
Current Landscape and Evolving Therapies for Primary Biliary Cholangitis

 ,
, 
 and
and
Abstract
:1. Introduction
2. Pathogenic Mechanisms in PBC
2.1. Genetics of PBC
2.2. Epigenetics and Environmental Factors
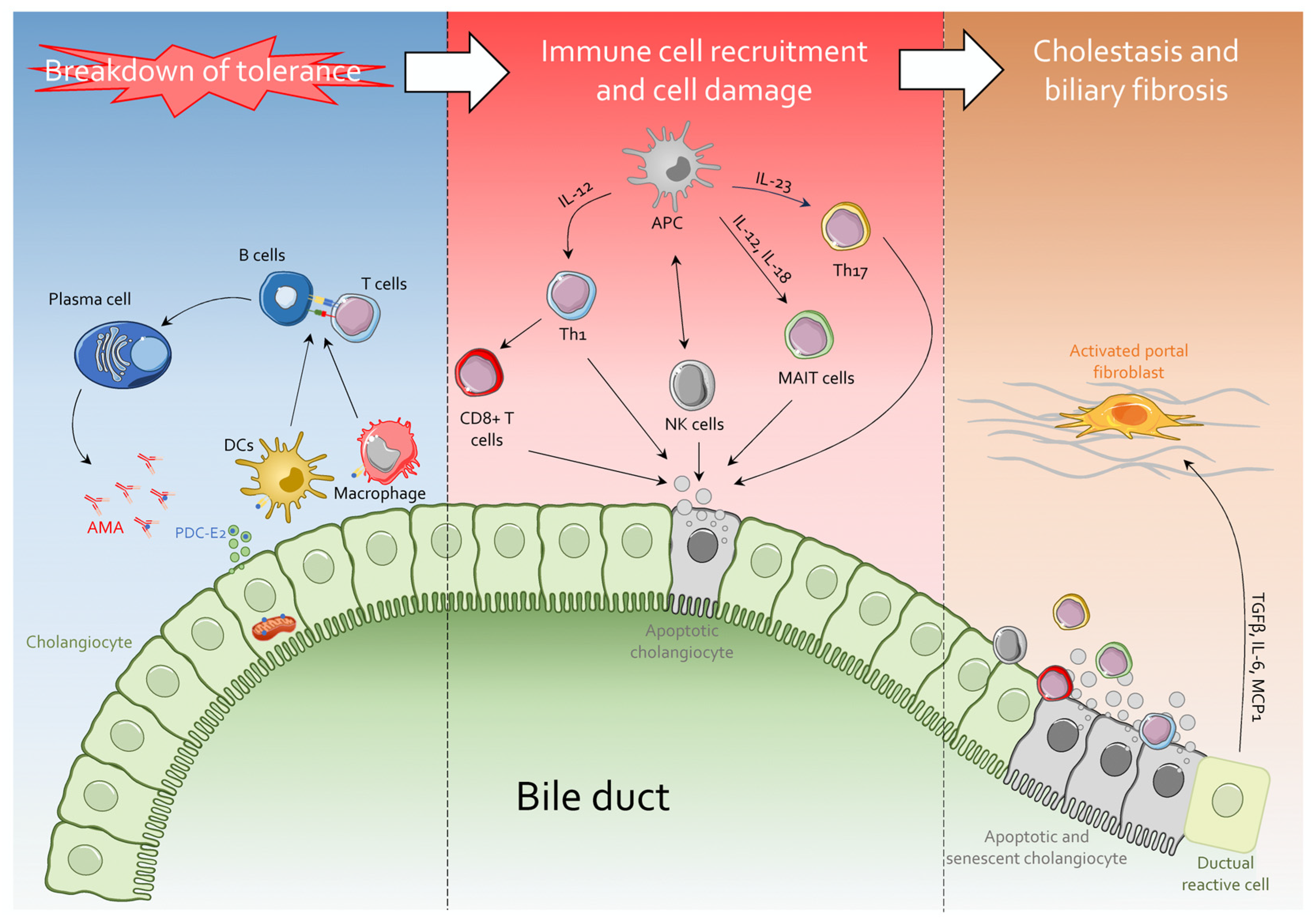
3. The Immunology of PBC
4. Immunobiology of Cholangiocytes in PBC
Apoptosis and Senescence of Cholangiocytes
5. Bile Acid-Regulated Receptors in PBC: From Pathogenesis to Therapy
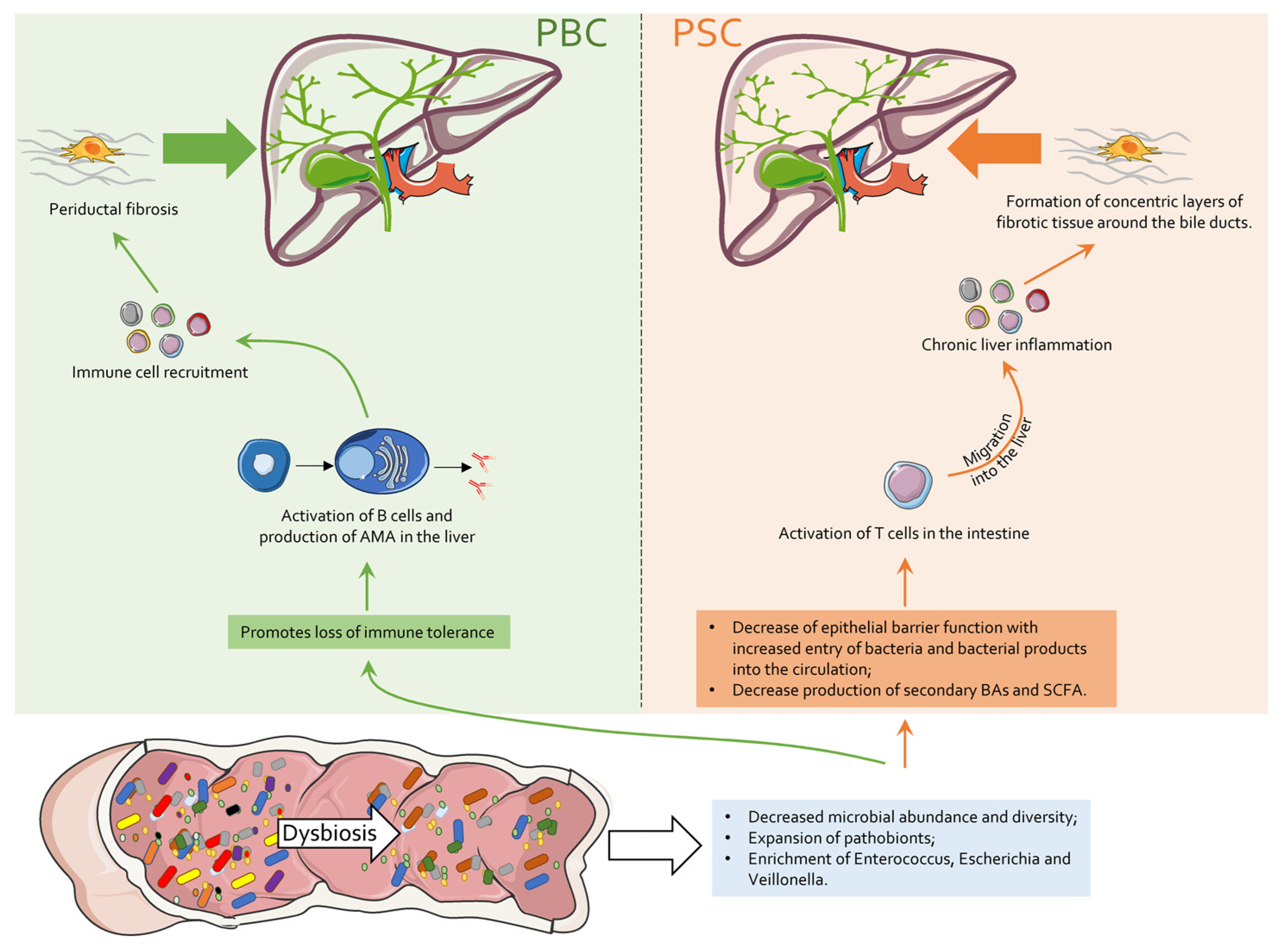
6. Intestinal Microbiota in PBC
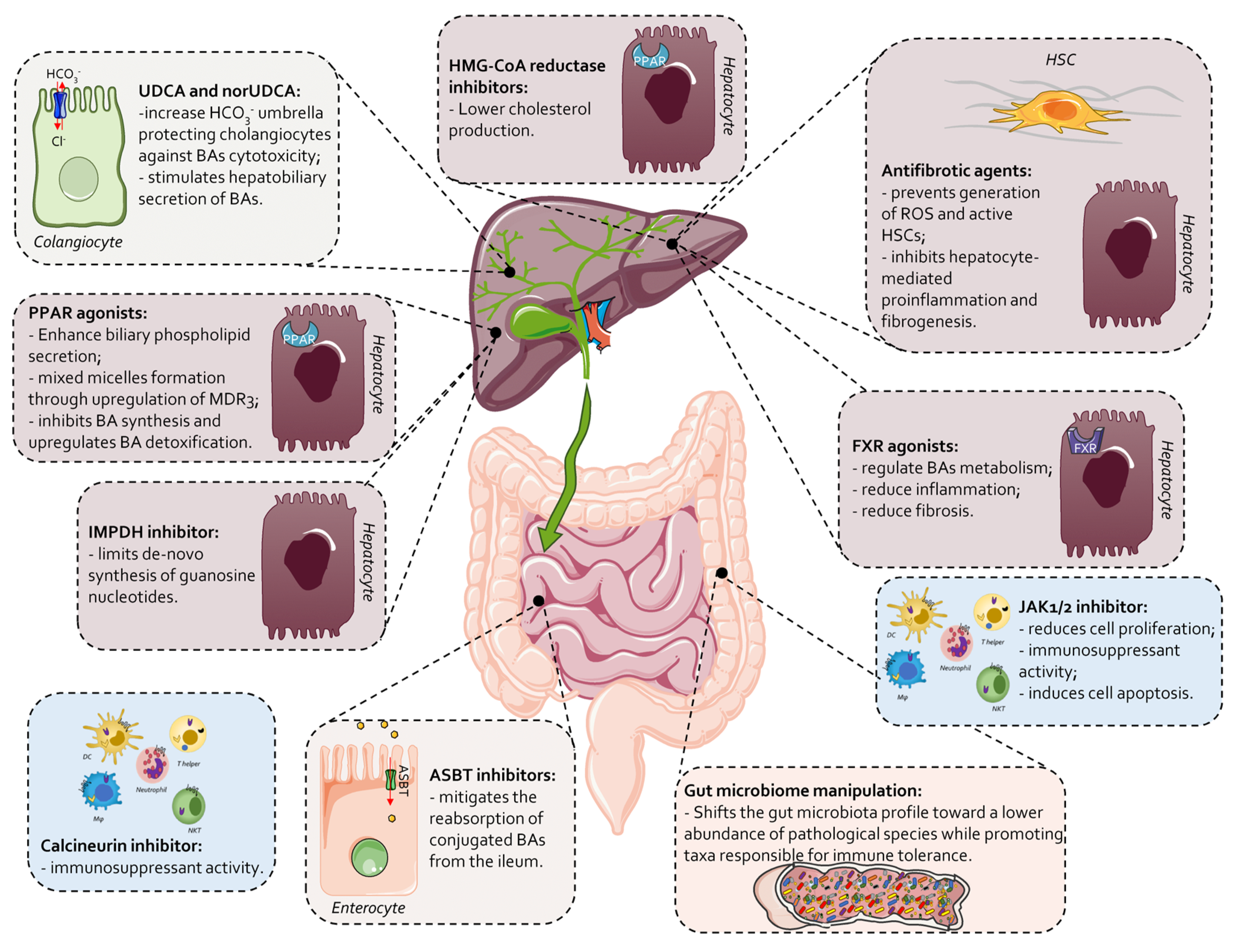
7. Current Therapeutic Landscape in PBC
7.1. UDCA
7.2. Second-Line Therapies and Novel Approaches in PBC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Clinical Trial | Treatments | End Points | Results | Ref. |
|---|---|---|---|---|---|
| Elafibrinor PPARα/δ | Phase 3 | 161 PBC adults, who had incomplete response to UDCA. Treatments: Group 1: elafibrinor 80 mg; Group 2: placebo. | Reduction in ALP levels (ALP ≥ 1.67-fold the upper limit of normal (ULN) at 52 weeks. | A biochemical response (the primary end point) was observed in 51% of the patients (55 of 108) who received elafibranor and in 4% (2 of 53) who received placebo, for a difference of 47 percentage points (95% confidence interval [CI], 32 to 57; p < 0.001). | [2] |
| Obeticolic acid (OCA) | Phase 2 | Patients with PBC from POISE cohort and external control patients from Global PBC cohort and UK-PBC cohort. Treatments: Group 1: OCA (POISE cohort n = 209); Group 2: non-OCA-treated external control (Global PBC cohort n = 1381 and UK-PBC cohort n = 2135). | Evaluate time to first occurrence of liver transplantation or death in patients with OCA vs. comparable non-OCA-treated external controls. | During the 6-year follow-up, there were 5 deaths or liver transplantations in Group 1 (2.4%), 135 in the Global PBC cohort control (10.0%) and 281 in the UK-PBC control (13.2%). | [128] |
