
Treg Cell Therapeutic Strategies for Breast Cancer: Holistic to Local Aspects




Abstract
:1. Introduction
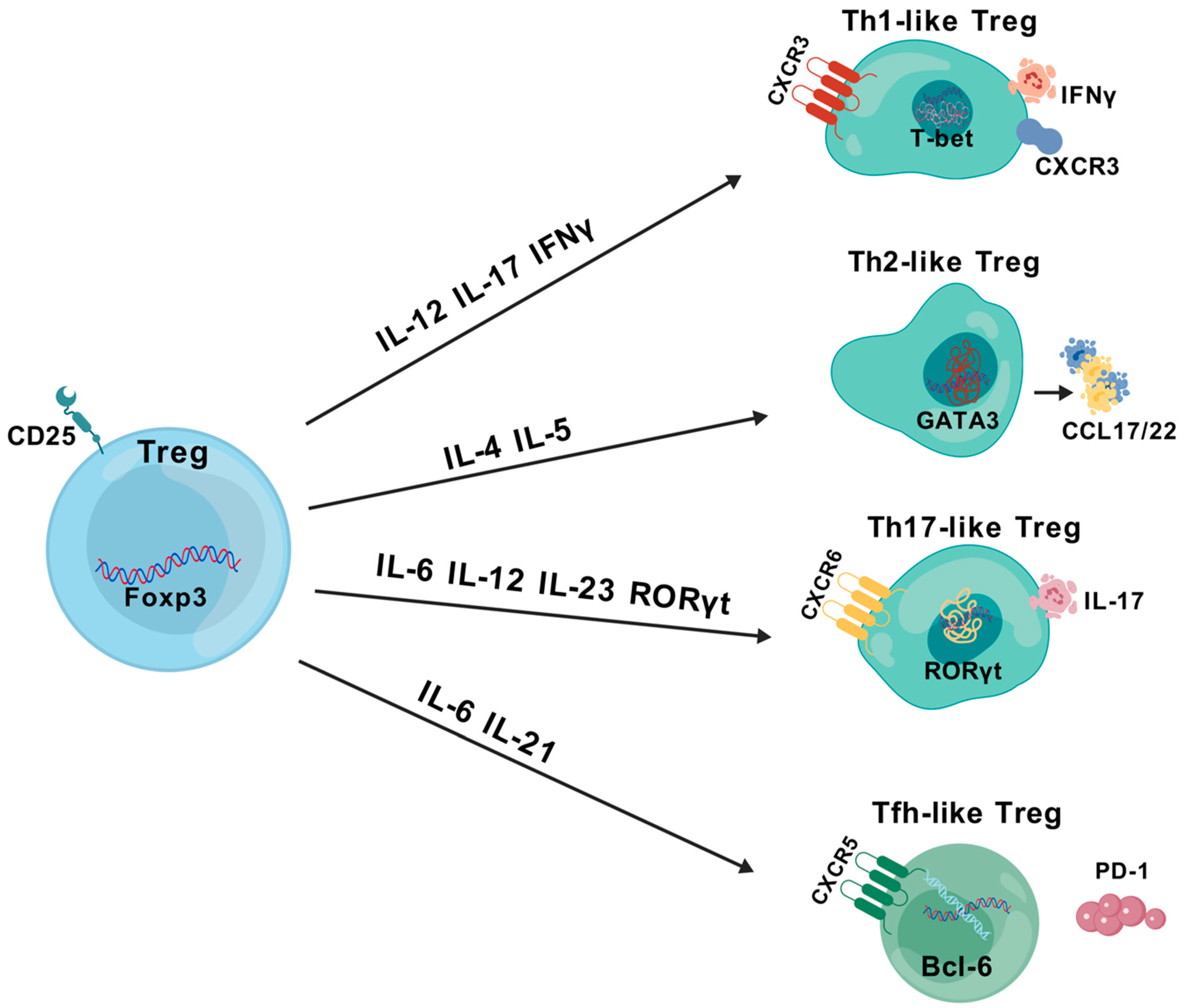
2. Treg Classification and Characteristics
2.1. Treg Classification
2.2. Treg Characteristics
3. Identification of Tregs
3.1. Flow Cytometry
3.2. Transcriptomics
3.3. Proteomics
4. Treg Interactions in BC
4.1. Chemokine Recruitment of Tregs
4.2. Treg Immunosuppression of Tumors
4.3. BC Metabolism and Treg Effects
4.4. Tregs Enhance Tumor Progression, Metastasis and Drug Resistance
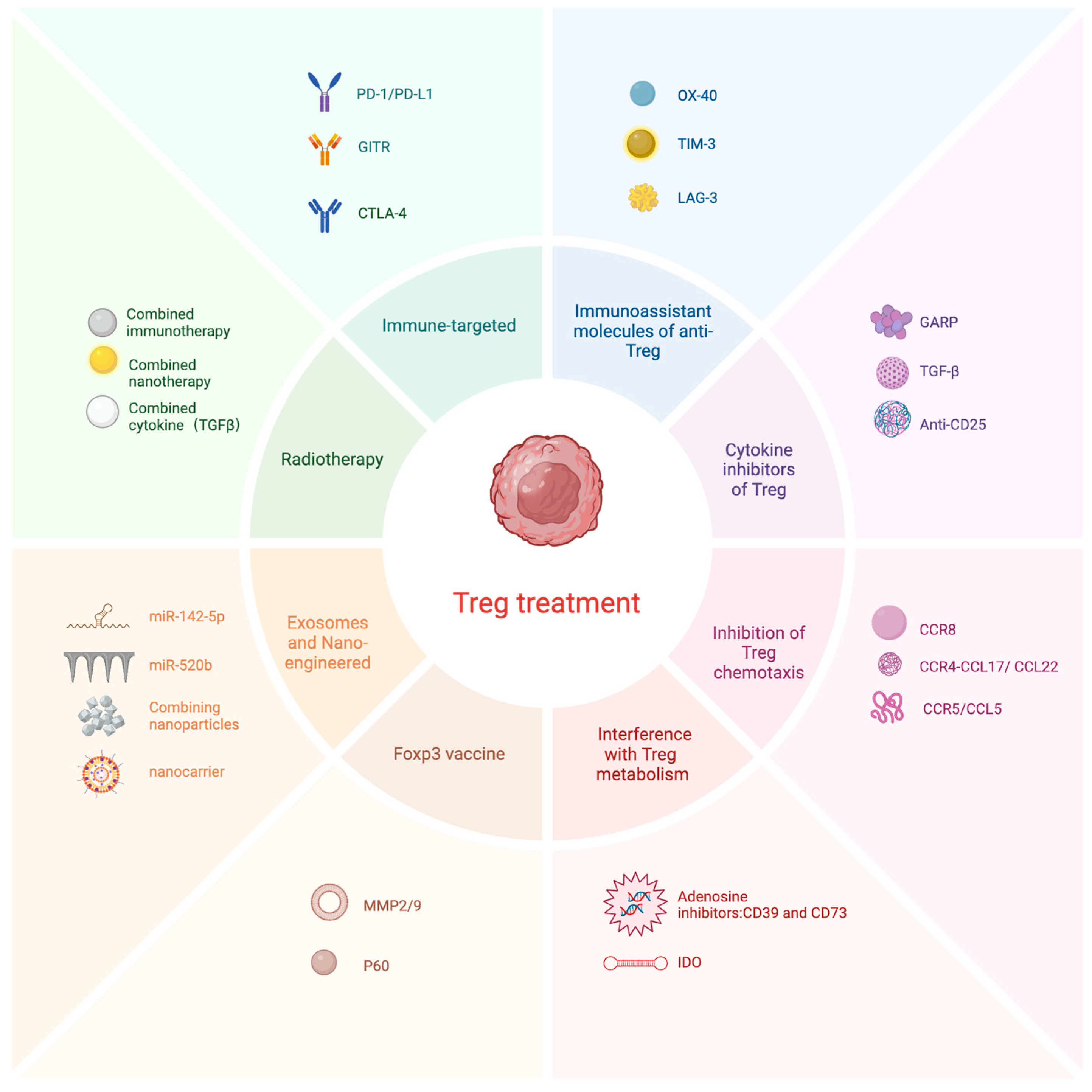
5. Immune-Targeted Therapy against Tregs
5.1. CTLA-4-Targeting Antibody
5.2. PD-1/PD-L1-Targeted Antibodies
5.3. Anti-GITR-Targeting Antibodies
5.4. Immunoassistant Molecules of Anti-Treg
5.5. Cytokine Inhibitors of Treg
5.5.1. Anti-CD25 Antibody
5.5.2. TGF-β and GARP
5.6. Inhibition of Treg Chemotaxis
5.6.1. CCR8 Inhibitors
5.6.2. CCR4-CCL17/CCL22 Antagonists
5.6.3. CCR5/CCL5
5.7. Interference with Treg Metabolism
5.7.1. Adenosine Inhibitors
5.7.2. Indoleamine 2,3-Dioxygenase (IDO) Inhibitors
5.8. Foxp3 Vaccine
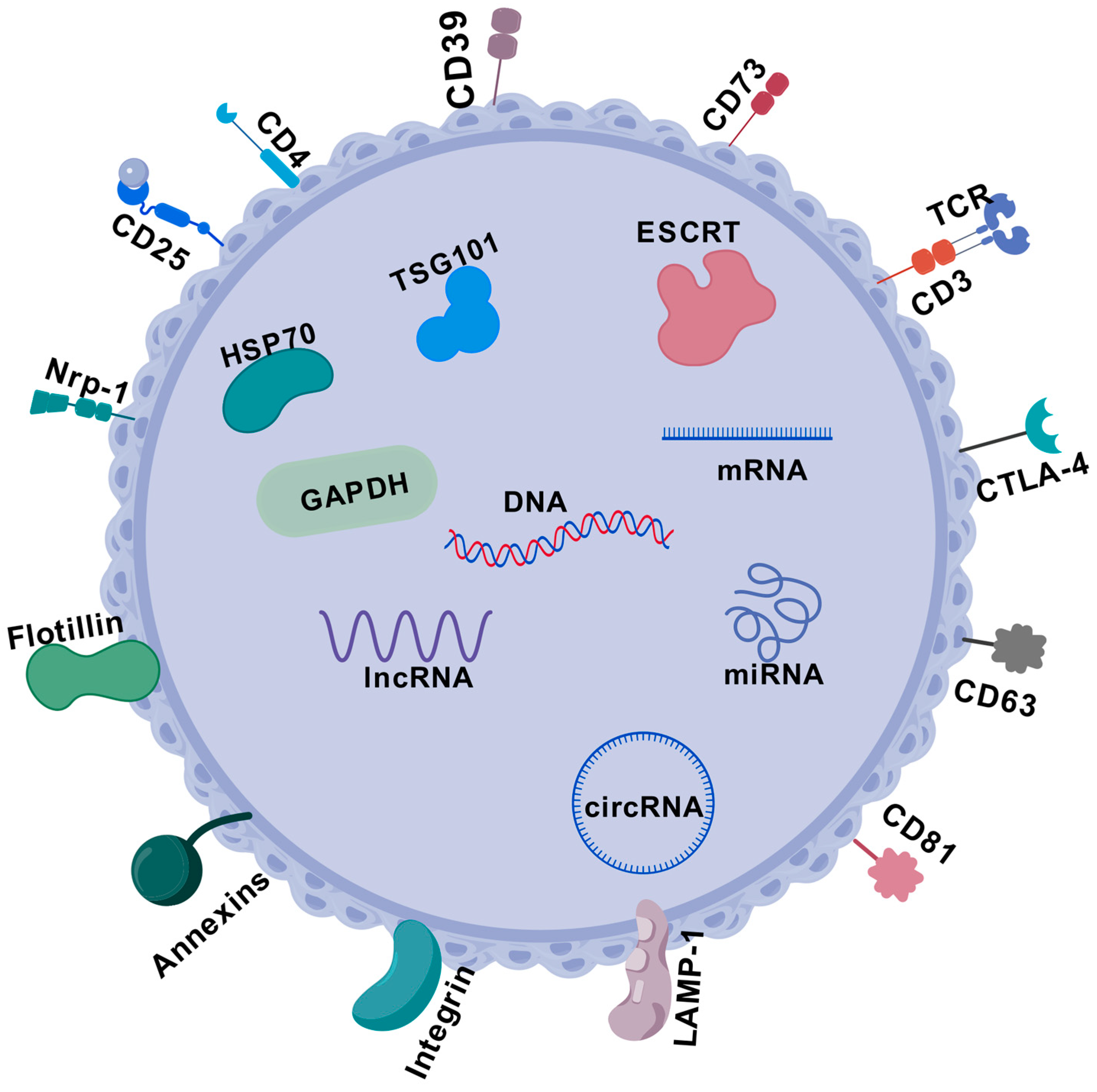
5.9. Exosomes and Tregs
5.10. Nano-Engineered Targeting of Tregs
5.11. Tregs in Combination with Radiotherapy
6. Challenges
7. Future Directions
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, U.; Hanlon, A.L.; Koshy, M.; Buras, R.; Chumsri, S.; Tkaczuk, K.H.; Cheston, S.B.; Regine, W.F.; Feigenberg, S.J. Increasing national mastectomy rates for the treatment of early stage breast cancer. Ann. Surg. Oncol. 2013, 20, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Kroman, N.; Holtveg, H.; Wohlfahrt, J.; Jensen, M.B.; Mouridsen, H.T.; Blichert-Toft, M.; Melbye, M. Effect of breast-conserving therapy versus radical mastectomy on prognosis for young women with breast carcinoma. Cancer 2004, 100, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.; Anderson, S.; Bryant, J.; Margolese, R.G.; Deutsch, M.; Fisher, E.R.; Jeong, J.H.; Wolmark, N. Twenty-year follow-up of a randomized trial comparing total mastectomy, lumpectomy, and lumpectomy plus irradiation for the treatment of invasive breast cancer. N. Engl. J. Med. 2002, 347, 1233–1241. [Google Scholar] [CrossRef]
- Regan, M.M.; Francis, P.A.; Pagani, O.; Fleming, G.F.; Walley, B.A.; Viale, G.; Colleoni, M.; Láng, I.; Gómez, H.L.; Tondini, C.; et al. Absolute Benefit of Adjuvant Endocrine Therapies for Premenopausal Women With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Early Breast Cancer: TEXT and SOFT Trials. J. Clin. Oncol. 2016, 34, 2221–2231. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Coussens, L.M. Inflammation and breast cancer. Balancing immune response: Crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007, 9, 212. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Sharma, N.; Vacher, J.; Allison, J.P. TLR1/2 ligand enhances antitumor efficacy of CTLA-4 blockade by increasing intratumoral Treg depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 10453–10462. [Google Scholar] [CrossRef]
- Coussens, L.M.; Pollard, J.W. Leukocytes in mammary development and cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, a003285. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Bohling, S.D.; Allison, K.H. Immunosuppressive regulatory T cells are associated with aggressive breast cancer phenotypes: A potential therapeutic target. Mod. Pathol. 2008, 21, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.G.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef]
- Sather, B.D.; Treuting, P.; Perdue, N.; Miazgowicz, M.; Fontenot, J.D.; Rudensky, A.Y.; Campbell, D.J. Altering the distribution of Foxp3+ regulatory T cells results in tissue-specific inflammatory disease. J. Exp. Med. 2007, 204, 1335–1347. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Chaudhry, A.; Rudensky, A.Y. Control of inflammation by integration of environmental cues by regulatory T cells. J. Clin. Investig. 2013, 123, 939–944. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Togashi, Y.; Nishikawa, H. Regulatory T Cells: Molecular and Cellular Basis for Immunoregulation. Curr. Top. Microbiol. Immunol. 2017, 410, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Toda, A.; Piccirillo, C.A. Development and function of naturally occurring CD4+CD25+ regulatory T cells. J. Leukoc. Biol. 2006, 80, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Takahashi, T.; Sakaguchi, S. Control of autoimmunity by naturally arising regulatory CD4+ T cells. Adv. Immunol. 2003, 81, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, C.A.; Thornton, A.M. Cornerstone of peripheral tolerance: Naturally occurring CD4+CD25+ regulatory T cells. Trends Immunol. 2004, 25, 374–380. [Google Scholar] [CrossRef]
- Picca, C.C.; Larkin, J., 3rd; Boesteanu, A.; Lerman, M.A.; Rankin, A.L.; Caton, A.J. Role of TCR specificity in CD4+ CD25+ regulatory T-cell selection. Immunol. Rev. 2006, 212, 74–85. [Google Scholar] [CrossRef]
- King, C.; Sarvetnick, N. Organ-specific autoimmunity. Curr. Opin. Immunol. 1997, 9, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Gavin, M.A.; Rasmussen, J.P.; Fontenot, J.D.; Vasta, V.; Manganiello, V.C.; Beavo, J.A.; Rudensky, A.Y. Foxp3-dependent programme of regulatory T-cell differentiation. Nature 2007, 445, 771–775. [Google Scholar] [CrossRef]
- Feuerer, M.; Hill, J.A.; Mathis, D.; Benoist, C. Foxp3+ regulatory T cells: Differentiation, specification, subphenotypes. Nat. Immunol. 2009, 10, 689–695. [Google Scholar] [CrossRef]
- Cassis, L.; Aiello, S.; Noris, M. Natural versus adaptive regulatory T cells. Contrib. Nephrol. 2005, 146, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, K.S.; Richards, E.; Srivastava, S.; Thomas, K.R.; Dudda, J.C.; Klonowski, K.D.; Campbell, D.J. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med. 2014, 211, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Panduro, M.; Benoist, C.; Mathis, D. Tissue Tregs. Annu. Rev. Immunol. 2016, 34, 609–633. [Google Scholar] [CrossRef]
- Burzyn, D.; Benoist, C.; Mathis, D. Regulatory T cells in nonlymphoid tissues. Nat. Immunol. 2013, 14, 1007–1013. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Y.; Liu, S.; Wang, H.; Zhu, J.; Ou, L.; Xu, X. Targeting regulatory T cells for immunotherapy in melanoma. Mol. Biomed. 2021, 2, 11. [Google Scholar] [CrossRef]
- Pere, H.; Montier, Y.; Bayry, J.; Quintin-Colonna, F.; Merillon, N.; Dransart, E.; Badoual, C.; Gey, A.; Ravel, P.; Marcheteau, E.; et al. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood 2011, 118, 4853–4862. [Google Scholar] [CrossRef]
- Ma, Y.; Xu, X.; Wang, H.; Liu, Y.; Piao, H. Non-coding RNA in tumor-infiltrating regulatory T cells formation and associated immunotherapy. Front. Immunol. 2023, 14, 1228331. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef]
- Di Pilato, M.; Kim, E.Y.; Cadilha, B.L.; Prüßmann, J.N.; Nasrallah, M.N.; Seruggia, D.; Usmani, S.M.; Misale, S.; Zappulli, V.; Carrizosa, E.; et al. Targeting the CBM complex causes T(reg) cells to prime tumours for immune checkpoint therapy. Nature 2019, 570, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Daniel, V.; Wang, H.; Sadeghi, M.; Opelz, G. Interferon-gamma producing regulatory T cells as a diagnostic and therapeutic tool in organ transplantation. Int. Rev. Immunol. 2014, 33, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Barrow, A.; Pettipher, R. Novel function of CRTH2 in preventing apoptosis of human Th2 cells through activation of the phosphatidylinositol 3-kinase pathway. J. Immunol. 2009, 182, 7580–7586. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Burton, O.T.; Wise, P.; Charbonnier, L.M.; Georgiev, P.; Oettgen, H.C.; Rachid, R.; Chatila, T.A. Regulatory T cell reprogramming toward a Th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity 2015, 42, 512–523. [Google Scholar] [CrossRef]
