
DNase II Can Efficiently Digest RNA and Needs to Be Redefined as a Nuclease


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Collection of Saliva
2.3. Degradation of Yeast RNA by Saliva
2.4. Degradation of Nucleic Acids by DNase II
2.5. Exonuclease Digestion of Products after RNA Digestion by DNase II
2.6. Dephosphorylation of RNA Products Digested by DNase II
2.7. Phosphorylation of Purchased DNA
2.8. Ligation of Products Digested by DNase II
3. Results
3.1. Efficient Degradation of RNA by DNase II under Acidic Conditions
3.2. RNA Is Digested by DNase II Similarly to ssDNA
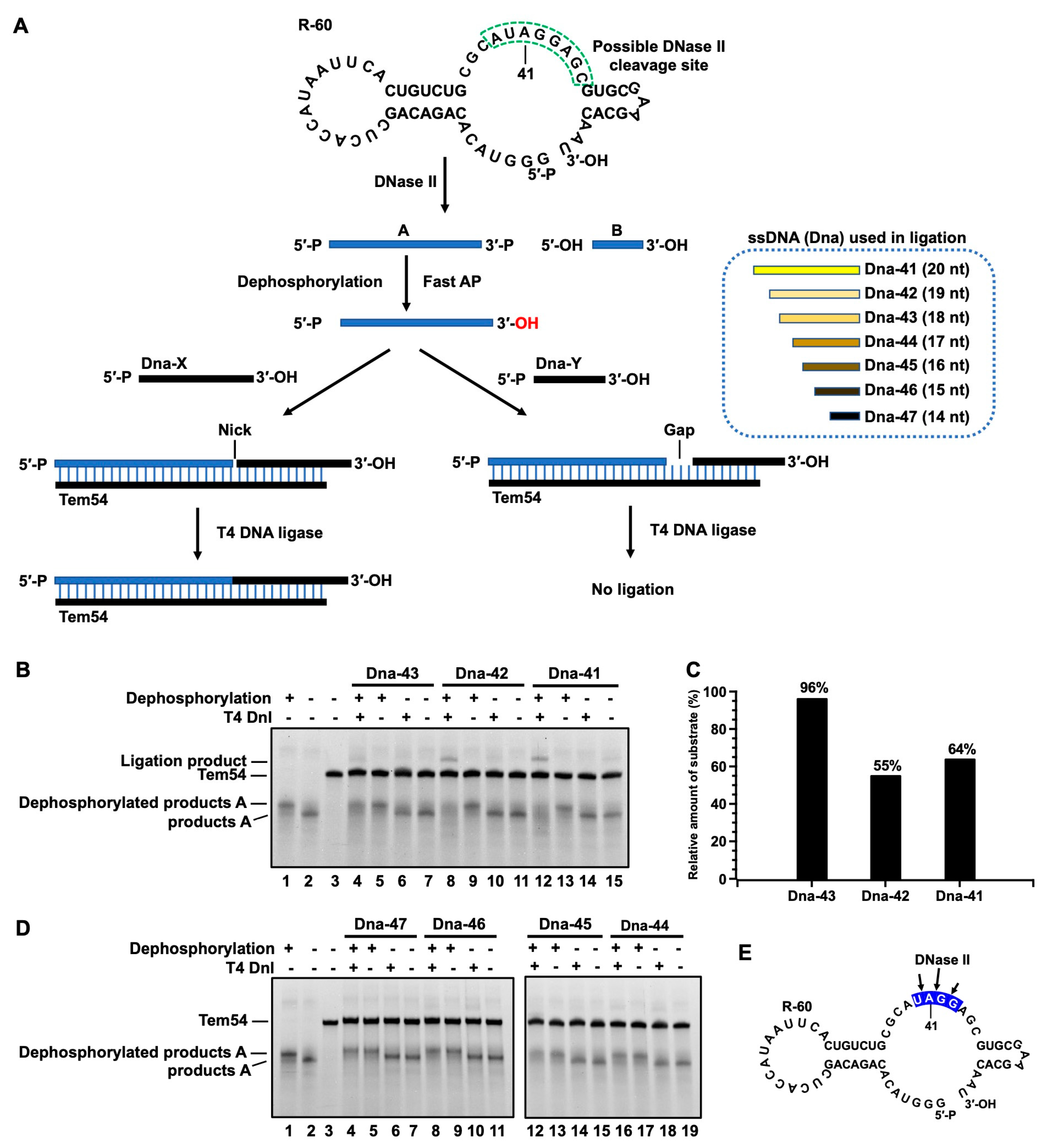
3.3. Determination of Terminal Structure of RNA Cleavage Products
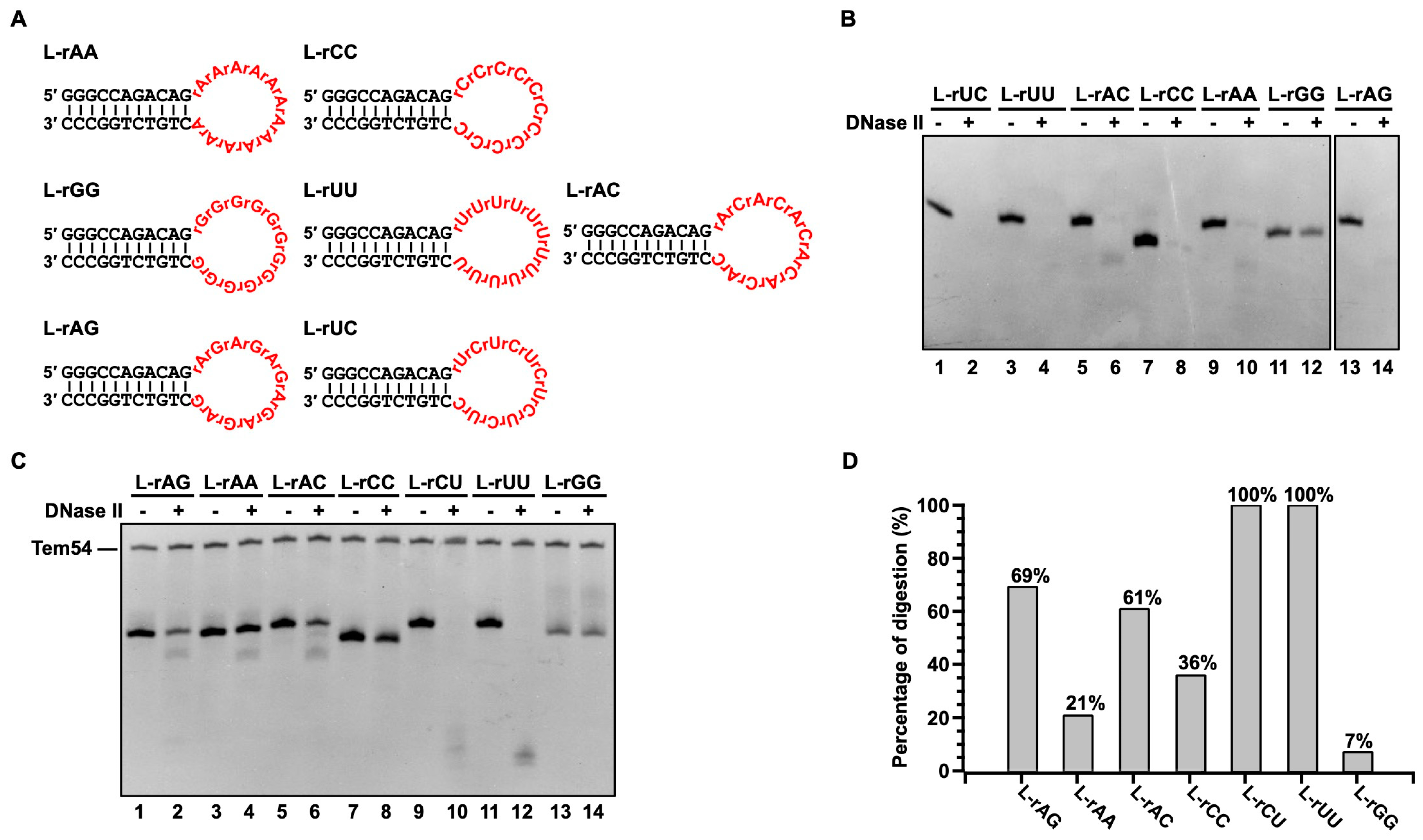
3.4. DNase II Only Has Weak Sequence Specificity for Cleaving RNA and Can Cleave Most RNA Sequences Efficiently under Relatively Strong Conditions
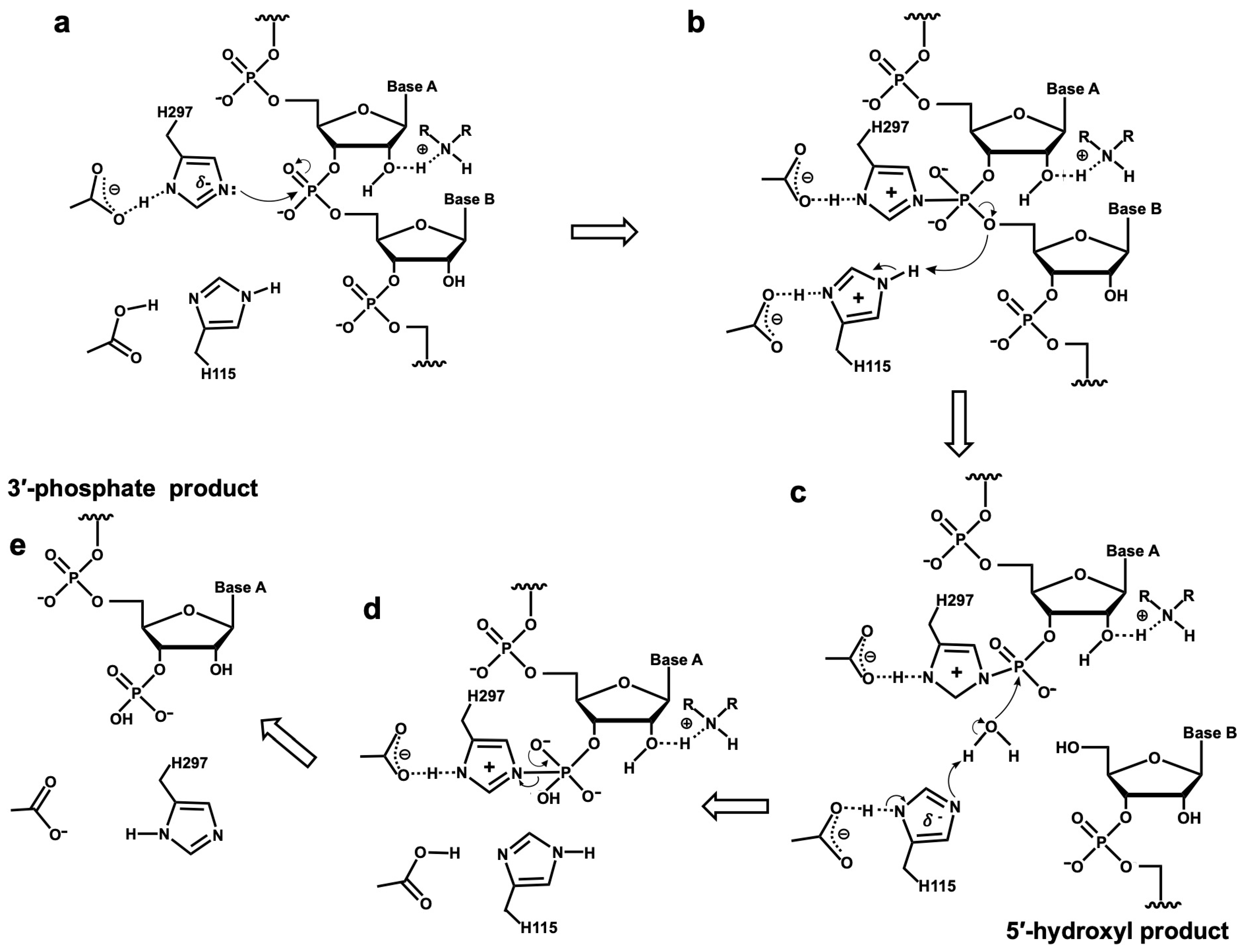
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catcheside, D.G.; Holmes, B. The action of enzymes on chromosomes. Symp. Soc. Exp. Biol. 1947, 1, 225–231. [Google Scholar]
- Bernardi, G. Spleen acid deoxyribonuclease. Enzymes 1971, 4, 271–287. [Google Scholar]
- Harosh, I.; Binninger, D.M.; Harris, P.V.; Mezzina, M.; Boyd, J.B. Mechanism of action of deoxyribonuclease II from human lymphoblasts. Eur. J. Biochem. 1991, 202, 479–484. [Google Scholar] [CrossRef]
- Cordonnier, C.; Bernardi, G. A comparative study of acid deoxyribonucleases extracted from different tissues and species. Can. J. Biochem. 1968, 46, 989–995. [Google Scholar] [CrossRef]
- Yasuda, T.; Takeshita, H.; Iida, R.; Nakajima, T.; Hosomi, O.; Nakashima, Y.; Kishi, K. Molecular cloning of the cDNA encoding human deoxyribonuclease II. J. Biol. Chem. 1998, 273, 2610–2616. [Google Scholar] [CrossRef] [PubMed]
