

Class Effect Unveiled: PPARγ Agonists and MEK Inhibitors in Cancer Cell Differentiation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cancer Trans-Differentiation into Adipocytes
2.3. Cell Proliferation Assay (EdU)
2.4. Ex Vivo Cultures of Tumor Tissues
2.5. Immunohistochemistry (IHC)
2.6. Immunofluorescence
2.7. Microscopy
2.8. Image Processing and Analysis
2.9. Statistical Analysis
3. Results
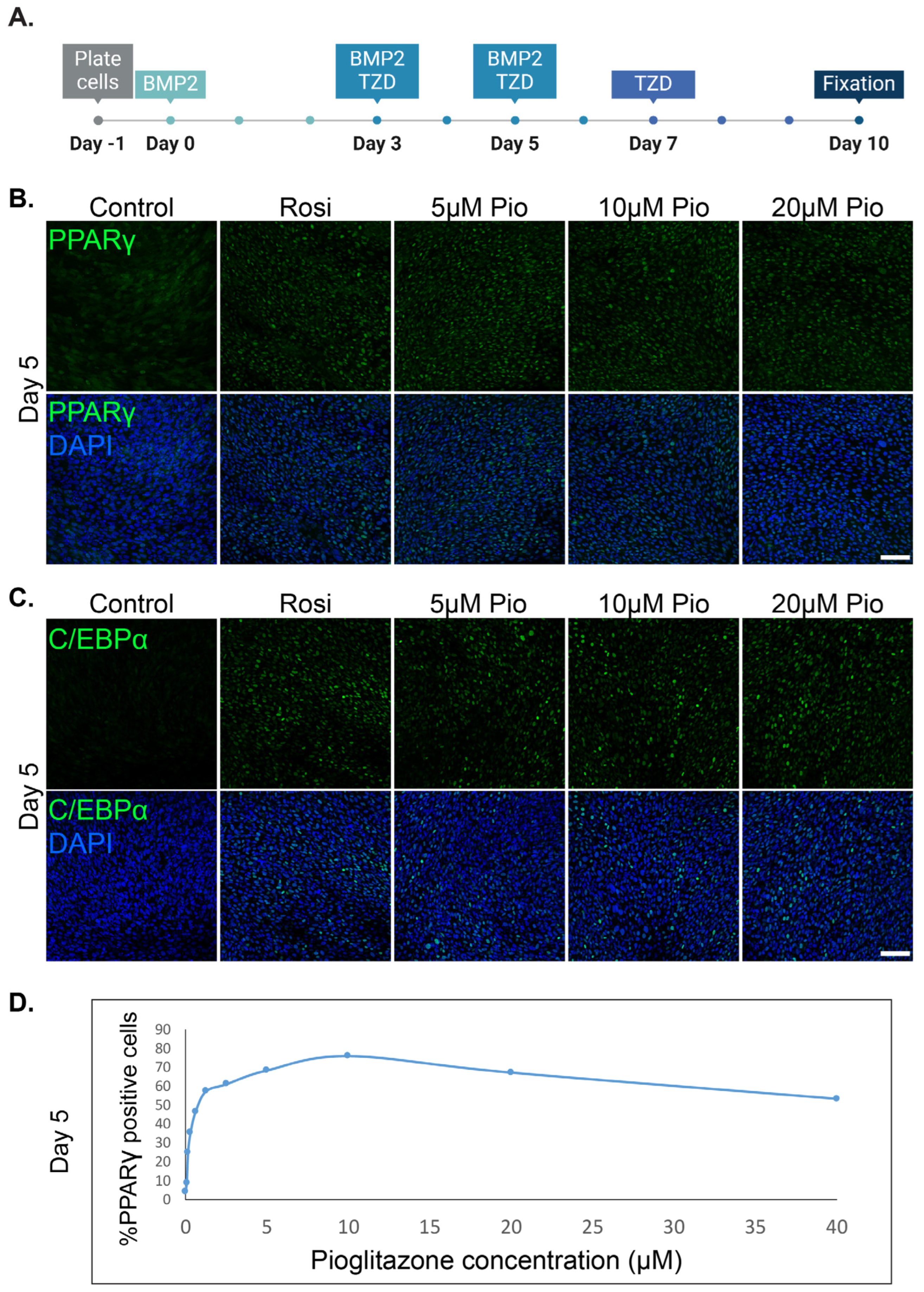
3.1. Pioglitazone Can Effectively Substitute Rosiglitazone and Induce the Trans-Differentiation of Cancer Cells into Adipocytes
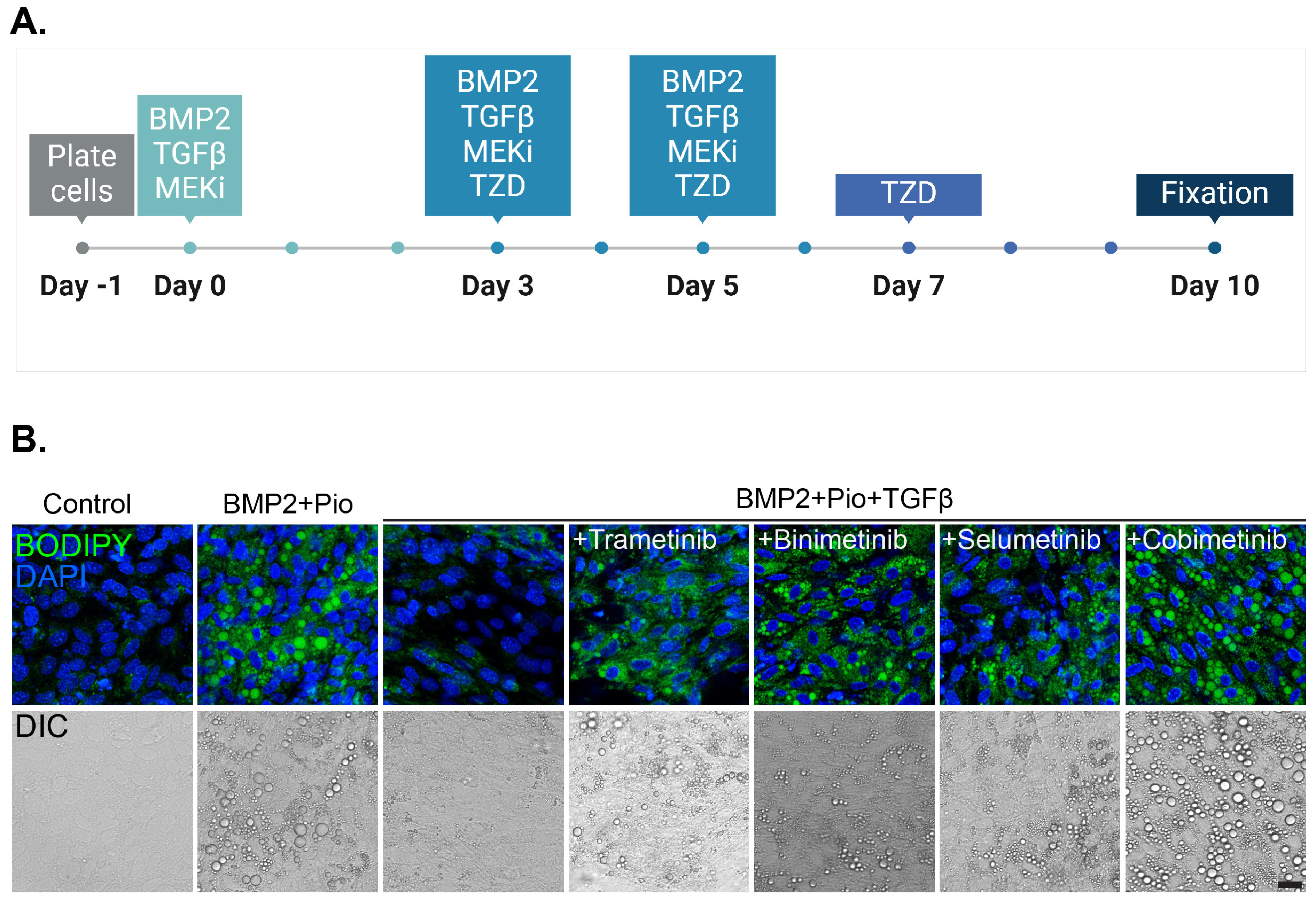
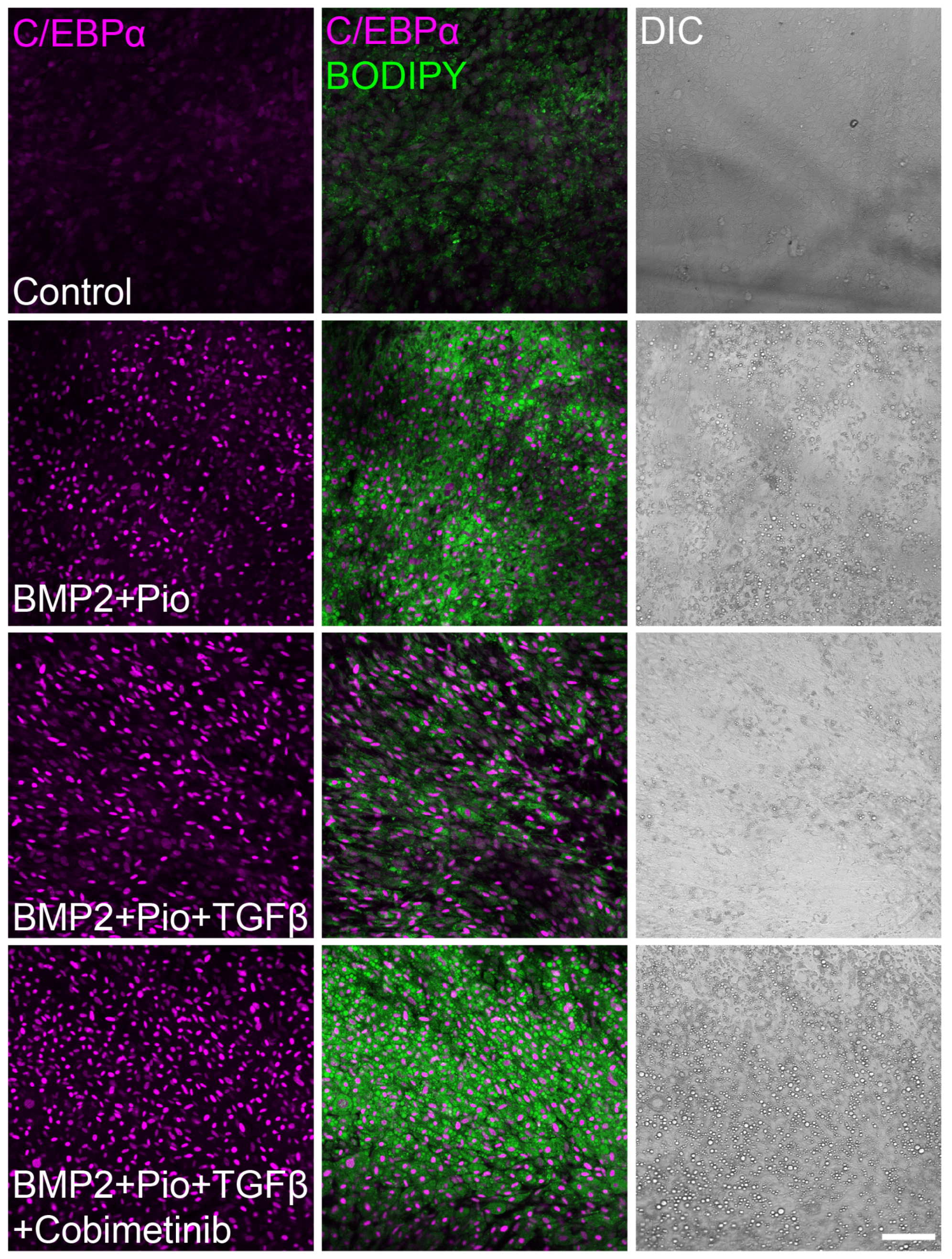
3.2. Class Effect of MEK Inhibitors Facilitates Cancer Trans-Differentiation in the Presence of TGFβ
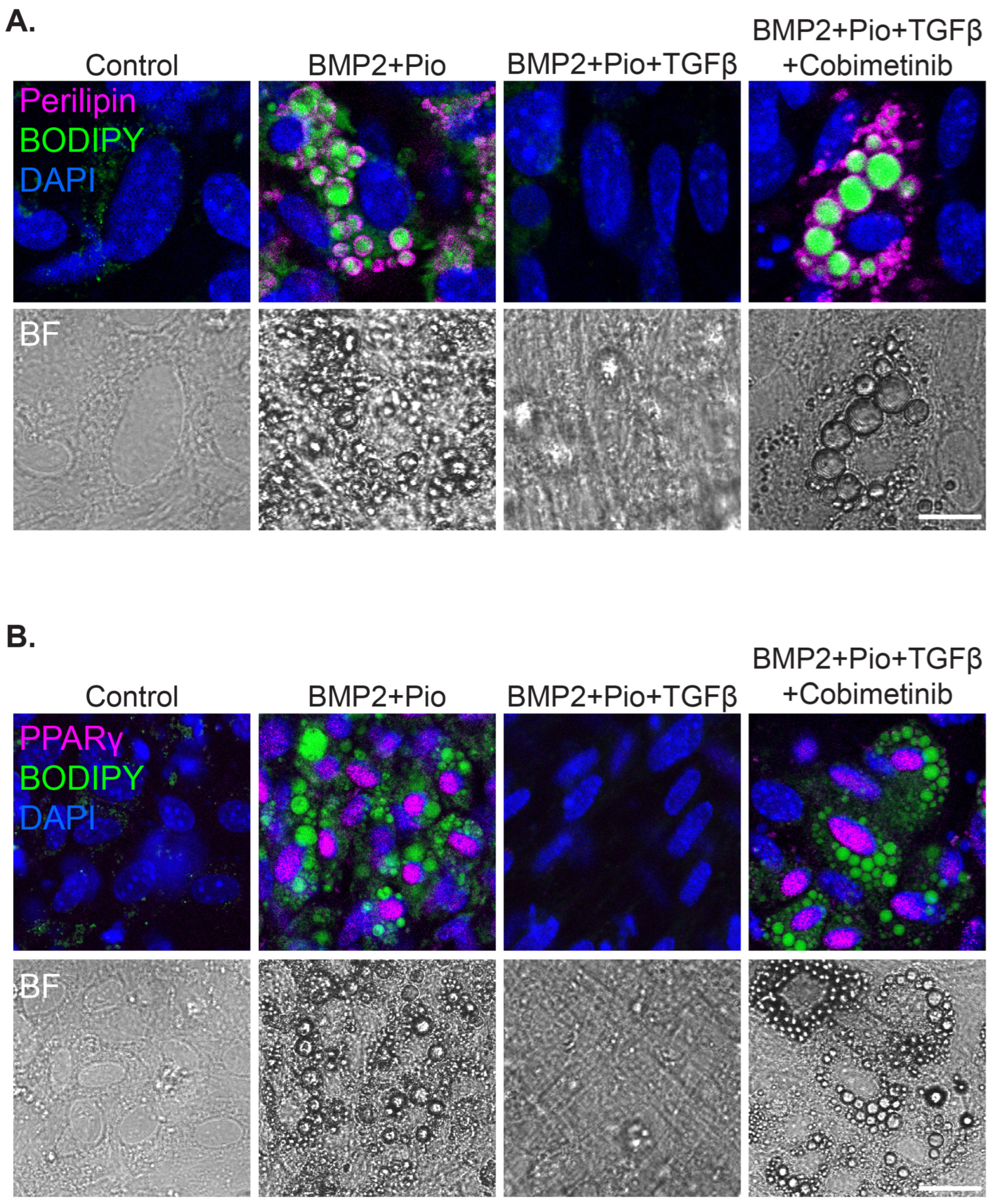
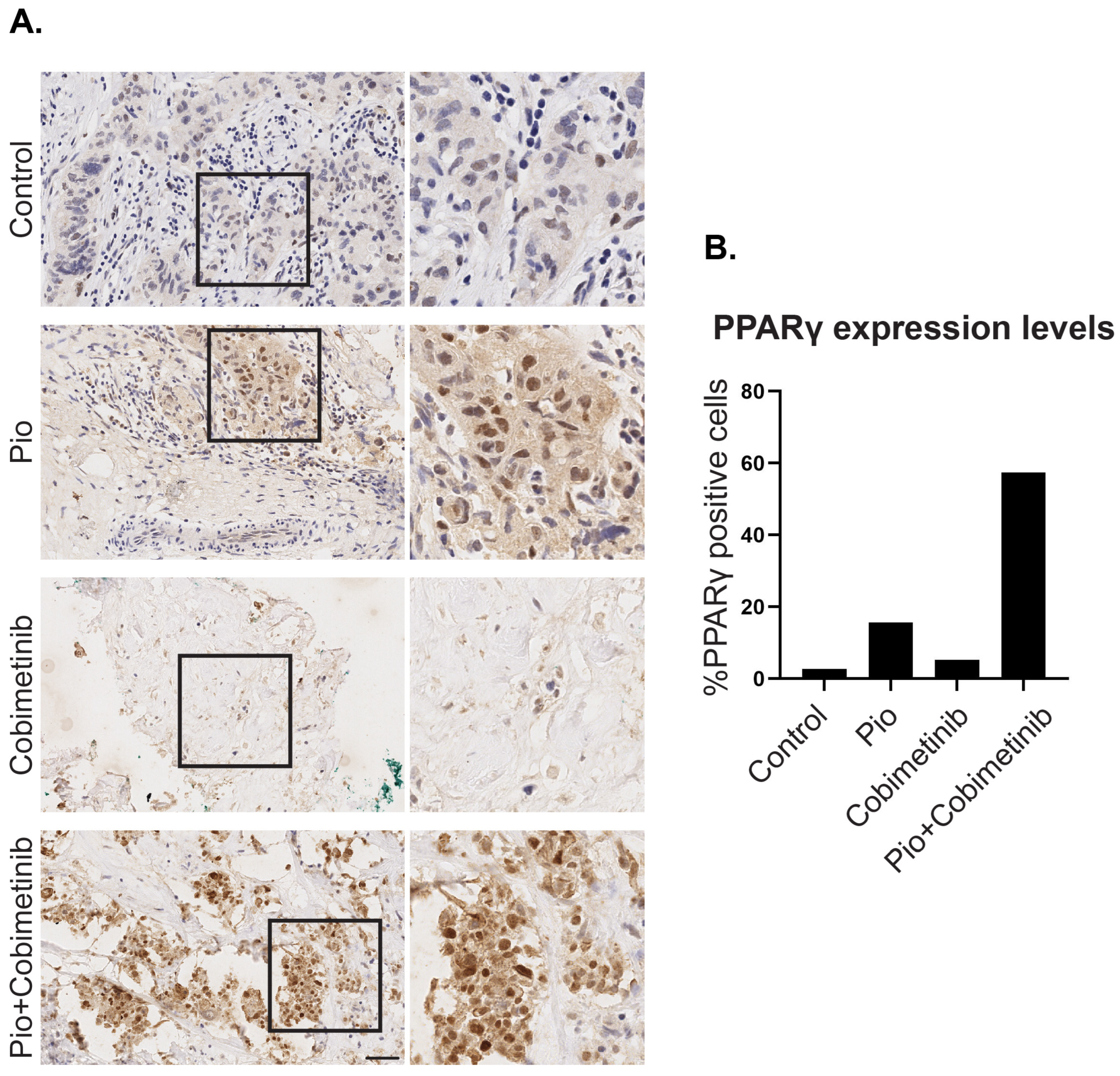