- Mizukami, Y.; Kono, K.; Kawaguchi, Y.; Akaike, H.; Kamimura, K.; Sugai, H.; Fujii, H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int. J. Cancer 2008, 122, 2286–2293. [Google Scholar] [CrossRef] [PubMed]
- Halim, L.; Romano, M.; McGregor, R.; Correa, I.; Pavlidis, P.; Grageda, N.; Hoong, S.J.; Yuksel, M.; Jassem, W.; Hannen, R.F.; et al. An Atlas of Human Regulatory T Helper-like Cells Reveals Features of Th2-like Tregs that Support a Tumorigenic Environment. Cell Rep. 2017, 20, 757–770. [Google Scholar] [CrossRef]
- Qianmei, Y.; Zehong, S.; Guang, W.; Hui, L.; Lian, G. Recent advances in the role of Th17/Treg cells in tumor immunity and tumor therapy. Immunol. Res. 2021, 69, 398–414. [Google Scholar] [CrossRef]
- Voo, K.S.; Wang, Y.H.; Santori, F.R.; Boggiano, C.; Wang, Y.H.; Arima, K.; Bover, L.; Hanabuchi, S.; Khalili, J.; Marinova, E.; et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 4793–4798. [Google Scholar] [CrossRef] [PubMed]
- Massoud, A.H.; Charbonnier, L.M.; Lopez, D.; Pellegrini, M.; Phipatanakul, W.; Chatila, T.A. An asthma-associated IL4R variant exacerbates airway inflammation by promoting conversion of regulatory T cells to TH17-like cells. Nat. Med. 2016, 22, 1013–1022. [Google Scholar] [CrossRef]
- O’Sullivan, C.C.; Davarpanah, N.N.; Abraham, J.; Bates, S.E. Current challenges in the management of breast cancer brain metastases. Semin. Oncol. 2017, 44, 85–100. [Google Scholar] [CrossRef]
- Wang, J.; Cai, D.; Ma, B.; Wu, G.; Wu, J. Skewing the balance of regulatory T-cells and T-helper 17 cells in breast cancer patients. J. Int. Med. Res. 2011, 39, 691–701. [Google Scholar] [CrossRef] [PubMed]
- King, C. New insights into the differentiation and function of T follicular helper cells. Nat. Rev. Immunol. 2009, 9, 757–766. [Google Scholar] [CrossRef]
- Maceiras, A.R.; Almeida, S.C.P.; Mariotti-Ferrandiz, E.; Chaara, W.; Jebbawi, F.; Six, A.; Hori, S.; Klatzmann, D.; Faro, J.; Graca, L. T follicular helper and T follicular regulatory cells have different TCR specificity. Nat. Commun. 2017, 8, 15067. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Liu, A.; Liu, G.; Wu, F.; Li, Z. T follicular regulatory cells suppress Tfh-mediated B cell help and synergistically increase IL-10-producing B cells in breast carcinoma. Immunol. Res. 2019, 67, 416–423. [Google Scholar] [CrossRef]
- Li, M.O.; Rudensky, A.Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016, 16, 220–233. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Pillars Article: Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Khattri, R.; Cox, T.; Yasayko, S.A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef]
- Rossetti, M.; Spreafico, R.; Consolaro, A.; Leong, J.Y.; Chua, C.; Massa, M.; Saidin, S.; Magni-Manzoni, S.; Arkachaisri, T.; Wallace, C.A.; et al. TCR repertoire sequencing identifies synovial Treg cell clonotypes in the bloodstream during active inflammation in human arthritis. Ann. Rheum. Dis. 2017, 76, 435–441. [Google Scholar] [CrossRef]
- Paparo, L.; Nocerino, R.; Cosenza, L.; Aitoro, R.; D’Argenio, V.; Del Monaco, V.; Di Scala, C.; Amoroso, A.; Di Costanzo, M.; Salvatore, F.; et al. Epigenetic features of FoxP3 in children with cow’s milk allergy. Clin. Epigenet. 2016, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Huss, D.J.; Mehta, D.S.; Sharma, A.; You, X.; Riester, K.A.; Sheridan, J.P.; Amaravadi, L.S.; Elkins, J.S.; Fontenot, J.D. In vivo maintenance of human regulatory T cells during CD25 blockade. J. Immunol. 2015, 194, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wang, J.; Thomson, A.W.; Gandhi, C.R. Hepatic stellate cells increase the immunosuppressive function of natural Foxp3+ regulatory T cells via IDO-induced AhR activation. J. Leukoc. Biol. 2017, 101, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, H.; Ohkura, N.; Vandenbon, A.; Itoh, M.; Nagao-Sato, S.; Kawaji, H.; Lassmann, T.; Carninci, P.; Hayashizaki, Y.; Forrest, A.R.; et al. Differential roles of epigenetic changes and Foxp3 expression in regulatory T cell-specific transcriptional regulation. Proc. Natl. Acad. Sci. USA 2014, 111, 5289–5294. [Google Scholar] [CrossRef]
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799. [Google Scholar] [CrossRef]
- Smiline Girija, A.S. Protean role of epigenetic mechanisms and their impact in regulating the Tregs in TME. Cancer Gene Ther. 2022, 29, 661–664. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Liu, B.; Shi, C.; Ma, X.; Ren, S.; Zhao, X.; Liu, Y. The expression landscape of FOXP3 and its prognostic value in breast cancer. Ann. Transl. Med. 2022, 10, 801. [Google Scholar] [CrossRef]
- Sahin, E.; Sahin, M. Epigenetical Targeting of the FOXP3 Gene by S-Adenosylmethionine Diminishes the Suppressive Capacity of Regulatory T Cells Ex Vivo and Alters the Expression Profiles. J. Immunother. 2019, 42, 11–22. [Google Scholar] [CrossRef]
- van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.; Beekman, J.M.; van Beekum, O.; Brenkman, A.B.; Hijnen, D.J.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef]
- Kwon, H.S.; Lim, H.W.; Wu, J.; Schnölzer, M.; Verdin, E.; Ott, M. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J. Immunol. 2012, 188, 2712–2721. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Nagai, Y.; Xiao, Y.; Greene, M.I.; Zhang, H. Lysosome-dependent p300/FOXP3 degradation and limits Treg cell functions and enhances targeted therapy against cancers. Exp. Mol. Pathol. 2013, 95, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance T(reg) cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yang, C.; Pan, F. Post-Translational Regulations of Foxp3 in Treg Cells and Their Therapeutic Applications. Front. Immunol. 2021, 12, 626172. [Google Scholar] [CrossRef]
- Tan, H.; Yang, K.; Li, Y.; Shaw, T.I.; Wang, Y.; Blanco, D.B.; Wang, X.; Cho, J.H.; Wang, H.; Rankin, S.; et al. Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 2017, 46, 488–503. [Google Scholar] [CrossRef]
- Kajal, K.; Bose, S.; Panda, A.K.; Chakraborty, D.; Chakraborty, S.; Pati, S.; Sarkar, T.; Dhar, S.; Roy, D.; Saha, S.; et al. Transcriptional regulation of VEGFA expression in T-regulatory cells from breast cancer patients. Cancer Immunol. Immunother. 2021, 70, 1877–1891. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Lu, F.; Zhao, X.; Nie, Z.; He, B. The prognostic values of FOXP3+ tumor-infiltrating T cells in breast cancer: A systematic review and meta-analysis. Clin. Transl. Oncol. 2023, 25, 1830–1843. [Google Scholar] [CrossRef]
- Liu, L.; Xiao, W.; Zhang, C.; Fan, P.; Zeng, J.; Yi, J. The Potential of FOXP3 in Predicting Survival and Treatment Response in Breast Cancer. Int. J. Gen. Med. 2024, 17, 1233–1251. [Google Scholar] [CrossRef]
- Zuo, T.; Wang, L.; Morrison, C.; Chang, X.; Zhang, H.; Li, W.; Liu, Y.; Wang, Y.; Liu, X.; Chan, M.W.; et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell 2007, 129, 1275–1286. [Google Scholar] [CrossRef]