- Krieser, R.J.; MacLea, K.S.; Park, J.P.; Eastman, A. The cloning, genomic structure, localization, and expression of human deoxyribonuclease IIbeta. Gene 2001, 269, 205–216. [Google Scholar] [CrossRef]
- Liao, T.H.; Liao, W.C.; Chang, H.C.; Lu, K.S. Deoxyribonuclease II purified from the isolated lysosomes of porcine spleen and from porcine liver homogenates. Comparison with deoxyribonuclease II purified from porcine spleen homogenates. Biochim. Biophys. Acta 1989, 1007, 15–22. [Google Scholar] [CrossRef]
- Shpak, M.; Kugelman, J.R.; Varela-Ramirez, A.; Aguilera, R.J. The phylogeny and evolution of deoxyribonuclease II: An enzyme essential for lysosomal DNA degradation. Mol. Phylogenet. Evol. 2008, 47, 841–854. [Google Scholar] [CrossRef]
- Yasuda, T.; Nadano, D.; Awazu, S.; Kishi, K. Human urine deoxyribonuclease II (DNase II) isoenzymes: A novel immunoaffinity purification, biochemical multiplicity, genetic heterogeneity and broad distribution among tissues and body fluids. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1992, 1119, 185–193. [Google Scholar] [CrossRef]
- Yamanaka, M.; Tsubota, Y.; Anai, M.; Ishimatsu, K.; Okumura, M.; Katsuki, S.; Takagi, Y. Purification and properties of acid deoxyribonucleases of human gastric mucosa and cervix uteri. J. Biol. Chem. 1974, 249, 3884–3889. [Google Scholar] [CrossRef]
- Evans, C.J.; Aguilera, R.J. DNase II: Genes, enzymes and function. Gene 2003, 322, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Oshima, R.G.; Price, P.A. Alkylation of an essential histidine residue in porcine spleen deoxyribonuclease. J. Biol. Chem. 1973, 248, 7522–7526. [Google Scholar] [CrossRef] [PubMed]
- Lyon, C.J.; Aguilera, R.J. Purification and characterization of the immunoglobulin switch sequence-specific endonuclease (Endo-SR) from bovine spleen. Mol. Immunol. 1997, 34, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Lyon, C.J.; Evans, C.J.; Bill, B.R.; Otsuka, A.J.; Aguilera, R.J. The C. elegans apoptotic nuclease NUC-1 is related in sequence and activity to mammalian DNase II. Gene 2000, 252, 147–154. [Google Scholar] [CrossRef]
