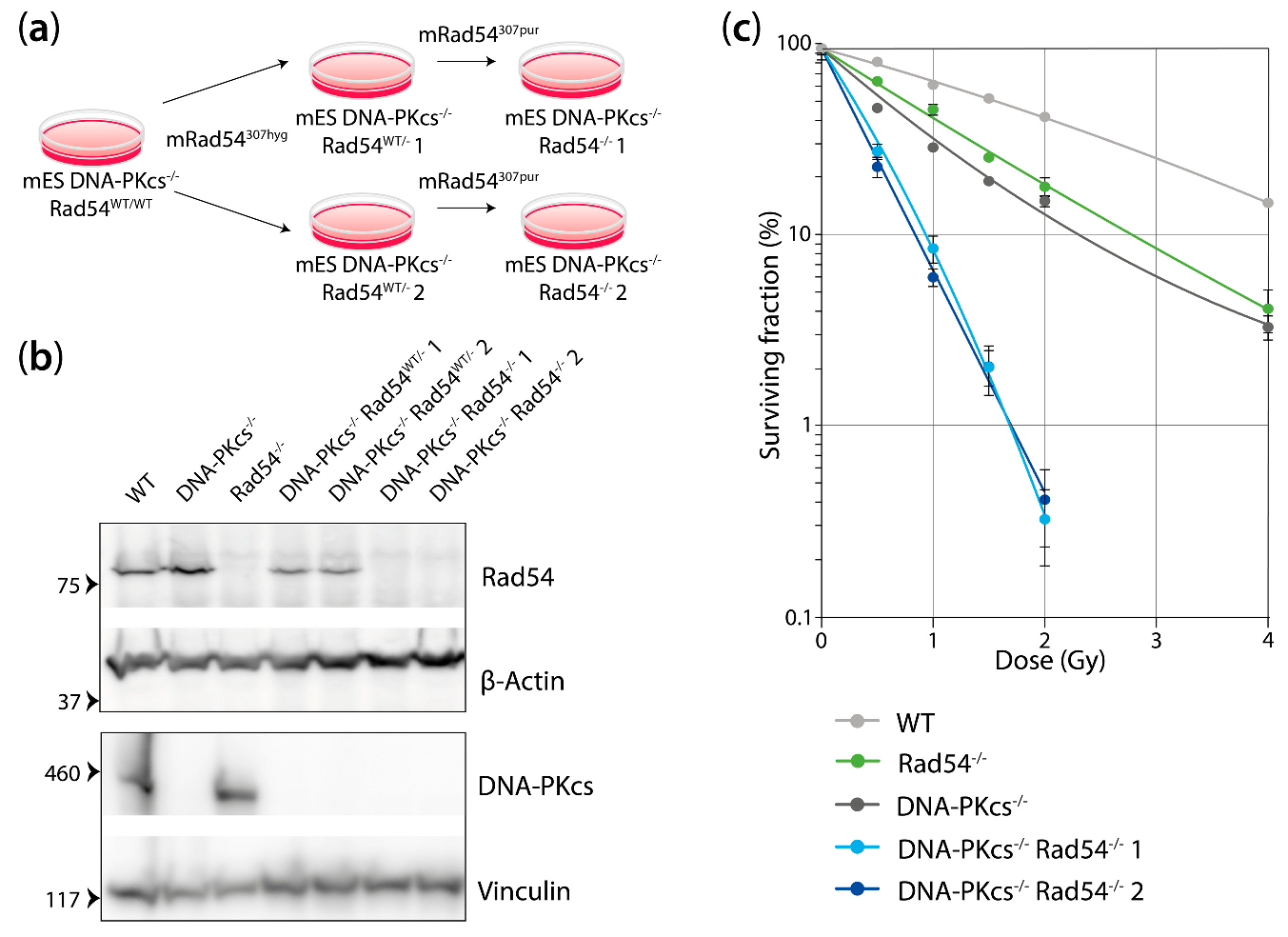
3.1. mES Cells Lacking DNA-Pkcs and Rad54 Are Hypersensitive to X-ray Radiation
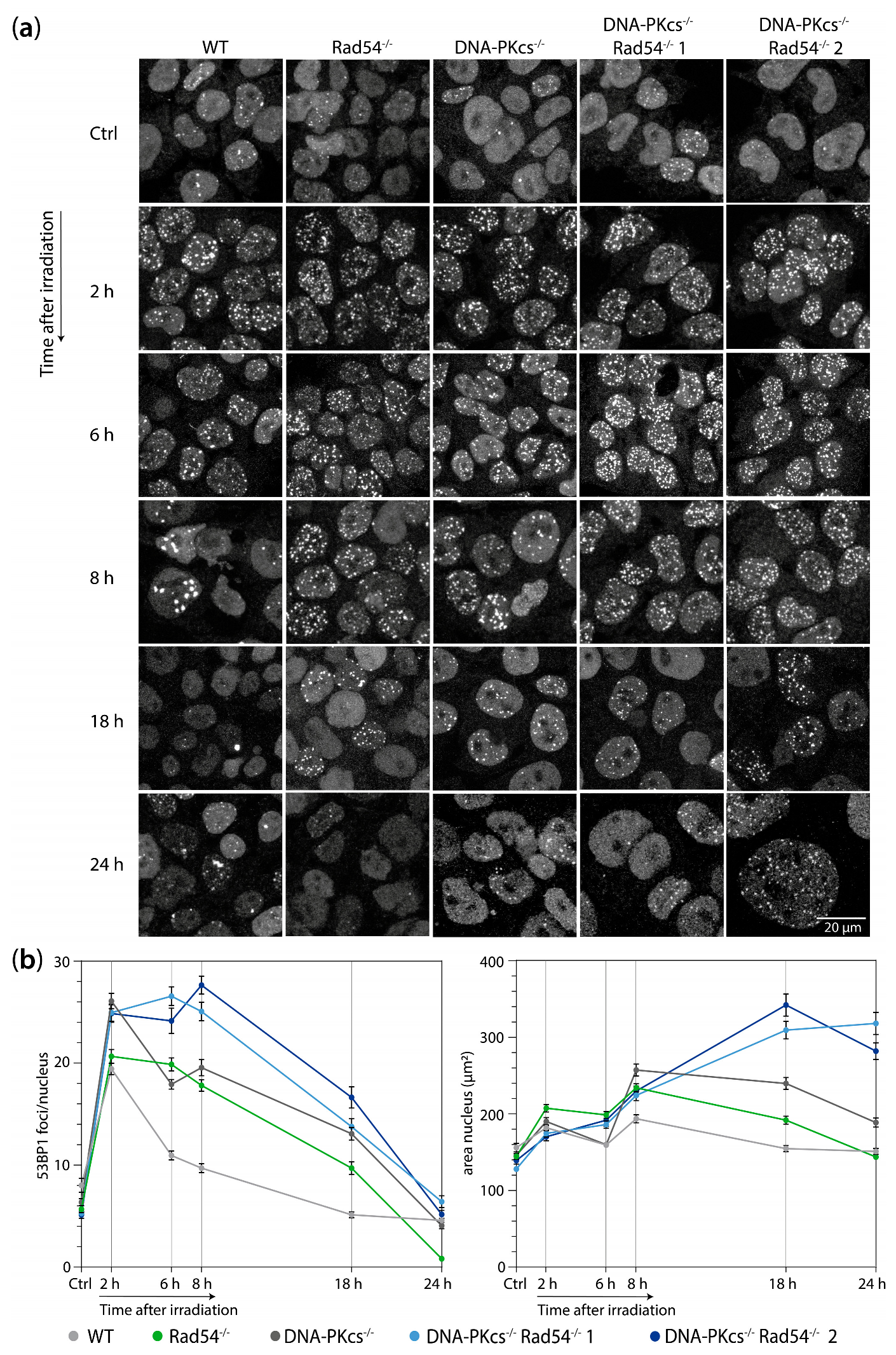
3.2. mES Lacking DNA-PKcs and Rad54 Cells Show Impaired 53BP1 Focus Resolution and an Increased Nuclear Size after 2 Gy of X-ray Radiation
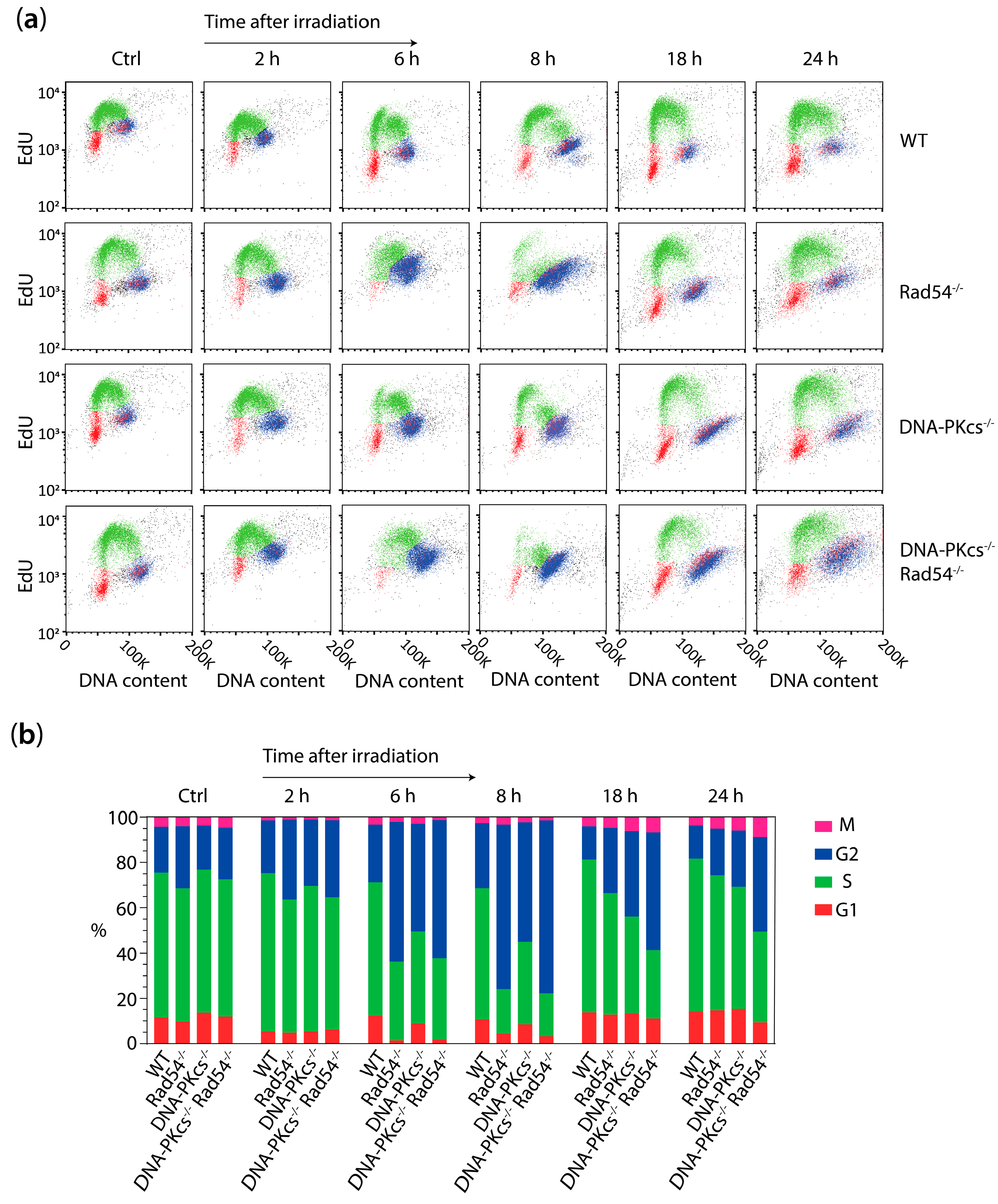
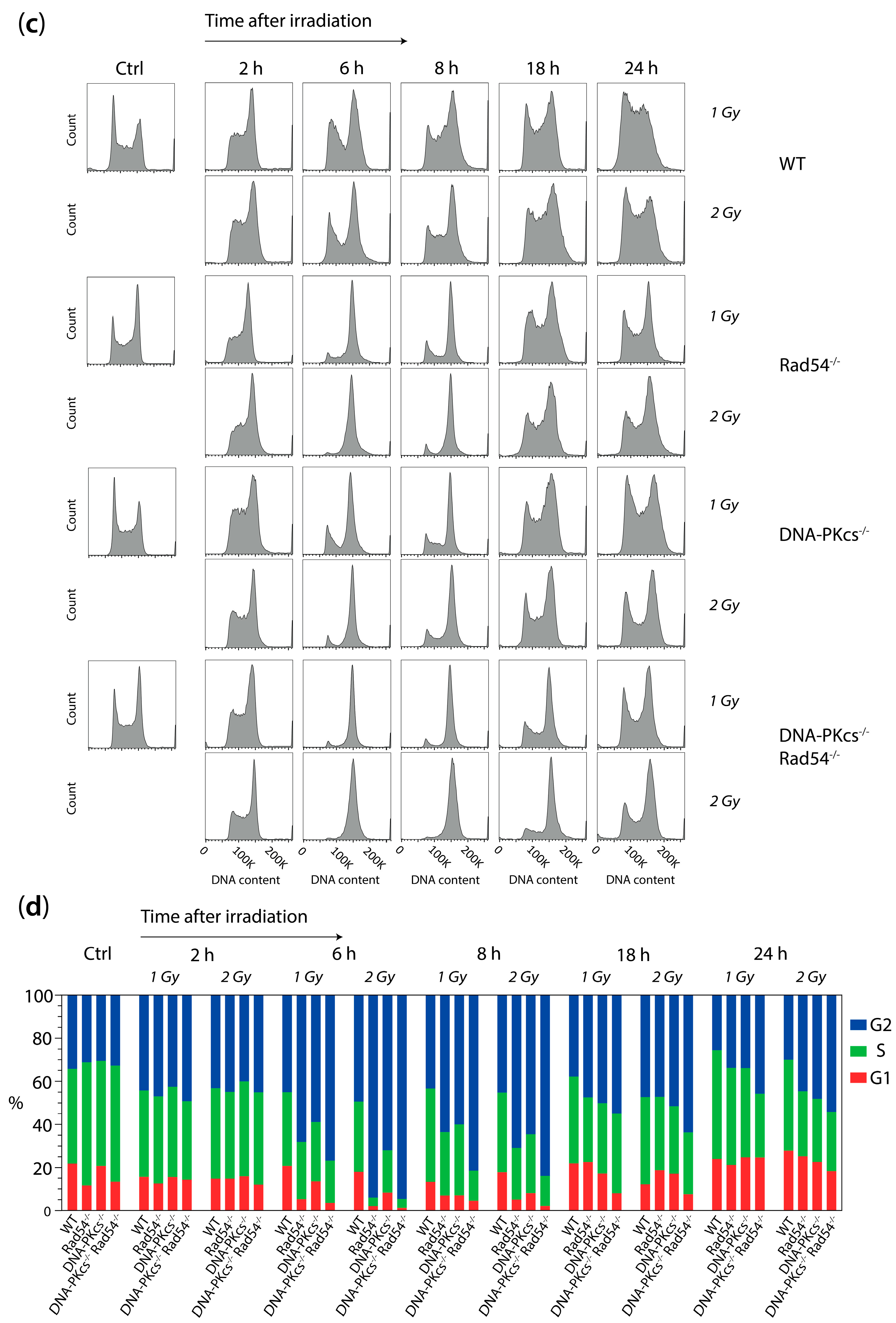
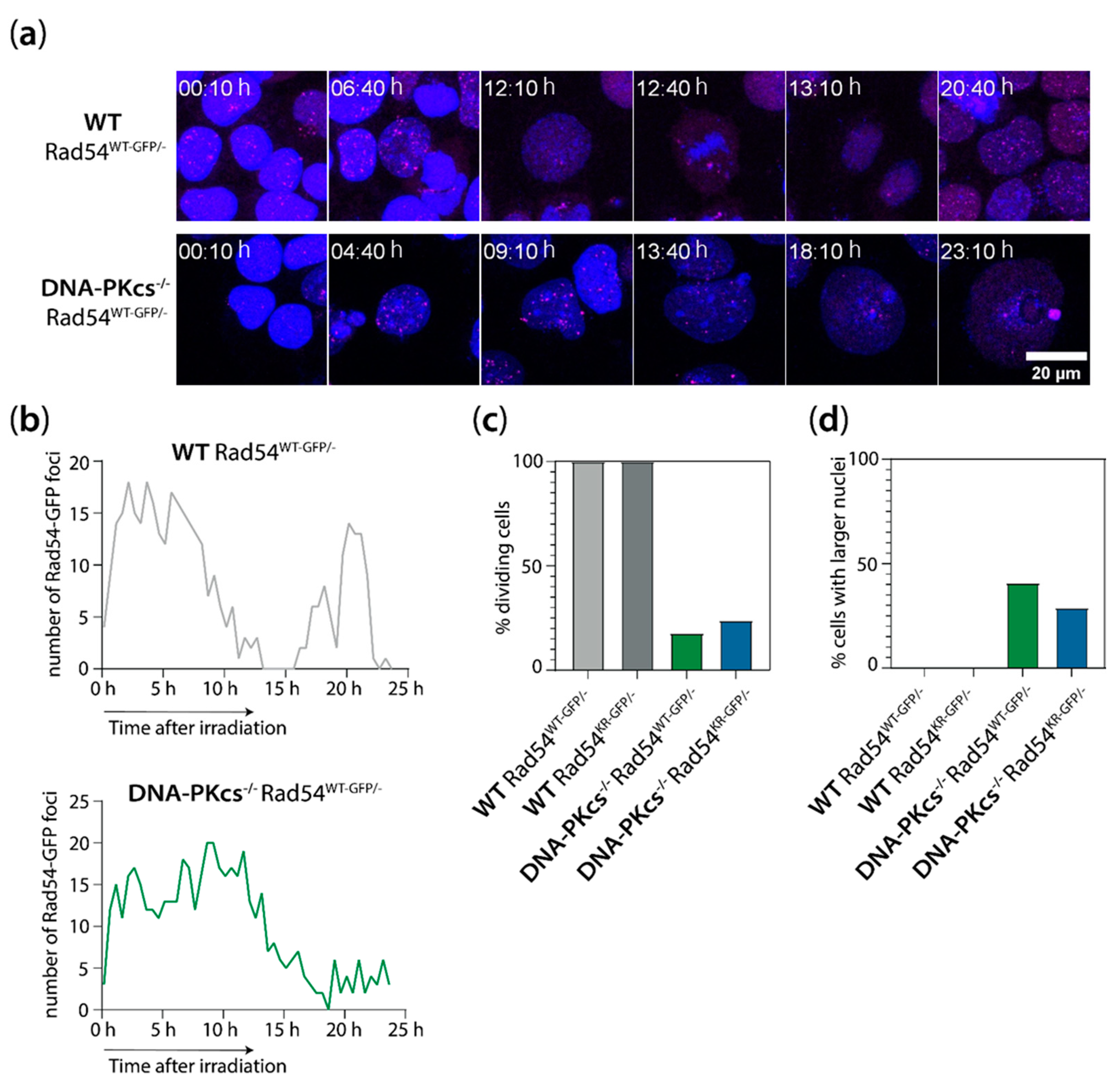
3.3. X-ray Irradiation Results in More Persistent G2 Phase Cell Cycle Block in mES Cells Lacking DNA-PKcs and Rad54
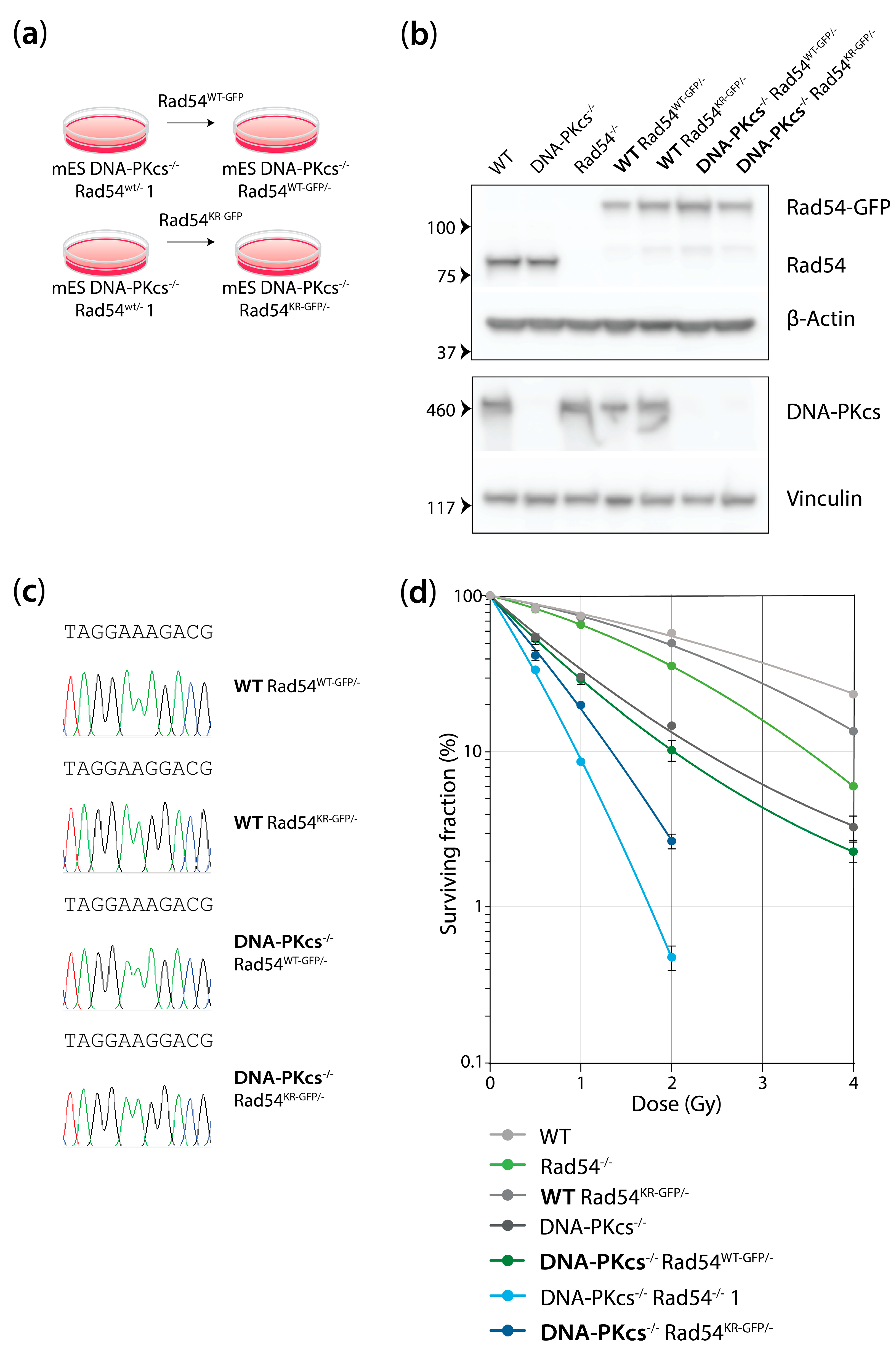
3.4. mES Cells Lacking DNA-PKcs and Rad54 or Expressing ATPase-Defective Rad54 Show Similar Sensitivity to X-Ray Radiation
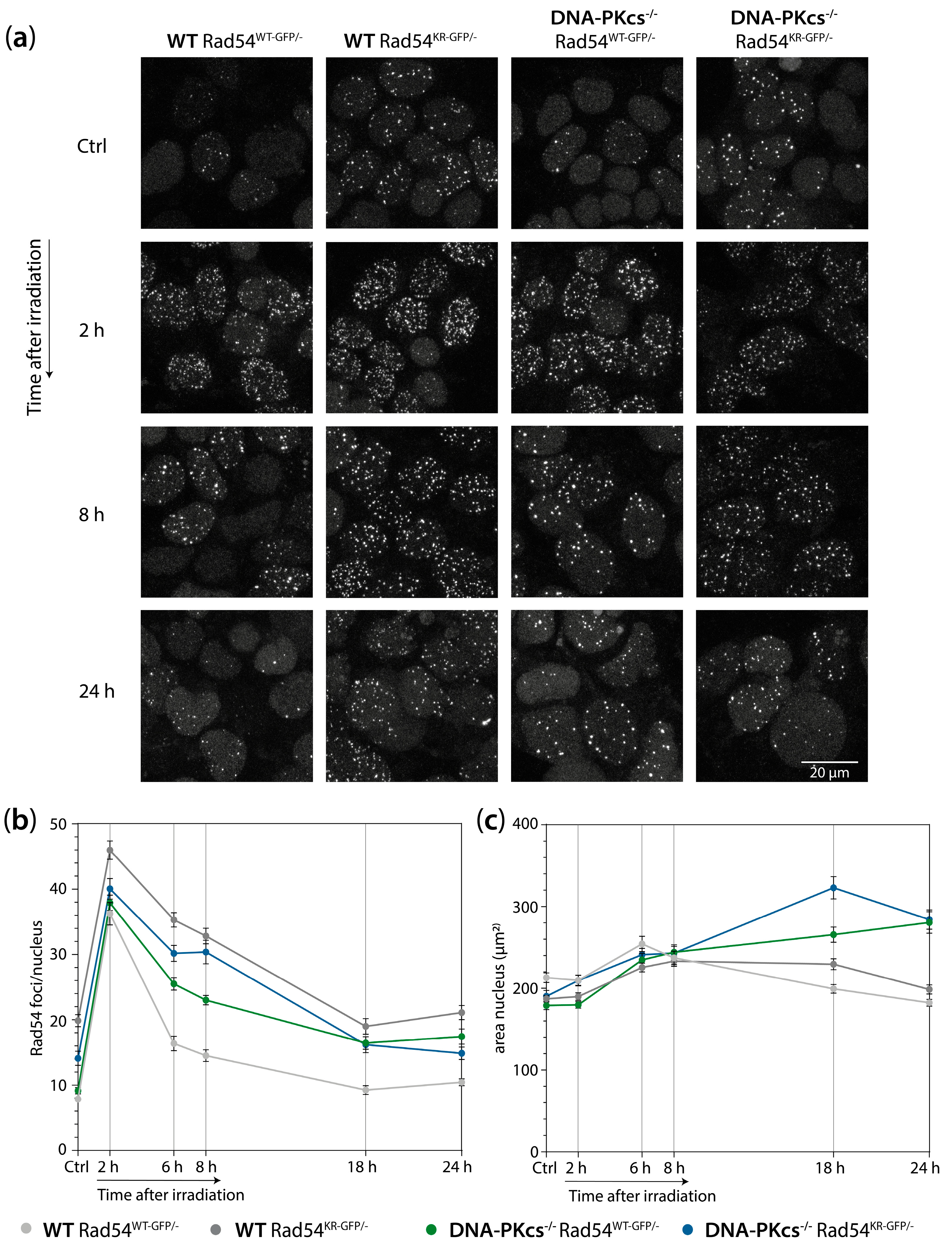
3.5. mES Cells Lacking DNA-PKcs Show Elevated Levels of Rad54 Foci after X-ray Irradiation
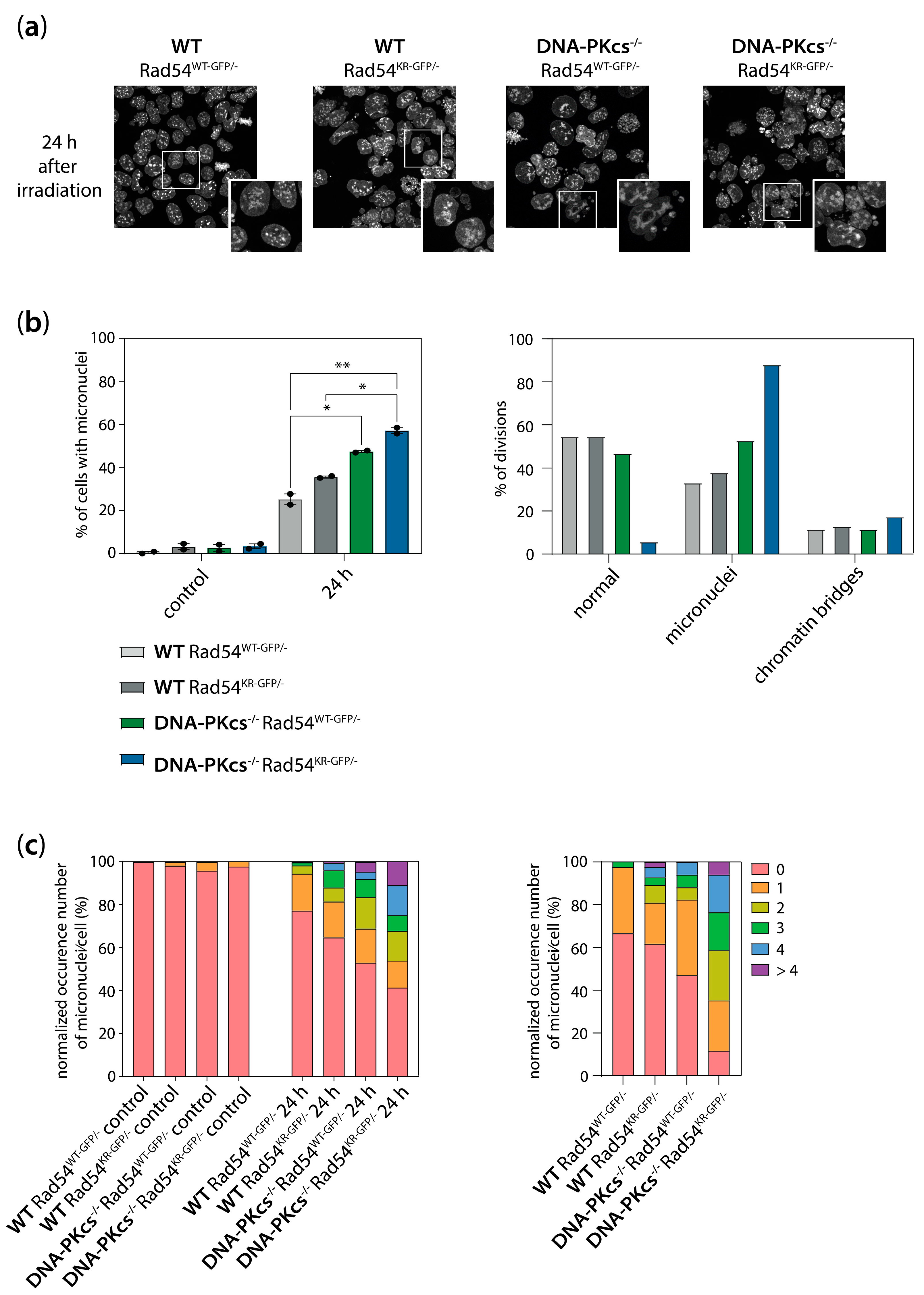
3.6. Increased Genomic Instability in Cells Lacking DNA-PKcs and Expressing ATPase-Defective Rad54
4. Discussion
4.1. Increased Nuclear Size of mES Cells Lacking DNA-PKcs
4.2. The Role of DNA-PKcs and Rad54 in Cell Cycle Regulation and Genome Stability Maintenance in mES Cells
4.3. Enhanced Activity of Rad54 in the Absence of DNA-PKcs
4.4. Therapeutic Applications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huen, M.S.Y.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- van de Kamp, G.; Heemskerk, T.; Kanaar, R.; Essers, J. DNA Double Strand Break Repair Pathways in Response to Different Types of Ionizing Radiation. Front. Genet. 2021, 12, 738230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Wyman, C.; Kanaar, R. DNA Double-Strand Break Repair: All’s Well that Ends Well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef]