- Stroukov, W.; Mastronicola, D.; Albany, C.J.; Catak, Z.; Lombardi, G.; Scottà, C. OMIP-090: A 20-parameter flow cytometry panel for rapid analysis of cell diversity and homing capacity in human conventional and regulatory T cells. Cytom. Part A 2023, 103, 362–367. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Brown, J.A.; Freeman, G.J.; Hafler, D.A. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 2001, 167, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Roncador, G.; Brown, P.J.; Maestre, L.; Hue, S.; Martínez-Torrecuadrada, J.L.; Ling, K.L.; Pratap, S.; Toms, C.; Fox, B.C.; Cerundolo, V.; et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur. J. Immunol. 2005, 35, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.; Dijkgraaf, E.M.; Battaglia, A.; Beckhove, P.; Britten, C.M.; Gallimore, A.; Godkin, A.; Gouttefangeas, C.; de Gruijl, T.D.; Koenen, H.J.; et al. Monitoring regulatory T cells in clinical samples: Consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. Cancer Immunol. Immunother. 2015, 64, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Perea, A.L.; Arcia, E.D.; Rueda, C.M.; Velilla, P.A. Phenotypical characterization of regulatory T cells in humans and rodents. Clin. Exp. Immunol. 2016, 185, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef]
- Barcenilla, H.; Pihl, M.; Sjögren, F.; Magnusson, L.; Casas, R. Regulatory T-Cell Phenotyping Using CyTOF. Methods Mol. Biol. 2023, 2559, 231–242. [Google Scholar] [CrossRef]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef]
- Hartmann, F.J.; Mrdjen, D.; McCaffrey, E.; Glass, D.R.; Greenwald, N.F.; Bharadwaj, A.; Khair, Z.; Verberk, S.G.S.; Baranski, A.; Baskar, R.; et al. Single-cell metabolic profiling of human cytotoxic T cells. Nat. Biotechnol. 2021, 39, 186–197. [Google Scholar] [CrossRef]
- Ali, H.R.; Jackson, H.W.; Zanotelli, V.R.T.; Danenberg, E.; Fischer, J.R.; Bardwell, H.; Provenzano, E.; Rueda, O.M.; Chin, S.F.; Aparicio, S.; et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat. Cancer 2020, 1, 163–175. [Google Scholar] [CrossRef]
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef]
- Yi, G.; Zhao, Y.; Xie, F.; Zhu, F.; Wan, Z.; Wang, J.; Wang, X.; Gao, K.; Cao, L.; Li, X.; et al. Single-cell RNA-seq unveils critical regulators of human FOXP3+ regulatory T cell stability. Sci. Bull. 2020, 65, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, V.; Maleki, L.A.; Esmaily, M.; Masjedi, A.; Ghalamfarsa, G.; Namdar, A.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Regulatory T cells in breast cancer as a potent anti-cancer therapeutic target. Int. Immunopharmacol. 2020, 78, 106087. [Google Scholar] [CrossRef] [PubMed]
- Neagu, A.N.; Whitham, D.; Buonanno, E.; Jenkins, A.; Alexa-Stratulat, T.; Tamba, B.I.; Darie, C.C. Proteomics and its applications in breast cancer. Am. J. Cancer Res. 2021, 11, 4006–4049. [Google Scholar] [PubMed]
- Qiu, J.; Xu, L.; Zeng, X.; Wu, H.; Liang, F.; Lv, Q.; Du, Z. CCL5 mediates breast cancer metastasis and prognosis through CCR5/Treg cells. Front. Oncol. 2022, 12, 972383. [Google Scholar] [CrossRef]
- Brett, E.; Duscher, D.; Pagani, A.; Daigeler, A.; Kolbenschlag, J.; Hahn, M. Naming the Barriers between Anti-CCR5 Therapy, Breast Cancer and Its Microenvironment. Int. J. Mol. Sci. 2022, 23, 14159. [Google Scholar] [CrossRef]
- Ahmadvand, S.; Faghih, Z.; Montazer, M.; Safaei, A.; Mokhtari, M.; Jafari, P.; Talei, A.R.; Tahmasebi, S.; Ghaderi, A. Importance of CD45RO+ tumor-infiltrating lymphocytes in post-operative survival of breast cancer patients. Cell. Oncol. 2019, 42, 343–356. [Google Scholar] [CrossRef]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: A systematic review and meta-analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Lee, A.H.; Ellis, I.O.; Green, A.R. An evaluation of the clinical significance of FOXP3+ infiltrating cells in human breast cancer. Breast Cancer Res. Treat. 2011, 127, 99–108. [Google Scholar] [CrossRef]
- Wang, Y.A.; Li, X.L.; Mo, Y.Z.; Fan, C.M.; Tang, L.; Xiong, F.; Guo, C.; Xiang, B.; Zhou, M.; Ma, J.; et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol. Cancer 2018, 17, 168. [Google Scholar] [CrossRef]
- Kondĕlková, K.; Vokurková, D.; Krejsek, J.; Borská, L.; Fiala, Z.; Ctirad, A. Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta Med. 2010, 53, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Bromley, S.K.; Mempel, T.R.; Luster, A.D. Orchestrating the orchestrators: Chemokines in control of T cell traffic. Nat. Immunol. 2008, 9, 970–980. [Google Scholar] [CrossRef]
- Kurose, K.; Ohue, Y.; Sato, E.; Yamauchi, A.; Eikawa, S.; Isobe, M.; Nishio, Y.; Uenaka, A.; Oka, M.; Nakayama, E. Increase in activated Treg in TIL in lung cancer and in vitro depletion of Treg by ADCC using an antihuman CCR4 mAb (KM2760). J. Thorac. Oncol. 2015, 10, 74–83. [Google Scholar] [CrossRef]
- Barsheshet, Y.; Wildbaum, G.; Levy, E.; Vitenshtein, A.; Akinseye, C.; Griggs, J.; Lira, S.A.; Karin, N. CCR8+FOXp3+ T(reg) cells as master drivers of immune regulation. Proc. Natl. Acad. Sci. USA 2017, 114, 6086–6091. [Google Scholar] [CrossRef]
- Eksteen, B.; Miles, A.; Curbishley, S.M.; Tselepis, C.; Grant, A.J.; Walker, L.S.; Adams, D.H. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J. Immunol. 2006, 177, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Carter, S.L.; Hofer, M.J.; Manders, P.; Getts, D.R.; Getts, M.T.; Dreykluft, A.; Lu, B.; Gerard, C.; King, N.J.; et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J. Immunol. 2007, 179, 2774–2786. [Google Scholar] [CrossRef]
- Lin, H.Y.; Sun, S.M.; Lu, X.F.; Chen, P.Y.; Chen, C.F.; Liang, W.Q.; Peng, C.Y. CCR10 activation stimulates the invasion and migration of breast cancer cells through the ERK1/2/MMP-7 signaling pathway. Int. Immunopharmacol. 2017, 51, 124–130. [Google Scholar] [CrossRef]
- Karnezis, T.; Farnsworth, R.H.; Harris, N.C.; Williams, S.P.; Caesar, C.; Byrne, D.J.; Herle, P.; Macheda, M.L.; Shayan, R.; Zhang, Y.F.; et al. CCL27/CCL28-CCR10 Chemokine Signaling Mediates Migration of Lymphatic Endothelial Cells. Cancer Res. 2019, 79, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Kidani, Y.; Nogami, W.; Yasumizu, Y.; Kawashima, A.; Tanaka, A.; Sonoda, Y.; Tona, Y.; Nashiki, K.; Matsumoto, R.; Hagiwara, M.; et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc. Natl. Acad. Sci. USA 2022, 119, e2114282119. [Google Scholar] [CrossRef]
- Olkhanud, P.B.; Baatar, D.; Bodogai, M.; Hakim, F.; Gress, R.; Anderson, R.L.; Deng, J.; Xu, M.; Briest, S.; Biragyn, A. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009, 69, 5996–6004. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Köhler, G.; Ecabert, B.; Mak, T.W.; Kopf, M. Cutting edge: Lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 1999, 163, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Gallimore, A.; Jones, E.; Ecabert, B.; Acha-Orbea, H.; Kopf, M. Normal pathogen-specific immune responses mounted by CTLA-4-deficient T cells: A paradigm reconsidered. Eur. J. Immunol. 2001, 31, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Gorski, J. Re-establishing peripheral tolerance in the absence of CTLA-4: Complementation by wild-type T cells points to an indirect role for CTLA-4. J. Immunol. 2002, 169, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.L.; Van Parijs, L.; Biuckians, A.; Zheng, X.X.; Strom, T.B.; Abbas, A.K. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity 1997, 6, 411–417. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef]
- Munari, E.; Mariotti, F.R.; Quatrini, L.; Bertoglio, P.; Tumino, N.; Vacca, P.; Eccher, A.; Ciompi, F.; Brunelli, M.; Martignoni, G.; et al. PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects. Int. J. Mol. Sci. 2021, 22, 5123. [Google Scholar] [CrossRef]