| Phase 2 | 59 PBC patients, intolerant to UDCA. Treatments: Group 1: placebo (n = 23) Group 2: OCA 10 mg (n = 20). Group 3: OCA 50 mg (n = 16) | The percent change in ALP from baseline to the end of the double-blind phase of the study. | ALP levels were reduced in both OCA groups, respectively, by −53.9% in 10 mg group and by −37.2% in 50 mg group compared with placebo −0.8% (p < 0.05). Similar reductions were observed through 6 years of open-label extension treatment. Side effects: pruritus increased dose-dependently with OCA treatment. 15% (OCA 10 mg) and 38% (OCA 50 mg) discontinued due to pruritus. | [129] | |
| Linerixibat IBAT inhibitor | Phase 2b | 147 adult PBC patients with moderate to severe pruritus, numerical rating scale (NRS) ≥ 3 after 4 week of placebo treatment. Treatments: Group 1: placebo (n = 36); Group 2: linerixibat at 20 mg/d (n = 16); 90 mg/d (n = 23) 180 mg/d (n = 27); 40 mg/b.d. (n = 23); 90 mg/b.d. (n = 22), for 12 weeks (from week 4 to week 16), followed by single-blind placebo (to week 20). | Investigate dose-related changes in Mean Worst Daily Itch (MWDI) score at week 16. | At week 16, MWDI analysis showed significant differences between placebo and and linerixibat 180 mg/d (p < 0.05), 40 mg/b.d. (p < 0.05) and 90 mg/b.d. (p < 0.05). Diarrhea was the most frequent adverse event, and incidence increased with dose. | [179] |
| Seladelpar PPARδ | Phase 3 | 193 PBC patients with an inadequate response or intolerance to UDCA were enrolled. Treatments: Group 1: sedalpar 10 mg/d (n = 89); Group 3: placebo for 12 months | Primary composite biochemical response (ALP < 1.67 × ULN and total bilirubin ≤ ULN) at month 12. | Primary 61.7 % improvement in Group 1 and 41.7% in the placebo p < 0.05. | [177] |