- Baker, K.P.; Baron, W.F.; Henzel, W.J.; Spencer, S.A. Molecular cloning and characterization of human and murine DNase II. Gene 1998, 215, 281–289. [Google Scholar] [CrossRef]
- Evans, C.J. The Identification and Characterization of Mammalian DNase II Homologs in Caenorhabditis Elegans and Drosophila; University of California: Los Angeles, CA, USA, 2002. [Google Scholar]
- Varela-Ramirez, A.; Abendroth, J.; Mejia, A.A.; Phan, I.Q.; Lorimer, D.D.; Edwards, T.E.; Aguilera, R.J. Structure of acid deoxyribonuclease. Nucleic Acids Res. 2017, 45, 6217–6227. [Google Scholar] [CrossRef]
- Bernardi, G. Competitive inhibition of acid deoxyribonuclease by polyribonucleotides. Biochem. Biophys. Res. Commun. 1964, 17, 573–577. [Google Scholar] [CrossRef]
- Jacquemin-Sablon, A.; Laval, J.; Letalaer, J.Y.; Lepecq, J.B.; Paoletti, C. Inhibition of splenic acid desoxyribonuclease (DNase II) by ribonucleic acids (RNA). C. R. Hebd. Seances L’academie Sci. 1964, 259, 2551–2554. [Google Scholar]
- Yang, W. Nucleases: Diversity of structure, function and mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Abelson, J.; Trotta, C.R.; Li, H. tRNA splicing. J. Biol. Chem. 1998, 273, 12685–12688. [Google Scholar] [CrossRef]
- Lauková, L.; Konečná, B.; Janovičová, Ľ.; Vlková, B.; Celec, P. Deoxyribonucleases and Their Applications in Biomedicine. Biomolecules 2020, 10, 1036. [Google Scholar] [CrossRef]
- Kawane, K.; Fukuyama, H.; Yoshida, H.; Nagase, H.; Ohsawa, Y.; Uchiyama, Y.; Okada, K.; Iida, T.; Nagata, S. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat. Immunol. 2003, 4, 138–144. [Google Scholar] [CrossRef]
- Kawane, K.; Fukuyama, H.; Kondoh, G.; Takeda, J.; Ohsawa, Y.; Uchiyama, Y.; Nagata, S. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 2001, 292, 1546–1549. [Google Scholar] [CrossRef]