- Crickard, J.B.; Moevus, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 Drives ATP Hydrolysis-Dependent DNA Sequence Alignment during Homologous Recombination. Cell 2020, 181, 1380–1394.e18. [Google Scholar] [CrossRef]
- Ceballos, S.J.; Heyer, W.-D. Functions of the Snf2/Swi2 family Rad54 motor protein in homologous recombination. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 2011, 1809, 509–523. [Google Scholar] [CrossRef]
- Agarwal, S.; van Cappellen, W.A.; Guénolé, A.; Eppink, B.; Linsen, S.E.V.; Meijering, E.; Houtsmuller, A.; Kanaar, R.; Essers, J. ATP-dependent and independent functions of Rad54 in genome maintenance. J. Cell Biol. 2011, 192, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.L.; Essers, J.; Citterio, E.; Swagemakers, S.M.; de Wit, J.; Benson, F.E.; Hoeijmakers, J.H.; Kanaar, R. Mouse Rad54 affects DNA conformation and DNA-damage-induced Rad51 foci formation. Curr. Biol. CB 1999, 9, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Wesoly, J.; Agarwal, S.; Sigurdsson, S.; Bussen, W.; Van Komen, S.; Qin, J.; van Steeg, H.; van Benthem, J.; Wassenaar, E.; Baarends, W.M.; et al. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol. Cell. Biol. 2006, 26, 976–989. [Google Scholar] [CrossRef]
- Ruis, B.L.; Fattah, K.R.; Hendrickson, E.A. The Catalytic Subunit of DNA-Dependent Protein Kinase Regulates Proliferation, Telomere Length, and Genomic Stability in Human Somatic Cells. Mol. Cell. Biol. 2008, 28, 6182–6195. [Google Scholar] [CrossRef]