| Saroglitazar PPARα/γ | Phase 2 | Phase 2 | 37 PBC patients with UDCA resistance or intolerance. Treatments: Group 1: saroglitazar 4 mg/d (n = 13); Group 2: saroglitazar 2 mg/d (n = 14): Group 3: placebo (n = 10). | At week 16, patients from Group 1 showed a reduction of ALP levels by −163.3 U/L and Group 2 by −155.8 U/L compared to placebo (−21.1 U/L) (p < 0.05). Study drug was discontinued in 4 patients (3 patients in Group 1 and 1 patient in the Group 2) due to ALP increases. | [180] |
| Fenofibrate PPAR | Phase 3 | 117 PBC treatment-naive patients. Treatments: Group 1: UDCA; Group 2: UDCA plus fenofibrate 200 mg/d | Biochemical response percentage, according to the Barcelona criterion at 12 months. | In Group 2, 81.4% of patients achieved the primary outcome and 64.3% in Group 1 achieved the primary outcome (p < 0.05). There was no difference between the 2 groups in liver fibrosis and biochemical markers. | [181] |
| Rituximab Anti-CD20 | Phase 3 | 57 aged 18-years-old or older patients with PBC and moderate to severe fatigue. Treatments: Group 1: rituximab 1000 mg/b.d.; Group 2: placebo | Primary Measurement of fatigue severity using the PBC-40 fatigue domain at 3 months. | Primary Improvement in fatigue score was seen in both groups No adverse events were registered. | [182] |
| OP-724 CREB-binding protein/β-catenin inhibitor | Phase 1 | 7 PBC patients median aged 68 years. Treatments: Group 1: OP-724 280 mg/m2/4 h/tw Group 2: OP-724 280 mg/m2/4 h/tw Only five of these completed twelve cycles of treatment. Consequently, the recommended dosage was determined to be 280 mg/m2/4 h. | Primary Assessment of the incidence of serious adverse events (SAEs). Secondary Measurement of the improvement