- Krieser, R.J.; MacLea, K.S.; Longnecker, D.S.; Fields, J.L.; Fiering, S.; Eastman, A. Deoxyribonuclease IIalpha is required during the phagocytic phase of apoptosis and its loss causes perinatal lethality. Cell Death Differ. 2002, 9, 956–962. [Google Scholar] [CrossRef]
- Yoshida, H.; Okabe, Y.; Kawane, K.; Fukuyama, H.; Nagata, S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat. Immunol. 2005, 6, 49–56. [Google Scholar] [CrossRef]
- Torriglia, A.; Chaudun, E.; Chany-Fournier, F.; Jeanny, J.C.; Courtois, Y.; Counis, M.F. Involvement of DNase II in nuclear degeneration during lens cell differentiation. J. Biol. Chem. 1995, 270, 28579–28585. [Google Scholar] [CrossRef]
- Nishimoto, S.; Kawane, K.; Watanabe-Fukunaga, R.; Fukuyama, H.; Ohsawa, Y.; Uchiyama, Y.; Hashida, N.; Ohguro, N.; Tano, Y.; Morimoto, T.; et al. Nuclear cataract caused by a lack of DNA degradation in the mouse eye lens. Nature 2003, 424, 1071–1074. [Google Scholar] [CrossRef]
- Hedgecock, E.M.; Sulston, J.E.; Thomson, J.N. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 1983, 220, 1277–1279. [Google Scholar] [CrossRef]
- Stone, J.C.; Dower, N.A.; Hauseman, J.; Cseko, Y.M.; Sederoff, R. The characterization of a mutant affecting DNA metabolism in the development of D. melanogaster. Can. J. Genet. Cytol. 1983, 25, 129–138. [Google Scholar] [CrossRef]
- McIlroy, D.; Tanaka, M.; Sakahira, H.; Fukuyama, H.; Suzuki, M.; Yamamura, K.; Ohsawa, Y.; Uchiyama, Y.; Nagata, S. An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes Dev. 2000, 14, 549–558. [Google Scholar] [CrossRef]
- Wu, Y.C.; Stanfield, G.M.; Horvitz, H.R. NUC-1, a caenorhabditis elegans DNase II homolog, functions in an intermediate step of DNA degradation during apoptosis. Genes Dev. 2000, 14, 536–548. [Google Scholar] [CrossRef]