- De Simone, M.; Arrigoni, A.; Rossetti, G.; Gruarin, P.; Ranzani, V.; Politano, C.; Bonnal, R.J.P.; Provasi, E.; Sarnicola, M.L.; Panzeri, I.; et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 2016, 45, 1135–1147. [Google Scholar] [CrossRef]
- Horwitz, D.A.; Zheng, S.G.; Gray, J.D. Natural and TGF-beta-induced Foxp3+CD4+ CD25+ regulatory T cells are not mirror images of each other. Trends Immunol. 2008, 29, 429–435. [Google Scholar] [CrossRef]
- Thornton, A.M.; Shevach, E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998, 188, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Larmonier, N.; Marron, M.; Zeng, Y.; Cantrell, J.; Romanoski, A.; Sepassi, M.; Thompson, S.; Chen, X.; Andreansky, S.; Katsanis, E. Tumor-derived CD4+CD25+ regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol. Immunother. 2007, 56, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; L’Italien, L.; Hodges, D.; Schebye, X.M. Pivotal roles of CD4+ effector T cells in mediating agonistic anti-GITR mAb-induced-immune activation and tumor immunity in CT26 tumors. J. Immunol. 2007, 179, 7365–7375. [Google Scholar] [CrossRef] [PubMed]
- Mascanfroni, I.D.; Yeste, A.; Vieira, S.M.; Burns, E.J.; Patel, B.; Sloma, I.; Wu, Y.; Mayo, L.; Ben-Hamo, R.; Efroni, S.; et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol. 2013, 14, 1054–1063. [Google Scholar] [CrossRef]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef]
- Chen, Y.; Kuchroo, V.K.; Inobe, J.; Hafler, D.A.; Weiner, H.L. Regulatory T cell clones induced by oral tolerance: Suppression of autoimmune encephalomyelitis. Science 1994, 265, 1237–1240. [Google Scholar] [CrossRef]
- Armstrong, J.M.; Chen, J.F.; Schwarzschild, M.A.; Apasov, S.; Smith, P.T.; Caldwell, C.; Chen, P.; Figler, H.; Sullivan, G.; Fink, S.; et al. Gene dose effect reveals no Gs-coupled A2A adenosine receptor reserve in murine T-lymphocytes: Studies of cells from A2A-receptor-gene-deficient mice. Biochem. J. 2001, 354, 123–130. [Google Scholar] [CrossRef]
- Friberg, M.; Jennings, R.; Alsarraj, M.; Dessureault, S.; Cantor, A.; Extermann, M.; Mellor, A.L.; Munn, D.H.; Antonia, S.J. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101, 151–155. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Zoso, A.; Mazza, E.M.; Bicciato, S.; Mandruzzato, S.; Bronte, V.; Serafini, P.; Inverardi, L. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur. J. Immunol. 2014, 44, 3307–3319. [Google Scholar] [CrossRef]
- Li, F.; Zhao, Y.; Wei, L.; Li, S.; Liu, J. Tumor-infiltrating Treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol. Ther. 2018, 19, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef]
- Rao, D.; Verburg, F.; Renner, K.; Peeper, D.S.; Lacroix, R.; Blank, C.U. Metabolic profiles of regulatory T cells in the tumour microenvironment. Cancer Immunol. Immunother. 2021, 70, 2417–2427. [Google Scholar] [CrossRef]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 2022, 40, 201–218.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Franco, F.; Tsui, Y.C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, X.; Qi, P.; Ye, Y.; Shen, W.; Leng, L.; Wang, L.; Li, X.; Luo, X.; Chen, Y.; et al. Sox2 Communicates with Tregs Through CCL1 to Promote the Stemness Property of Breast Cancer Cells. Stem Cells 2017, 35, 2351–2365. [Google Scholar] [CrossRef]
- Kos, K.; Aslam, M.A.; van de Ven, R.; Wellenstein, M.D.; Pieters, W.; van Weverwijk, A.; Duits, D.E.M.; van Pul, K.; Hau, C.S.; Vrijland, K.; et al. Tumor-educated T(regs) drive organ-specific metastasis in breast cancer by impairing NK cells in the lymph node niche. Cell Rep. 2022, 38, 110447. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Cao, Y.; Yuan, J.; Hu, Y. Adipose-derived stem cell exosomes promote tumor characterization and immunosuppressive microenvironment in breast cancer. Cancer Immunol. Immunother. 2024, 73, 39. [Google Scholar] [CrossRef] [PubMed]
- Bolandi, Z.; Mokhberian, N.; Eftekhary, M.; Sharifi, K.; Soudi, S.; Ghanbarian, H.; Hashemi, S.M. Adipose derived mesenchymal stem cell exosomes loaded with miR-10a promote the differentiation of Th17 and Treg from naive CD4+ T cell. Life Sci. 2020, 259, 118218. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Fang, Q.Q.; Chen, W.Z.; Jiang, J.X.; Jiang, Z.; Ye, J.; Zhang, T.; Yang, L.; Meng, F.B.; Xia, W.J.; et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+γδ1 Treg cells. Signal Transduct. Target. Ther. 2020, 5, 41. [Google Scholar] [CrossRef]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef]
- Li, P.; Liu, C.; Yu, Z.; Wu, M. New Insights into Regulatory T Cells: Exosome- and Non-Coding RNA-Mediated Regulation of Homeostasis and Resident Treg Cells. Front. Immunol. 2016, 7, 574. [Google Scholar] [CrossRef]
- Chen, L.; Huang, H.; Zhang, W.; Ding, F.; Fan, Z.; Zeng, Z. Exosomes Derived From T Regulatory Cells Suppress CD8+ Cytotoxic T Lymphocyte Proliferation and Prolong Liver Allograft Survival. Med. Sci. Monit. 2019, 25, 4877–4884. [Google Scholar] [CrossRef]
- Hu, H.; Wu, J.; Cao, C.; Ma, L. Exosomes derived from regulatory T cells ameliorate acute myocardial infarction by promoting macrophage M2 polarization. IUBMB Life 2020, 72, 2409–2419. [Google Scholar] [CrossRef]
- Mao, H.; Zhang, L.; Yang, Y.; Zuo, W.; Bi, Y.; Gao, W.; Deng, B.; Sun, J.; Shao, Q.; Qu, X. New insights of CTLA-4 into its biological function in breast cancer. Curr. Cancer Drug Targets 2010, 10, 728–736. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Kirchhof, M.G.; Madrenas, J. A molecular perspective of CTLA-4 function. Annu. Rev. Immunol. 2006, 24, 65–97. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Madrenas, J. Molecular determinants of inverse agonist activity of biologicals targeting CTLA-4. J. Immunol. 2007, 179, 3631–3637. [Google Scholar] [CrossRef]
- Knorr, D.A.; Blanchard, L.; Leidner, R.S.; Jensen, S.M.; Meng, R.; Jones, A.; Ballesteros-Merino, C.; Bell, R.B.; Baez, M.; Marino, A.; et al. FcγRIIB Is an Immune Checkpoint Limiting the Activity of Treg-Targeting Antibodies in the Tumor Microenvironment. Cancer Immunol. Res. 2024, 12, 322–333. [Google Scholar] [CrossRef]
- Gan, X.; Shan, Q.; Li, H.; Janssens, R.; Shen, Y.; He, Y.; Chen, F.; van Haperen, R.; Drabek, D.; Li, J.; et al. An anti-CTLA-4 heavy chain-only antibody with enhanced T(reg) depletion shows excellent preclinical efficacy and safety profile. Proc. Natl. Acad. Sci. USA 2022, 119, e2200879119. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Y.; Yang, L.; Lei, L.; He, B.; Cao, J.; Gao, H. Challenges Coexist with Opportunities: Spatial Heterogeneity Expression of PD-L1 in Cancer Therapy. Adv. Sci. 2024, 11, e2303175. [Google Scholar] [CrossRef]
- Lazarus, G.; Audrey, J.; Iskandar, A.W.B. Efficacy and safety profiles of programmed cell death-1/programmed cell death ligand-1 inhibitors in the treatment of triple-negative breast cancer: A comprehensive systematic review. Oncol. Rev. 2019, 13, 425. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Wang, Y.; Li, Y.; Yang, S.; Sheng, W.; Liu, X. The triple-drug combination DBDx enhances the antitumor efficacy of PD-1 antibody associated with Treg modulation. J. Cancer Res. Ther. 2023, 19, 1603–1609. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Yang, Y.; Deng, Y.; Ni, X.; Zhao, B.; Yan, Z.; He, W.; Li, Y.; Li, S.; et al. Synergistic efficacy of PI3Kδ inhibitor with anti-PD-1 mAbs in immune-humanized PDX model of endocrine resistance hormone receptor-positive advanced breast cancer. Heliyon 2023, 9, e18498. [Google Scholar] [CrossRef]