3.3. A Synergistic Effect of Pioglitazone and Cobimetinib in PPARγ Upregulation in Patient-Derived Ex Vivo Tumor Culture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haerinck, J.; Goossens, S.; Berx, G. The epithelial–mesenchymal plasticity landscape: Principles of design and mechanisms of regulation. Nat. Rev. Genet. 2023, 24, 590–609. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Jiang, Q.; Ganesh, K. Metastasis. Cell 2023, 186, 1564–1579. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2019, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef]
- Ishay-Ronen, D.; Diepenbruck, M.; Kalathur, R.K.R.; Sugiyama, N.; Tiede, S.; Ivanek, R.; Bantug, G.; Morini, M.F.; Wang, J.; Hess, C.; et al. Gain Fat—Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell 2019, 35, 17–32.e6. [Google Scholar] [CrossRef]
- Bar-Hai, N.; Ishay-Ronen, D. Engaging plasticity: Differentiation therapy in solid tumors. Front. Pharmacol. 2022, 13, 944773. [Google Scholar] [CrossRef]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef]
- Ishay-Ronen, D.; Christofori, G. Targeting Cancer Cell Metastasis by Converting Cancer Cells into Fat. Cancer Res. 2019, 79, 5471–5475. [Google Scholar] [CrossRef]
- Visan, I.; Visan, I. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity 2018, 49, 312–325.e5. [Google Scholar] [CrossRef]
- Gubelmann, C.; Schwalie, P.C.; Raghav, S.K.; Röder, E.; Delessa, T.; Kiehlmann, E.; Waszak, S.M.; Corsinotti, A.; Udin, G.; Holcombe, W.; et al. Identification of the transcription factor ZEB1 as a central component of the adipogenic gene regulatory network. eLife 2014, 3, e03346. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.G.; Zushin, P.-J.H.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.P.; Spiegelman, B.M. An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.F.; Farmer, S.R. Role of PPARgamma in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18(INK4c) and p21(Waf1/Cip1), during adipogenesis. J. Biol. Chem. 1999, 274, 17088–17097. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Infante, J.R.; A Fecher, L.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Gonzalez, R.; Pavlick, A.; Hamid, O.; Gajewski, T.F.; Daud, A.; Flaherty, L.; Logan, T.; Chmielowski, B.; Lewis, K.; et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAFV600-mutated melanoma: A phase 1b study. Lancet Oncol. 2014, 15, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-Mediated Angiogenesis Links EMT-Induced Cancer Stemness to Tumor Initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.J.; Sinn, E.; Pattengale, P.K.; Wallace, R.; Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54, 105–115. [Google Scholar] [CrossRef]
- Lehembre, F.; Yilmaz, M.; Wicki, A.; Schomber, T.; Strittmatter, K.; Ziegler, D.; Kren, A.; Went, P.; Derksen, P.W.B.; Berns, A.; et al. NCAM-induced focal adhesion assembly: A functional switch upon loss of E-cadherin. EMBO J. 2008, 27, 2603–2615. [Google Scholar] [CrossRef]
- Gavert, N.; Zwang, Y.; Weiser, R.; Greenberg, O.; Halperin, S.; Jacobi, O.; Mallel, G.; Sandler, O.; Berger, A.J.; Stossel, E.; et al. Ex vivo organotypic cultures for synergistic therapy prioritization identify patient-specific responses to combined MEK and Src inhibition in colorectal cancer. Nat. Cancer 2022, 3, 219–231. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben-Yishay, R.; Globus, O.; Balint-Lahat, N.; Arbili-Yarhi, S.; Bar-Hai, N.; Bar, V.; Aharon, S.; Kosenko, A.; Zundelevich, A.; Berger, R.; et al. Class Effect Unveiled: PPARγ Agonists and MEK Inhibitors in Cancer Cell Differentiation. Cells 2024, 13, 1506. https://doi.org/10.3390/cells13171506