- Mukae, N.; Yokoyama, H.; Yokokura, T.; Sakoyama, Y.; Nagata, S. Activation of the innate immunity in Drosophila by endogenous chromosomal DNA that escaped apoptotic degradation. Genes Dev. 2002, 16, 2662–2671. [Google Scholar] [CrossRef]
- Potenza, N.; Salvatore, V.; Migliozzi, A.; Martone, V.; Nobile, V.; Russo, A. Hybridase activity of human ribonuclease-1 revealed by a real-time fluorometric assay. Nucleic Acids Res. 2006, 34, 2906–2913. [Google Scholar] [CrossRef]
- Sorrentino, S.; Libonati, M. Structure-function relationships in human ribonucleases: Main distinctive features of the major RNase types. FEBS. Lett. 1997, 404, 1–5. [Google Scholar] [CrossRef]
- An, R.; Jia, Y.; Wan, B.; Zhang, Y.; Dong, P.; Li, J.; Liang, X. Non-enzymatic depurination of nucleic acids: Factors and mechanisms. PLoS ONE 2014, 9, e115950. [Google Scholar] [CrossRef]
- Stevens, A. Purification and characterization of a Saccharomyces cerevisiae exoribonuclease which yields 5′-mononucleotides by a 5′ leads to 3′ mode of hydrolysis. J. Biol. Chem. 1980, 255, 3080–3085. [Google Scholar] [CrossRef]
- Zuo, Y.; Deutscher, M.P. The physiological role of RNase T can be explained by its unusual substrate specificity. J. Biol. Chem. 2002, 277, 29654–29661. [Google Scholar] [CrossRef]
- Drew, H.R.; Travers, A.A. DNA structural variations in the E. coli tyrT promoter. Cell 1984, 37, 491–502. [Google Scholar] [CrossRef]
- Drew, H.R.; Travers, A.A. Structural junctions in DNA: The influence of flanking sequence on nuclease digestion specificities. Nucleic Acids Res. 1985, 13, 4445–4467. [Google Scholar] [CrossRef]
- Peracaula, R.; Cleary, K.R.; Lorenzo, J.; de Llorens, R.; Frazier, M.L. Human pancreatic ribonuclease 1: Expression and distribution in pancreatic adenocarcinoma. Cancer 2000, 89, 1252–1258. [Google Scholar] [CrossRef]