- Gao, Y.; Chaudhuri, J.; Zhu, C.; Davidson, L.; Weaver, D.T.; Alt, F.W. A Targeted DNA-PKcs-Null Mutation Reveals DNA-PK-Independent Functions for KU in V(D)J Recombination. Immunity 1998, 9, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bezzubova, O.; Silbergleit, A.; Yamaguchi-Iwai, Y.; Takeda, S.; Buerstedde, J.-M. Reduced X-Ray Resistance and Homologous Recombination Frequencies in a RAD54−/− Mutant of the Chicken DT40 Cell Line. Cell 1997, 89, 185–193. [Google Scholar] [CrossRef]
- Essers, J.; Hendriks, R.W.; Swagemakers, S.M.A.; Troelstra, C.; de Wit, J.; Bootsma, D.; Hoeijmakers, J.H.J.; Kanaar, R. Disruption of Mouse RAD54 Reduces Ionizing Radiation Resistance and Homologous Recombination. Cell 1997, 89, 195–204. [Google Scholar] [CrossRef]
- Neal, J.A.; Meek, K. Deciphering phenotypic variance in different models of DNA-PKcs deficiency. DNA Repair 2019, 73, 7–16. [Google Scholar] [CrossRef]
- Taccioli, G.E.; Amatucci, A.G.; Beamish, H.J.; Gell, D.; Xiang, X.H.; Arzayus, M.I.T.; Priestley, A.; Jackson, S.P.; Rothstein, A.M.; Jeggo, P.A.; et al. Targeted Disruption of the Catalytic Subunit of the DNA-PK Gene in Mice Confers Severe Combined Immunodeficiency and Radiosensitivity. Immunity 1998, 9, 355–366. [Google Scholar] [CrossRef]
- Essers, J.; van Steeg, H.; de Wit, J.; Swagemakers, S.M.A.; Vermeij, M.; Hoeijmakers, J.H.J.; Kanaar, R. Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J. 2000, 19, 1703–1710. [Google Scholar] [CrossRef]
- Couëdel, C.; Mills, K.D.; Barchi, M.; Shen, L.; Olshen, A.; Johnson, R.D.; Nussenzweig, A.; Essers, J.; Kanaar, R.; Li, G.C.; et al. Collaboration of homologous recombination and nonhomologous end-joining factors for the survival and integrity of mice and cells. Genes Dev. 2004, 18, 1293–1304. [Google Scholar] [CrossRef]