- Fattori, S.; Le Roy, A.; Houacine, J.; Robert, L.; Abes, R.; Gorvel, L.; Granjeaud, S.; Rouvière, M.S.; Ben Amara, A.; Boucherit, N.; et al. CD25high Effector Regulatory T Cells Hamper Responses to PD-1 Blockade in Triple-Negative Breast Cancer. Cancer Res. 2023, 83, 3026–3044. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Kim, B.S.; Koh, C.H.; Seok, J.W.; Park, J.S.; Shin, K.S.; Bae, E.A.; Lee, G.E.; Jeon, H.; Cho, J.; et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat. Med. 2015, 21, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Coe, D.; Begom, S.; Addey, C.; White, M.; Dyson, J.; Chai, J.G. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1367–1377. [Google Scholar] [CrossRef]
- Xiao, X.; Shi, X.; Fan, Y.; Zhang, X.; Wu, M.; Lan, P.; Minze, L.; Fu, Y.X.; Ghobrial, R.M.; Liu, W.; et al. GITR subverts Foxp3+ Tregs to boost Th9 immunity through regulation of histone acetylation. Nat. Commun. 2015, 6, 8266. [Google Scholar] [CrossRef]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Mitsui, J.; Nishikawa, H.; Muraoka, D.; Wang, L.; Noguchi, T.; Sato, E.; Kondo, S.; Allison, J.P.; Sakaguchi, S.; Old, L.J.; et al. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin. Cancer Res. 2010, 16, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef]
- Song, J.Y.; Han, M.G.; Kim, Y.; Kim, M.J.; Kang, M.H.; Jeon, S.H.; Kim, I.A. Combination of local radiotherapy and anti-glucocorticoid-induced tumor necrosis factor receptor (GITR) therapy augments PD-L1 blockade-mediated anti-tumor effects in murine breast cancer model. Radiother. Oncol. 2024, 190, 109981. [Google Scholar] [CrossRef]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell-cell contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Teng, M.W.; Smyth, M.J. Prospects for TIM3-Targeted Antitumor Immunotherapy. Cancer Res. 2011, 71, 6567–6571. [Google Scholar] [CrossRef]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Kashiwagi, S.; Takada, K.; Ishihara, S.; Goto, W.; Morisaki, T.; Shibutani, M.; Tanaka, H.; Hirakawa, K.; Ohira, M. Clinical Significance of Expression of Immunoadjuvant Molecules (LAG-3, TIM-3, OX-40) in Neoadjuvant Chemotherapy for Breast Cancer. Anticancer Res. 2022, 42, 125–136. [Google Scholar] [CrossRef]
- Brignone, C.; Gutierrez, M.; Mefti, F.; Brain, E.; Jarcau, R.; Cvitkovic, F.; Bousetta, N.; Medioni, J.; Gligorov, J.; Grygar, C.; et al. First-line chemoimmunotherapy in metastatic breast carcinoma: Combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J. Transl. Med. 2010, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Dirix, L.; Triebel, F. AIPAC: A Phase IIb study of eftilagimod alpha (IMP321 or LAG-3Ig) added to weekly paclitaxel in patients with metastatic breast cancer. Future Oncol. 2019, 15, 1963–1973. [Google Scholar] [CrossRef]
- Wildiers, H.; Armstrong, A.; Cuypere, E.; Dalenc, F.; Dirix, L.; Chan, S.; Marme, F.; Schröder, C.P.; Huober, J.; Duhoux, F.P.; et al. Paclitaxel plus Eftilagimod Alpha, a Soluble LAG-3 Protein, in Metastatic, HR+ Breast Cancer: Results from AIPAC, a Randomized, Placebo Controlled Phase IIb Trial. Clin. Cancer Res. 2024, 30, 532–541. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Wu, H.; Zhang, B.; Zhang, X.; Wu, X. Blockade of TIM-3 and PD-1 enhances the antitumor effects of MAGE-A11 antigen-specific cytotoxic T lymphocytes. Bull. Cancer 2022, 109, 895–908. [Google Scholar] [CrossRef]
- Weinberg, A.D.; Lemon, M.; Jones, A.J.; Vainiene, M.; Celnik, B.; Buenafe, A.C.; Culbertson, N.; Bakke, A.; Vandenbark, A.A.; Offner, H. OX-40 antibody enhances for autoantigen specific V beta 8.2+ T cells within the spinal cord of Lewis rats with autoimmune encephalomyelitis. J. Neurosci. Res. 1996, 43, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, A.D.; Rivera, M.M.; Prell, R.; Morris, A.; Ramstad, T.; Vetto, J.T.; Urba, W.J.; Alvord, G.; Bunce, C.; Shields, J. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J. Immunol. 2000, 164, 2160–2169. [Google Scholar] [CrossRef]
- Campos Carrascosa, L.; van Beek, A.A.; de Ruiter, V.; Doukas, M.; Wei, J.; Fisher, T.S.; Ching, K.; Yang, W.; van Loon, K.; Boor, P.P.C.; et al. FcγRIIB engagement drives agonistic activity of Fc-engineered αOX40 antibody to stimulate human tumor-infiltrating T cells. J. Immunother. Cancer 2020, 8, e000816. [Google Scholar] [CrossRef]
- Setoguchi, R.; Hori, S.; Takahashi, T.; Sakaguchi, S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005, 201, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Gambineri, E.; Torgerson, T.R.; Ochs, H.D. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr. Opin. Rheumatol. 2003, 15, 430–435. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- Onizuka, S.; Tawara, I.; Shimizu, J.; Sakaguchi, S.; Fujita, T.; Nakayama, E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999, 59, 3128–3133. [Google Scholar]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar] [CrossRef]
- Kreijveld, E.; Koenen, H.J.; Klasen, I.S.; Hilbrands, L.B.; Joosten, I. Following anti-CD25 treatment, a functional CD4+CD25+ regulatory T-cell pool is present in renal transplant recipients. Am. J. Transplant. 2007, 7, 249–255. [Google Scholar] [CrossRef]
- Baldassari, L.E.; Rose, J.W. Daclizumab: Development, Clinical Trials, and Practical Aspects of Use in Multiple Sclerosis. Neurotherapeutics 2017, 14, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Kapic, E.; Becic, F.; Kusturica, J. Basiliximab, mechanism of action and pharmacological properties. Med. Arh. 2004, 58, 373–376. [Google Scholar]
- Solomon, I.; Amann, M.; Goubier, A.; Arce Vargas, F.; Zervas, D.; Qing, C.; Henry, J.Y.; Ghorani, E.; Akarca, A.U.; Marafioti, T.; et al. CD25-T(reg)-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat. Cancer 2020, 1, 1153–1166. [Google Scholar] [CrossRef]
- Zammarchi, F.; Havenith, K.; Bertelli, F.; Vijayakrishnan, B.; Chivers, S.; van Berkel, P.H. CD25-targeted antibody-drug conjugate depletes regulatory T cells and eliminates established syngeneic tumors via antitumor immunity. J. Immunother. Cancer 2020, 8, e000860. [Google Scholar] [CrossRef]
- Dehbashi, M.; Hojati, Z.; Motovali-Bashi, M.; Ganjalikhany, M.R.; Cho, W.C.; Shimosaka, A.; Navabi, P.; Ganjalikhani-Hakemi, M. A Novel CAR Expressing NK Cell Targeting CD25 With the Prospect of Overcoming Immune Escape Mechanism in Cancers. Front. Oncol. 2021, 11, 649710. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massagué, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Lainé, A.; Labiad, O.; Hernandez-Vargas, H.; This, S.; Sanlaville, A.; Léon, S.; Dalle, S.; Sheppard, D.; Travis, M.A.; Paidassi, H.; et al. Regulatory T cells promote cancer immune-escape through integrin αvβ8-mediated TGF-β activation. Nat. Commun. 2021, 12, 6228. [Google Scholar] [CrossRef]
- De Martino, M.; Daviaud, C.; Vanpouille-Box, C. Activin A backs-up TGF-ß to promote regulatory T cells. Oncoimmunology 2021, 10, 1883288. [Google Scholar] [CrossRef]
- De Martino, M.; Daviaud, C.; Diamond, J.M.; Kraynak, J.; Alard, A.; Formenti, S.C.; Miller, L.D.; Demaria, S.; Vanpouille-Box, C. Activin A Promotes Regulatory T-cell-Mediated Immunosuppression in Irradiated Breast Cancer. Cancer Immunol. Res. 2021, 9, 89–102. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tapia-Galisteo, A.; Sánchez-Rodríguez, I.; Narbona, J.; Iglesias-Hernández, P.; Aragón-García, S.; Jiménez-Reinoso, A.; Compte, M.; Khan, S.; Tsuda, T.; Chames, P.; et al. Combination of T cell-redirecting strategies with a bispecific antibody blocking TGF-β and PD-L1 enhances antitumor responses. Oncoimmunology 2024, 13, 2338558. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, Y.; Zhang, X.; Lu, B.; Sun, P.; Wu, Q.; Ding, X.; Huang, J. GARP Correlates With Tumor-Infiltrating T-Cells and Predicts the Outcome of Gastric Cancer. Front. Immunol. 2021, 12, 660397. [Google Scholar] [CrossRef]