- Li, M.; Chen, L.; Tian, S.; Lin, Y.; Tang, Q.; Zhou, X.; Li, D.; Yeung, C.K.L.; Che, T.; Jin, L.; et al. Comprehensive variation discovery and recovery of missing sequence in the pig genome using multiple de novo assemblies. Genome Res. 2017, 27, 865–874. [Google Scholar] [CrossRef]
- Buckle, A.M.; Fersht, A.R. Subsite binding in an RNase: Structure of a barnase-tetranucleotide complex at 1.76-A resolution. Biochemistry 1994, 33, 1644–1653. [Google Scholar] [CrossRef]
- Calvin, K.; Li, H. RNA-splicing endonuclease structure and function. Cell Mol. Life Sci. 2008, 65, 1176–1185. [Google Scholar] [CrossRef]
- Cochrane, J.C.; Strobel, S.A. Catalytic strategies of self-cleaving ribozymes. Acc. Chem. Res. 2008, 41, 1027–1035. [Google Scholar] [CrossRef]
- Correll, C.C.; Yang, X.; Gerczei, T.; Beneken, J.; Plantinga, M.J. RNA recognition and base flipping by the toxin sarcin. J. Synchrotron Radiat. 2004, 11, 93–96. [Google Scholar] [CrossRef]
- Schäfer, P.; Cymerman, I.A.; Bujnicki, J.M.; Meiss, G. Human lysosomal DNase IIalpha contains two requisite PLD-signature (HxK) motifs: Evidence for a pseudodimeric structure of the active enzyme species. Protein Sci. 2007, 16, 82–91. [Google Scholar] [CrossRef]
- Gottlin, E.B.; Rudolph, A.E.; Zhao, Y.; Matthews, H.R.; Dixon, J.E. Catalytic mechanism of the phospholipase D superfamily proceeds via a covalent phosphohistidine intermediate. Proc. Natl. Acad. Sci. USA 1998, 95, 9202–9207. [Google Scholar] [CrossRef]
- Stuckey, J.A.; Dixon, J.E. Crystal structure of a phospholipase D family member. Nat. Struct. Biol. 1999, 6, 278–284. [Google Scholar]
- Waite, M. The PLD superfamily: Insights into catalysis. Biochim. Biophys. Acta 1999, 1439, 187–197. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Hsueh, C.C.; Lu, S.C.; Liao, T.H. Identification of three crucial histidine residues (His115, His132 and His297) in porcine deoxyribonuclease II. Biochem. J. 2006, 398, 177–185. [Google Scholar] [CrossRef]
- Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. [Google Scholar] [PubMed]