- Mills, K.D.; Ferguson, D.O.; Essers, J.; Eckersdorff, M.; Kanaar, R.; Alt, F.W. Rad54 and DNA Ligase IV cooperate to maintain mammalian chromatid stability. Genes Dev. 2004, 18, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Pluth, J.M.; Fried, L.M.; Kirchgessner, C.U. Severe combined immunodeficient cells expressing mutant hRAD54 exhibit a marked DNA double-strand break repair and error-prone chromosome repair defect. Cancer Res. 2001, 61, 2649–2655. [Google Scholar]
- Hooper, M.; Hardy, K.; Handyside, A.; Hunter, S.; Monk, M. HPRT-deficient (Lesch–Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 1987, 326, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement With The Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- van Royen, M.E.; Cunha, S.M.; Brink, M.C.; Mattern, K.A.; Nigg, A.L.; Dubbink, H.J.; Verschure, P.J.; Trapman, J.; Houtsmuller, A.B. Compartmentalization of androgen receptor protein–protein interactions in living cells. J. Cell Biol. 2007, 177, 63–72. [Google Scholar] [CrossRef]
- Fox, M.H. A model for the computer analysis of synchronous DNA distributions obtained by flow cytometry. Cytometry 1980, 1, 71–77. [Google Scholar] [CrossRef]
- Kieffer, S.R.; Lowndes, N.F. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front. Genet. 2022, 13. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef]
- Hendzel, M.J.; Wei, Y.; Mancini, M.A.; Van Hooser, A.; Ranalli, T.; Brinkley, B.R.; Bazett-Jones, D.P.; Allis, C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997, 106, 348–360. [Google Scholar] [CrossRef]
- Juan, G.; Traganos, F.; James, W.M.; Ray, J.M.; Roberge, M.; Sauve, D.M.; Anderson, H.; Darzynkiewicz, Z. Histone H3 phosphorylation and expression of cyclins A and B1 measured in individual cells during their progression through G2 and mitosis. Cytometry 1998, 32, 71–77. [Google Scholar] [CrossRef]