in the modified Histological Activity Index (mHAI) score. | Primary SAEs did not occur. Secondary The most common AEs were abdominal discomfort (29%) and abdominal hepatic function (43%). Histological improvements in the fibrosis stage (2/5 40%) and mHAI score (3/5 60%). | [183] |
| Setanaxib NADP oxidase 1/4 inhibitor | Phase 2 | 111 patients with ≥6 months of UDCA treatment. Treatments: Group 1: oral setanaxib 400 mg/d (n = 38) Group 2 oral setanaxib 400 mg/b.d. (n = 36) Group 3: placebo (n = 37). | Primary Assessment of percentage change from baseline in GGT at Week 24. | Primary 104/111 patients completed Week 24. The primary end point was not met: change in GGT to Week 24 was −4.9% for Group 1 patients, −19.0% for Group 2 and −8.4% for placebo. | [184] |
| Ursodeoxycholic acid (UDCA) | 73 PBC patients with poor response or who did not respond completely to a standard dose of UDCA. Treatments: Group 1: standard dosage of 13–15 mg/kg/d Group 2: higher dosage of 18–22 mg/kg/d. | Primary Evaluation of the rate of response at 6 months and drug side effects. Secondary Evaluation of the rate of response at 12 months and drug side effects. | Primary At 6 months, Group 2 patients achieved a response rate of 59.4% compared with 36.1% in the first group (p < 0.05) Secondary At 12 months, the Group 2 achieved a response rate of 59.4% compared with 47.2% in the Group 1 (p > 0.05). | [185] | |
| Budesonide/UDCA | Phase 3 | 62 PBC patients after at least 6 months of UDCA terapy and hepatic inflammatory activity as assessed by Ishak score, and ALP >1.5 × ULN. Treatments: Group 1: budesonide 9 mg/d plus UDCA 12–16 mg/kg/d Group 2: placebo plus UDCA 12–16 mg/kg/d. | Primary Assessment of an improvement in liver histology with respect to inflammation and no progression of