- Basner-Tschakarjan, E.; Mirmohammadsadegh, A.; Baer, A.; Hengge, U.R. Uptake and trafficking of DNA in keratinocytes: Evidence for DNA-binding proteins. Gene Ther. 2004, 11, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Wada, K.; Kabuta, T. Lysosomal degradation of intracellular nucleic acids-multiple autophagic pathways. J. Biochem. 2017, 161, 145–154. [Google Scholar] [CrossRef]
- Huang, H.; Kawamata, T.; Horie, T.; Tsugawa, H.; Nakayama, Y.; Ohsumi, Y.; Fukusaki, E. Bulk RNA degradation by nitrogen starvation-induced autophagy in yeast. EMBO J. 2015, 34, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Pisoni, R.L.; Thoene, J.G. Detection and characterization of a nucleoside transport system in human fibroblast lysosomes. J. Biol. Chem. 1989, 264, 4850–4856. [Google Scholar] [CrossRef]
- Luhtala, N.; Parker, R. T2 Family ribonucleases: Ancient enzymes with diverse roles. Trends Biochem. Sci. 2010, 35, 253–259. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, J.; Du, X.; Liu, K.; Hao, J.; Wang, H.; An, R.; Liang, X. DNase II Can Efficiently Digest RNA and Needs to Be Redefined as a Nuclease. Cells 2024, 13, 1525. https://doi.org/10.3390/cells13181525
Zhuang J, Du X, Liu K, Hao J, Wang H, An R, Liang X. DNase II Can Efficiently Digest RNA and Needs to Be Redefined as a Nuclease. Cells. 2024; 13(18):1525. https://doi.org/10.3390/cells13181525
Chicago/Turabian StyleZhuang, Jingyun, Xinmei Du, Kehan Liu, Jing Hao, Haoyu Wang, Ran An, and Xingguo Liang. 2024. "DNase II Can Efficiently Digest RNA and Needs to Be Redefined as a Nuclease" Cells 13, no. 18: 1525. https://doi.org/10.3390/cells13181525
APA StyleZhuang, J., Du, X., Liu, K., Hao, J., Wang, H., An, R., & Liang, X. (2024). DNase II Can Efficiently Digest RNA and Needs to Be Redefined as a Nuclease. Cells, 13(18), 1525. https://doi.org/10.3390/cells13181525