- Giunta, S.; Jackson, S.P. Give me a break, but not in mitosis: The mitotic DNA damage response marks DNA double-strand breaks with early signaling events. Cell Cycle 2011, 10, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-J.; Lin, Y.-F.; Chou, H.-Y.; Yajima, H.; Fattah, K.R.; Lee, S.-C.; Chen, B.P.C. Involvement of DNA-dependent Protein Kinase in Normal Cell Cycle Progression through Mitosis*. J. Biol. Chem. 2011, 286, 12796–12802. [Google Scholar] [CrossRef]
- Lee, K.-J.; Shang, Z.-F.; Lin, Y.-F.; Sun, J.; Morotomi-Yano, K.; Saha, D.; Chen, B.P.C. The Catalytic Subunit of DNA-Dependent Protein Kinase Coordinates with Polo-Like Kinase 1 to Facilitate Mitotic Entry. Neoplasia 2015, 17, 329–338. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, A.K.; Jodkowska, K.; Teloni, F.; Bizard, A.H.; Zellweger, R.; Herrador, R.; Ortega, S.; Hickson, I.D.; Altmeyer, M.; Mendez, J.; et al. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 2016, 7, 10660. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, Á.; Cook, A.W.; Gough, R.E.; Schilling, M.; Olszok, N.A.; Brown, I.; Wang, L.; Aaron, J.; Martin-Fernandez, M.L.; Rehfeldt, F.; et al. DNA damage alters nuclear mechanics through chromatin reorganization. Nucleic Acids Res. 2021, 49, 340–353. [Google Scholar] [CrossRef]
- Suvorova, I.I.; Grigorash, B.B.; Chuykin, I.A.; Pospelova, T.V.; Pospelov, V.A. G1 checkpoint is compromised in mouse ESCs due to functional uncoupling of p53-p21Waf1 signaling. Cell Cycle 2016, 15, 52–63. [Google Scholar] [CrossRef]
- Malashicheva, A.B.; Kislyakova, T.V.; Aksenov, N.D.; Osipov, K.A.; Pospelov, V.A. F9 embryonal carcinoma cells fail to stop at G1/S boundary of the cell cycle after γ-irradiation due to p21WAF1/CIP1 degradation. Oncogene 2000, 19, 3858–3865. [Google Scholar] [CrossRef]