- Pandey, J.P.; Namboodiri, A.M.; Armeson, K.E.; Iwasaki, M.; Kasuga, Y.; Hamada, G.S.; Tsugane, S. IGHG, IGKC, and FCGR genes and endogenous antibody responses to GARP in patients with breast cancer and matched controls. Hum. Immunol. 2018, 79, 632–637. [Google Scholar] [CrossRef]
- Probst-Kepper, M.; Geffers, R.; Kröger, A.; Viegas, N.; Erck, C.; Hecht, H.J.; Lünsdorf, H.; Roubin, R.; Moharregh-Khiabani, D.; Wagner, K.; et al. GARP: A key receptor controlling FOXP3 in human regulatory T cells. J. Cell. Mol. Med. 2009, 13, 3343–3357. [Google Scholar] [CrossRef]
- Dedobbeleer, O.; Stockis, J.; van der Woning, B.; Coulie, P.G.; Lucas, S. Cutting Edge: Active TGF-β1 Released from GARP/TGF-β1 Complexes on the Surface of Stimulated Human B Lymphocytes Increases Class-Switch Recombination and Production of IgA. J. Immunol. 2017, 199, 391–396. [Google Scholar] [CrossRef]
- Italiano, A.; Cassier, P.A.; Lin, C.C.; Alanko, T.; Peltola, K.J.; Gazzah, A.; Shiah, H.S.; Calvo, E.; Cervantes, A.; Roda, D.; et al. First-in-human phase 1 study of budigalimab, an anti-PD-1 inhibitor, in patients with non-small cell lung cancer and head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 2022, 71, 417–431. [Google Scholar] [CrossRef]
- Satoh, K.; Kobayashi, Y.; Fujimaki, K.; Hayashi, S.; Ishida, S.; Sugiyama, D.; Sato, T.; Lim, K.; Miyamoto, M.; Kozuma, S.; et al. Novel anti-GARP antibody DS-1055a augments anti-tumor immunity by depleting highly suppressive GARP+ regulatory T cells. Int. Immunol. 2021, 33, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, Y.; Li, T.; Cao, Y.; Hu, B.; Liu, Y.; Zhang, Y.; Li, X.; Liu, J.; Zhang, W.; et al. Suppression of lysosome metabolism-meditated GARP/TGF-β1 complexes specifically depletes regulatory T cells to inhibit breast cancer metastasis. Oncogene 2024, 43, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Edwards, E.W.; Tacke, F.; Angeli, V.; Llodrá, J.; Sanchez-Schmitz, G.; Garin, A.; Haque, N.S.; Peters, W.; van Rooijen, N.; et al. Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J. Exp. Med. 2004, 200, 1231–1241. [Google Scholar] [CrossRef]
- Whiteside, S.K.; Grant, F.M.; Alvisi, G.; Clarke, J.; Tang, L.; Imianowski, C.J.; Zhang, B.; Evans, A.C.; Wesolowski, A.J.; Conti, A.G.; et al. Acquisition of suppressive function by conventional T cells limits antitumor immunity upon T(reg) depletion. Sci. Immunol. 2023, 8, eabo5558. [Google Scholar] [CrossRef]
- Knipfer, L.; Schulz-Kuhnt, A.; Kindermann, M.; Greif, V.; Symowski, C.; Voehringer, D.; Neurath, M.F.; Atreya, I.; Wirtz, S. A CCL1/CCR8-dependent feed-forward mechanism drives ILC2 functions in type 2-mediated inflammation. J. Exp. Med. 2019, 216, 2763–2777. [Google Scholar] [CrossRef]
- Kim, N.; Kim, M.H.; Pyo, J.; Lee, S.M.; Jang, J.S.; Lee, D.W.; Kim, K.W. CCR8 as a Therapeutic Novel Target: Omics-Integrated Comprehensive Analysis for Systematically Prioritizing Indications. Biomedicines 2023, 11, 2910. [Google Scholar] [CrossRef]
- Campbell, J.R.; McDonald, B.R.; Mesko, P.B.; Siemers, N.O.; Singh, P.B.; Selby, M.; Sproul, T.W.; Korman, A.J.; Vlach, L.M.; Houser, J.; et al. Fc-Optimized Anti-CCR8 Antibody Depletes Regulatory T Cells in Human Tumor Models. Cancer Res. 2021, 81, 2983–2994. [Google Scholar] [CrossRef]
- Weaver, J.D.; Stack, E.C.; Buggé, J.A.; Hu, C.; McGrath, L.; Mueller, A.; Wong, M.; Klebanov, B.; Rahman, T.; Kaufman, R.; et al. Differential expression of CCR8 in tumors versus normal tissue allows specific depletion of tumor-infiltrating T regulatory cells by GS-1811, a novel Fc-optimized anti-CCR8 antibody. Oncoimmunology 2022, 11, 2141007. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xi, J.; Li, Y.; Li, Z.; Zhang, Y.; Wang, J.; Fan, G.H. Discovery of a Potent and Selective CCR8 Small Molecular Antagonist IPG7236 for the Treatment of Cancer. J. Med. Chem. 2023, 66, 4548–4564. [Google Scholar] [CrossRef] [PubMed]
- Hoelzinger, D.B.; Smith, S.E.; Mirza, N.; Dominguez, A.L.; Manrique, S.Z.; Lustgarten, J. Blockade of CCL1 inhibits T regulatory cell suppressive function enhancing tumor immunity without affecting T effector responses. J. Immunol. 2010, 184, 6833–6842. [Google Scholar] [CrossRef]
- Sarkar, T.; Dhar, S.; Chakraborty, D.; Pati, S.; Bose, S.; Panda, A.K.; Basak, U.; Chakraborty, S.; Mukherjee, S.; Guin, A.; et al. FOXP3/HAT1 Axis Controls Treg Infiltration in the Tumor Microenvironment by Inducing CCR4 Expression in Breast Cancer. Front. Immunol. 2022, 13, 740588. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef]
- Moore, D.C.; Elmes, J.B.; Shibu, P.A.; Larck, C.; Park, S.I. Mogamulizumab: An Anti-CC Chemokine Receptor 4 Antibody for T-Cell Lymphomas. Ann. Pharmacother. 2020, 54, 371–379. [Google Scholar] [CrossRef]
- Bogacka, J.; Pawlik, K.; Ciapała, K.; Ciechanowska, A.; Mika, J. CC Chemokine Receptor 4 (CCR4) as a Possible New Target for Therapy. Int. J. Mol. Sci. 2022, 23, 15638. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef]
- Nishikawa, H.; Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J. Immunother. Cancer 2021, 9, e002591. [Google Scholar] [CrossRef]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef]
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J.; et al. Tumors establish resistance to immunotherapy by regulating T(reg) recruitment via CCR4. J. Immunother. Cancer 2020, 8, e000764. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Bai, Z.; Srinoulprasert, Y.; Yang, B.G.; Hayasaka, H.; Miyasaka, M. Chemokines in tumor progression and metastasis. Cancer Sci. 2005, 96, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Lin, Y.C.; Mahalingam, J.; Huang, C.T.; Chen, T.W.; Kang, C.W.; Peng, H.M.; Chu, Y.Y.; Chiang, J.M.; Dutta, A.; et al. Tumor-derived chemokine CCL5 enhances TGF-β-mediated killing of CD8+ T cells in colon cancer by T-regulatory cells. Cancer Res. 2012, 72, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, A.; Zepp, M.; Mahmood, S.; Ali, D.M.; Berger, M.R.; Adwan, H. CCR5 blockage by maraviroc: A potential therapeutic option for metastatic breast cancer. Cell. Oncol. 2019, 42, 93–106. [Google Scholar] [CrossRef]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. Clin. Cancer Res. 2019, 25, 3873–3886. [Google Scholar] [CrossRef]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef]
- Giannone, G.; Ghisoni, E.; Genta, S.; Scotto, G.; Tuninetti, V.; Turinetto, M.; Valabrega, G. Immuno-Metabolism and Microenvironment in Cancer: Key Players for Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4414. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Ohta, A. The ‘danger’ sensors that STOP the immune response: The A2 adenosine receptors? Trends Immunol. 2005, 26, 299–304. [Google Scholar] [CrossRef]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef]
- Sitkovsky, M.V. T regulatory cells: Hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 2009, 30, 102–108. [Google Scholar] [CrossRef]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef]
- Stagg, J.; Beavis, P.A.; Divisekera, U.; Liu, M.C.; Möller, A.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef]
- Jackson, S.W.; Hoshi, T.; Wu, Y.; Sun, X.; Enjyoji, K.; Cszimadia, E.; Sundberg, C.; Robson, S.C. Disordered purinergic signaling inhibits pathological angiogenesis in cd39/Entpd1-null mice. Am. J. Pathol. 2007, 171, 1395–1404. [Google Scholar] [CrossRef]