Ben-Yishay R, Globus O, Balint-Lahat N, Arbili-Yarhi S, Bar-Hai N, Bar V, Aharon S, Kosenko A, Zundelevich A, Berger R, et al. Class Effect Unveiled: PPARγ Agonists and MEK Inhibitors in Cancer Cell Differentiation. Cells. 2024; 13(17):1506. https://doi.org/10.3390/cells13171506
Chicago/Turabian StyleBen-Yishay, Rakefet, Opher Globus, Nora Balint-Lahat, Sheli Arbili-Yarhi, Neta Bar-Hai, Vered Bar, Sara Aharon, Anna Kosenko, Adi Zundelevich, Raanan Berger, and et al. 2024. "Class Effect Unveiled: PPARγ Agonists and MEK Inhibitors in Cancer Cell Differentiation" Cells 13, no. 17: 1506. https://doi.org/10.3390/cells13171506
APA StyleBen-Yishay, R., Globus, O., Balint-Lahat, N., Arbili-Yarhi, S., Bar-Hai, N., Bar, V., Aharon, S., Kosenko, A., Zundelevich, A., Berger, R., & Ishay-Ronen, D. (2024). Class Effect Unveiled: PPARγ Agonists and MEK Inhibitors in Cancer Cell Differentiation. Cells, 13(17), 1506. https://doi.org/10.3390/cells13171506