- ter Huurne, M.; Chappell, J.; Dalton, S.; Stunnenberg, H.G. Distinct Cell-Cycle Control in Two Different States of Mouse Pluripotency. Cell Stem Cell 2017, 21, 449–455.e4. [Google Scholar] [CrossRef]
- Deckbar, D.; Jeggo, P.A.; Löbrich, M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 271–283. [Google Scholar] [CrossRef]
- Deckbar, D.; Birraux, J.; Krempler, A.; Tchouandong, L.; Beucher, A.; Walker, S.; Stiff, T.; Jeggo, P.; Löbrich, M. Chromosome breakage after G2 checkpoint release. J. Cell Biol. 2007, 176, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Mohebi, S.; Mizuno, K.; Watson, A.; Carr, A.M.; Murray, J.M. Checkpoints are blind to replication restart and recombination intermediates that result in gross chromosomal rearrangements. Nat. Commun. 2015, 6, 6357. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Hanaoka, F.; Mazin, A.V. Rad54 dissociates homologous recombination intermediates by branch migration. Nat. Struct. Mol. Biol. 2007, 14, 746–753. [Google Scholar] [CrossRef]
- De Marco Zompit, M.; Stucki, M. Mechanisms of genome stability maintenance during cell division. DNA Repair 2021, 108, 103215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rohde, L.H.; Emami, K.; Hammond, D.; Casey, R.; Mehta, S.K.; Jeevarajan, A.S.; Pierson, D.L.; Wu, H. Suppressed expression of non-DSB repair genes inhibits gamma-radiation-induced cytogenetic repair and cell cycle arrest. DNA Repair 2008, 7, 1835–1845. [Google Scholar] [CrossRef]
- Oliveira, N.G.; Castro, M.; Rodrigues, A.S.; Gonçalves, I.C.; Gil, O.M.; Fernandes, A.P.; Toscano-Rico, J.M.; Rueff, J. Wortmannin enhances the induction of micronuclei by low and high LET radiation. Mutagenesis 2003, 18, 37–44. [Google Scholar] [CrossRef]