- Zohair, B.; Chraa, D.; Rezouki, I.; Benthami, H.; Razzouki, I.; Elkarroumi, M.; Olive, D.; Karkouri, M.; Badou, A. The immune checkpoint adenosine 2A receptor is associated with aggressive clinical outcomes and reflects an immunosuppressive tumor microenvironment in human breast cancer. Front. Immunol. 2023, 14, 1201632. [Google Scholar] [CrossRef]
- Jadidi-Niaragh, F.; Atyabi, F.; Rastegari, A.; Kheshtchin, N.; Arab, S.; Hassannia, H.; Ajami, M.; Mirsanei, Z.; Habibi, S.; Masoumi, F.; et al. CD73 specific siRNA loaded chitosan lactate nanoparticles potentiate the antitumor effect of a dendritic cell vaccine in 4T1 breast cancer bearing mice. J. Control. Release 2017, 246, 46–59. [Google Scholar] [CrossRef]
- Menzel, S.; Duan, Y.; Hambach, J.; Albrecht, B.; Wendt-Cousin, D.; Winzer, R.; Tolosa, E.; Rissiek, A.; Guse, A.H.; Haag, F.; et al. Generation and characterization of antagonistic anti-human CD39 nanobodies. Front. Immunol. 2024, 15, 1328306. [Google Scholar] [CrossRef]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef]
- de Leve, S.; Wirsdörfer, F.; Jendrossek, V. Targeting the Immunomodulatory CD73/Adenosine System to Improve the Therapeutic Gain of Radiotherapy. Front. Immunol. 2019, 10, 698. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, P. Role of indoleamine 2, 3-dioxygenase 1 in immunosuppression of breast cancer. Cancer Pathog. Ther. 2023. [Google Scholar] [CrossRef]
- Yu, C.P.; Fu, S.F.; Chen, X.; Ye, J.; Ye, Y.; Kong, L.D.; Zhu, Z. The Clinicopathological and Prognostic Significance of IDO1 Expression in Human Solid Tumors: Evidence from a Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2018, 49, 134–143. [Google Scholar] [CrossRef]
- Guan, J.; Wu, Y.; Liu, X.; Wang, H.; Ye, N.; Li, Z.; Xiao, C.; Zhang, Z.; Li, Z.; Yang, X. A novel prodrug and its nanoformulation suppress cancer stem cells by inducing immunogenic cell death and inhibiting indoleamine 2, 3-dioxygenase. Biomaterials 2021, 279, 121180. [Google Scholar] [CrossRef]
- Nair, S.; Boczkowski, D.; Fassnacht, M.; Pisetsky, D.; Gilboa, E. Vaccination against the forkhead family transcription factor Foxp3 enhances tumor immunity. Cancer Res. 2007, 67, 371–380. [Google Scholar] [CrossRef]
- Nicola Candia, A.J.; Garcia Fallit, M.; Peña Agudelo, J.A.; Pérez Küper, M.; Gonzalez, N.; Moreno Ayala, M.A.; De Simone, E.; Giampaoli, C.; Casares, N.; Seilicovich, A.; et al. Targeting FOXP3 Tumor-Intrinsic Effects Using Adenoviral Vectors in Experimental Breast Cancer. Viruses 2023, 15, 1813. [Google Scholar] [CrossRef]
- Ding, X.; Peng, C.; Li, Y.; Liu, J.; Song, Y.; Cai, B.; Xiang, M.; Zhang, J.; Wang, Z.; Wang, L. Targeting Inhibition of Foxp3 by MMP2/9 Sensitive Short Peptide Linked P60 Fusion Protein 6(P60-MMPs) to Enhance Antitumor Immunity. Macromol. Biosci. 2020, 20, e2000098. [Google Scholar] [CrossRef]
- Esteller, M.; Pandolfi, P.P. The Epitranscriptome of Noncoding RNAs in Cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Moallemi-Rad, L.; Ghorbani, A.; Dadyar, M.; Hussen, B.M.; Rasul, M.F.; Eslami, S.; Taheri, M.; Jamali, E.; Ghafouri-Fard, S. Expression of Treg-associated lncRNAs in breast cancer. Pathol. Res. Pract. 2023, 241, 154270. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity 2014, 41, 89–103. [Google Scholar] [CrossRef]
- Anandagoda, N.; Willis, J.C.; Hertweck, A.; Roberts, L.B.; Jackson, I.; Gökmen, M.R.; Jenner, R.G.; Howard, J.K.; Lord, G.M. microRNA-142-mediated repression of phosphodiesterase 3B critically regulates peripheral immune tolerance. J. Clin. Investig. 2019, 129, 1257–1271. [Google Scholar] [CrossRef]
- Pei, X.; Wang, X.; Li, H. LncRNA SNHG1 regulates the differentiation of Treg cells and affects the immune escape of breast cancer via regulating miR-448/IDO. Int. J. Biol. Macromol. 2018, 118, 24–30. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, C.; Li, Y.; Zhao, J.; Chen, C.; Zhou, Y.; Tao, Y.; Guo, M.; Qin, N.; Ren, T.; et al. MiR-21 controls in situ expansion of CCR6⁺ regulatory T cells through PTEN/AKT pathway in breast cancer. Immunol. Cell Biol. 2015, 93, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Yuan, J.; He, Y.; Hu, Y. The Effect of miR-520b on Macrophage Polarization and T Cell Immunity by Targeting PTEN in Breast Cancer. J. Oncol. 2021, 2021, 5170496. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, S.; Shi, S.; Chen, Y.; Xu, F.; Wei, X.; Xu, Y. Solubilization and delivery of Ursolic-acid for modulating tumor microenvironment and regulatory T cell activities in cancer immunotherapy. J. Control. Release 2020, 320, 168–178. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Z.; Zhan, X.; Chen, K.; Pu, Y.; Liang, Y.; He, B. In situ injection of dual-delivery PEG based MMP-2 sensitive hydrogels for enhanced tumor penetration and chemo-immune combination therapy. Nanoscale 2021, 13, 9577–9589. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zhai, Z.; Xu, J.; Li, S.; Chen, C.; Lu, B. IL36 Cooperates With Anti-CTLA-4 mAbs to Facilitate Antitumor Immune Responses. Front. Immunol. 2020, 11, 634. [Google Scholar] [CrossRef]
- He, S.; Wang, L.; Wu, D.; Tong, F.; Zhao, H.; Li, H.; Gong, T.; Gao, H.; Zhou, Y. Dual-responsive supramolecular photodynamic nanomedicine with activatable immunomodulation for enhanced antitumor therapy. Acta Pharm. Sin. B 2024, 14, 765–780. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, R.; Huang, Y.; Zhao, W.; Li, J.; Zhang, X.; Wang, P.; Venkataramanan, R.; Fan, J.; Xie, W.; et al. An immunostimulatory dual-functional nanocarrier that improves cancer immunochemotherapy. Nat. Commun. 2016, 7, 13443. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, M.; Voilin, E.; Gros, G.; Corgnac, S.; de Montpréville, V.; Validire, P.; Bismuth, G.; Mami-Chouaib, F. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat. Commun. 2019, 10, 3345. [Google Scholar] [CrossRef]
- Du, B.; Du, Q.; Bai, Y.; Yu, L.; Wang, Y.; Huang, J.; Zheng, M.; Shen, G.; Zhou, J.; Yao, H. Chemotherapy based on “Domino-effect” combined with immunotherapy amplifying the efficacy of an anti-metastatic treatment. J. Mater. Chem. B 2020, 8, 9139–9150. [Google Scholar] [CrossRef] [PubMed]
- Domvri, K.; Petanidis, S.; Zarogoulidis, P.; Anestakis, D.; Charalampidis, C.; Tsavlis, D.; Huang, H.; Freitag, L.; Hohenforst-Schmidt, W.; Matthaios, D.; et al. Engineered Hybrid Treg-Targeted Nanosomes Restrain Lung Immunosuppression by Inducing Intratumoral CD8+T Cell Immunity. Int. J. Nanomed. 2022, 17, 4449–4468. [Google Scholar] [CrossRef]
- Ou, W.; Jiang, L.; Thapa, R.K.; Soe, Z.C.; Poudel, K.; Chang, J.H.; Ku, S.K.; Choi, H.G.; Yong, C.S.; Kim, J.O. Combination of NIR therapy and regulatory T cell modulation using layer-by-layer hybrid nanoparticles for effective cancer photoimmunotherapy. Theranostics 2018, 8, 4574–4590. [Google Scholar] [CrossRef]
- Chen, H.; Luan, X.; Paholak, H.J.; Burnett, J.P.; Stevers, N.O.; Sansanaphongpricha, K.; He, M.; Chang, A.E.; Li, Q.; Sun, D. Depleting tumor-associated Tregs via nanoparticle-mediated hyperthermia to enhance anti-CTLA-4 immunotherapy. Nanomedicine 2020, 15, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Song, J.; Yi, X.; Piyu, J. Polymeric indoximod based prodrug nanoparticles with doxorubicin entrapment for inducing immunogenic cell death and improving the immunotherapy of breast cancer. J. Mater. Chem. B 2022, 10, 2019–2027. [Google Scholar] [CrossRef]