- Schimmel, J.; Kool, H.; van Schendel, R.; Tijsterman, M. Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J. 2017, 36, 3634–3649. [Google Scholar] [CrossRef]
- van Vugt, M.A.T.M.; Tijsterman, M. POLQ to the rescue for double-strand break repair during mitosis. Nat. Struct. Mol. Biol. 2023, 30, 1828–1830. [Google Scholar] [CrossRef]
- Llorens-Agost, M.; Ensminger, M.; Le, H.P.; Gawai, A.; Liu, J.; Cruz-García, A.; Bhetawal, S.; Wood, R.D.; Heyer, W.-D.; Löbrich, M. POLθ-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis. Nat. Cell Biol. 2021, 23, 1095–1104. [Google Scholar] [CrossRef]
- Gelot, C.; Kovacs, M.T.; Miron, S.; Mylne, E.; Haan, A.; Boeffard-Dosierre, L.; Ghouil, R.; Popova, T.; Dingli, F.; Loew, D.; et al. Polθ is phosphorylated by PLK1 to repair double-strand breaks in mitosis. Nature 2023, 621, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Yu, Y.; Gupta, S.; Cho, Y.-M.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of DNA-Dependent Protein Kinase Regulates DNA End Processing and May Also Alter Double-Strand Break Repair Pathway Choice. Mol. Cell. Biol. 2005, 25, 10842–10852. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of Homologous Recombination by DNA-Dependent Protein Kinase Requires Kinase Activity, Is Titratable, and Is Modulated by Autophosphorylation ▿. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Willoughby, C.E.; Jiang, Y.; Thomas, H.D.; Willmore, E.; Kyle, S.; Wittner, A.; Phillips, N.; Zhao, Y.; Tudhope, S.J.; Prendergast, L.; et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J. Clin. Invest. 2020, 130, 258–271. [Google Scholar] [CrossRef]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef]
- Gordhandas, S.B.; Manning-Geist, B.; Henson, C.; Iyer, G.; Gardner, G.J.; Sonoda, Y.; Moore, K.N.; Aghajanian, C.; Chui, M.H.; Grisham, R.N. Pre-clinical activity of the oral DNA-PK inhibitor, peposertib (M3814), combined with radiation in xenograft models of cervical cancer. Sci. Rep. 2022, 12, 974. [Google Scholar] [CrossRef]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef]
- Samuels, M.; Falkenius, J.; Bar-Ad, V.; Dunst, J.; van Triest, B.; Yachnin, J.; Rodriguez-Gutierrez, A.; Kuipers, M.; You, X.; Sarholz, B.; et al. A Phase 1 Study of the DNA-PK Inhibitor Peposertib in Combination With Radiation Therapy With or Without Cisplatin in Patients With Advanced Head and Neck Tumors. Int. J. Radiat. Oncol. 2024, 118, 743–756. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van de Kamp, G.; Heemskerk, T.; Kanaar, R.; Essers, J. Synergistic Roles of Non-Homologous End Joining and Homologous Recombination in Repair of Ionizing Radiation-Induced DNA Double Strand Breaks in Mouse Embryonic Stem Cells. Cells 2024, 13, 1462. https://doi.org/10.3390/cells13171462
van de Kamp G, Heemskerk T, Kanaar R, Essers J. Synergistic Roles of Non-Homologous End Joining and Homologous Recombination in Repair of Ionizing Radiation-Induced DNA Double Strand Breaks in Mouse Embryonic Stem Cells. Cells. 2024; 13(17):1462. https://doi.org/10.3390/cells13171462
Chicago/Turabian Stylevan de Kamp, Gerarda, Tim Heemskerk, Roland Kanaar, and Jeroen Essers. 2024. "Synergistic Roles of Non-Homologous End Joining and Homologous Recombination in Repair of Ionizing Radiation-Induced DNA Double Strand Breaks in Mouse Embryonic Stem Cells" Cells 13, no. 17: 1462. https://doi.org/10.3390/cells13171462