- Zhang, L.; Dou, X.; Zheng, Z.; Ye, C.; Lu, T.X.; Liang, H.L.; Wang, L.; Weichselbaum, R.R.; He, C. YTHDF2/m(6) A/NF-κB axis controls anti-tumor immunity by regulating intratumoral Tregs. EMBO J. 2023, 42, e113126. [Google Scholar] [CrossRef]
- Tian, L.R.; Lin, M.Z.; Zhong, H.H.; Cai, Y.J.; Li, B.; Xiao, Z.C.; Shuai, X.T. Nanodrug regulates lactic acid metabolism to reprogram the immunosuppressive tumor microenvironment for enhanced cancer immunotherapy. Biomater. Sci. 2022, 10, 3892–3900. [Google Scholar] [CrossRef]
- Hu, N.; Xue, H.; Zhang, T.; Fan, Y.; Guo, F.; Li, Z.; Huo, M.; Guan, X.; Chen, G. Harnessing PD-1 cell membrane-coated paclitaxel dimer nanoparticles for potentiated chemoimmunotherapy. Biomed. Pharmacother. 2024, 174, 116482. [Google Scholar] [CrossRef]
- Chang, H.C.; Zou, Z.Z.; Wang, Q.H.; Li, J.; Jin, H.; Yin, Q.X.; Xing, D. Targeting and Specific Activation of Antigen-Presenting Cells by Endogenous Antigen-Loaded Nanoparticles Elicits Tumor-Specific Immunity. Adv. Sci. 2020, 7, 1900069. [Google Scholar] [CrossRef] [PubMed]
- Masjedi, A.; Hassannia, H.; Atyabi, F.; Rastegari, A.; Hojjat-Farsangi, M.; Namdar, A.; Soleimanpour, H.; Azizi, G.; Nikkhoo, A.; Ghalamfarsa, G.; et al. Downregulation of A2AR by siRNA loaded PEG-chitosan-lactate nanoparticles restores the T cell mediated anti-tumor responses through blockage of PKA/CREB signaling pathway. Int. J. Biol. Macromol. 2019, 133, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Radiation therapy to convert the tumor into an in situ vaccine. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef]
- Schmidt, M.A.; Förtsch, C.; Schmidt, M.; Rau, T.T.; Fietkau, R.; Distel, L.V. Circulating regulatory T cells of cancer patients receiving radiochemotherapy may be useful to individualize cancer treatment. Radiother. Oncol. 2012, 104, 131–138. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef]
- Guo, W.; Jia, L.; Xie, L.; Kiang, J.G.; Wang, Y.; Sun, F.; Lin, Z.; Wang, E.; Zhang, Y.; Huang, P.; et al. Turning anecdotal irradiation-induced anticancer immune responses into reproducible in situ cancer vaccines via disulfiram/copper-mediated enhanced immunogenic cell death of breast cancer cells. Cell Death Dis. 2024, 15, 298. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef]
- Kumar, P.; Saini, S.; Prabhakar, B.S. Cancer immunotherapy with check point inhibitor can cause autoimmune adverse events due to loss of Treg homeostasis. Semin. Cancer Biol. 2020, 64, 29–35. [Google Scholar] [CrossRef]
- Gordon, I.O.; Wade, T.; Chin, K.; Dickstein, J.; Gajewski, T.F. Immune-mediated red cell aplasia after anti-CTLA-4 immunotherapy for metastatic melanoma. Cancer Immunol. Immunother. 2009, 58, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.F.; Proverbs-Singh, T.A.; Postow, M.A. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors: A Review. JAMA Oncol. 2016, 2, 1346–1353. [Google Scholar] [CrossRef]
- Bertrand, A.; Kostine, M.; Barnetche, T.; Truchetet, M.E.; Schaeverbeke, T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC Med. 2015, 13, 211. [Google Scholar] [CrossRef]
- Huang, X.; Rudensky, A.Y. Regulatory T cells in the context: Deciphering the dynamic interplay with the tissue environment. Curr. Opin. Immunol. 2024, 89, 102453. [Google Scholar] [CrossRef] [PubMed]
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of Co-stimulation in Cancer Immunotherapy Directed Against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Ménétrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment-How to Target Them? Cancers 2021, 13, 1850. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Bhattacharya, P.; Prabhakar, B.S. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J. Autoimmun. 2018, 95, 77–99. [Google Scholar] [CrossRef]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef]
- Du, X.; Liu, M.; Su, J.; Zhang, P.; Tang, F.; Ye, P.; Devenport, M.; Wang, X.; Zhang, Y.; Liu, Y.; et al. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res. 2018, 28, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Blake, S.J.; Harjunpää, H.; Fairfax, K.A.; Yong, M.C.; Allen, S.; Kohrt, H.E.; Takeda, K.; Smyth, M.J.; Teng, M.W. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res. 2016, 76, 5288–5301. [Google Scholar] [CrossRef] [PubMed]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Regulatory T cell targeting in cancer: Emerging strategies in immunotherapy. Eur. J. Immunol. 2021, 51, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Shams, F.; Golchin, A.; Azari, A.; Mohammadi Amirabad, L.; Zarein, F.; Khosravi, A.; Ardeshirylajimi, A. Nanotechnology-based products for cancer immunotherapy. Mol. Biol. Rep. 2022, 49, 1389–1412. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Sanders, K.L.; Edwards, J.L.; Ye, J.; Si, F.; Gao, A.; Huang, L.; Hsueh, E.C.; Ford, D.A. TLR8-mediated metabolic control of human Treg function: A mechanistic target for cancer immunotherapy. Cell Metab. 2019, 29, 103–123.e5. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanocarrier | Size (nm) | Supportive Measures | Treg Level | Other Marker Levels | Breast Cancer Model | Ref. |
|---|---|---|---|---|---|---|
| Lipid | 176.5 ± 62.23 nm | UA | Down | pSTAT5; IL-10; IL-6 ↓ | 4T1 mice | [267] |
| PEG2k-Fmoc-NLG | 90 nm | PTX | Down | IFN-γ+ CD4+; CD8+ T; M1 Macrophages; G-MDSC ↑ M2 Macrophages ↓ | 4T1.2 mice | [259] |
| DOX/IND@NPs | 104 ± 3.21 nm | IND+DOX | Down | CD8+ T ↑; VEGF; MMP9; CD31 ↓ | 4T1 mice | [259] |
| PNT/DOX NPs | 232.0 ± 11.5 nm | DOX+R837 | Down | IL-6; IL-12; TNF-α; IFN-γ; DC ↑ | 4T1 mice | [267] |
| L@S/L | 100 nm | LND+Sy | Down | M1 Macrophages; NK ↑ M2 Macrophages; Lactic acid ↓ | 4T1 mice | [267] |
| PEG2k-Fmoc-IL36 | <450 nm | Anti-CTLA-4mAb | Down | IFN-γ+ CD4+T ↑ IFN-γ+ CD8+ T ↑ | 4T1.2 Lung metastasis mice | [259] |
| ChLa NPs | 100 nm | CD73-specific siRNA+DC vaccine | Down | IFN-γ ↑ CD73; A2AR; IL-10; IL-17 ↓ | 4T1 mice | [259] |
| PCL NPs | 100 nm | A2AR-specific siRNA | Down | IFN-γ ↑ IL-10; A2AR ↓ | 4T1 mice | [259] |
| PD-1@PTX 2 NPs | 203.7 nm | PTX | Down | TNF-α; IFN-γ ↑; CD8+ T ↑ (3.2 times) | 4T1 mice | [259] |
| DACss | 73.67 ± 1.80 nm | DMC+Anti-PD-1mAb+PDT | Down | ROS ↑ CD80+ CD86+ DC ↑ | 4T1 mice | [270] |
| CA@α-LA | 175.0 ± 8.4 nm | Anti-Nrp-1 mAb | Down | Caspase-3; Bcl-2 ↑ VEGF ↓ | 4T1 mice | [259] |
| Polymer-coated IONPs | 30.7 ± 1.8 nm | Anti-CTLA-4 mAb+PTT | Down | CD8+ T ↑ | 4T1 mice | [270] |
| CuS | 84 ± 3.0 nm | EPDA +PTT | Down | ROS ↑ Ki-67; IDO-1; c-KIT; EPHA2; PDGFRβ ↓ | 4T1 mice | [259] |
| PCL-Hyd-PEG | 150 nm | Tumor endogenous antigens (HCP)+ adjuvants CpG ODN+ Anti-CD80 mAb | Down | IFN-γ; CD4+ T; CD8+ T ↑ | 4T1 rat | [259] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Felthaus, O.; Eigenberger, A.; Klein, S.; Prantl, L. Treg Cell Therapeutic Strategies for Breast Cancer: Holistic to Local Aspects. Cells 2024, 13, 1526. https://doi.org/10.3390/cells13181526
Zhang H, Felthaus O, Eigenberger A, Klein S, Prantl L. Treg Cell Therapeutic Strategies for Breast Cancer: Holistic to Local Aspects. Cells. 2024; 13(18):1526. https://doi.org/10.3390/cells13181526
Chicago/Turabian StyleZhang, Hanwen, Oliver Felthaus, Andreas Eigenberger, Silvan Klein, and Lukas Prantl. 2024. "Treg Cell Therapeutic Strategies for Breast Cancer: Holistic to Local Aspects" Cells 13, no. 18: 1526. https://doi.org/10.3390/cells13181526
APA StyleZhang, H., Felthaus, O., Eigenberger, A., Klein, S., & Prantl, L. (2024). Treg Cell Therapeutic Strategies for Breast Cancer: Holistic to Local Aspects. Cells, 13(18), 1526. https://doi.org/10.3390/cells13181526