fibrosis. Secondary Measurement of changes in biochemical markers of liver injury. | Primary Comparing patients with paired biopsies only (n = 43), the primary histologic endpoint was not met (p > 0.05). Secondary Group 1 patients had a reduction of mean ALP and 35% of them achieved normalization of ALP (placebo 9%) (p < 0.05). Serious adverse events occurred in 10 patients receiving budesonide and 7 patients receiving placebo. | [186] |
| A4250 IBAT inhibitor | Phase 2 | 9 patients with PBC, after a two-week whash out of bile acid sequestrant, treatment of cholestatic pruritus. Treatments: Group 1: A4250 0.75 mg (n = 4); Group 2: A2450 1.5 mg (n = 5). | After 4 weeks, evaluation of the effect of A4250 on pruritus, assessed by Visual Analogue Scale (VAS), 5D-itch scale and the pruritus module of the PBC40 questionnaire. | All 9 patients had an improvement in pruritus, until none or mild according to 5D-itch, VAS and PBC40 pruritus. Study was not completed due to abdominal pain (5/5) and diarrhoea (4/5). | [187] |
| Bezafibrate PPAR | Phase 2 | 74 cholestatic patients (24 PBC, 44 PSC, 2 SSC) with moderate to severe pruritus (≥5 of 10 on VAS). Treatments: Group 1: benzafibrate 400 mg/d Group 2: placebo. | Primary After 21 days, reduction of pruritus ≥ 50% in Group 1 patients. Secondary Evaluation of pruritus changes through VAS and 5D-Itch questionnaire. Evaluation of biochemical features changes. | 70/74 patients completed the trial Primary Group 1 patients had a reduction of 45% (41% PSC, 55% PBC) and Group 2 of 11% to ≥50% reduction of severe or moderate pruritus (p < 0.05). Secondary Group 1 exhibited a reduction of morning (p < 0.05 vs. placebo) and evening (p < 0.05) VAS and improved the validated 5D-Itch questionnaire (p < 0.05 vs. placebo) compared with Group 2 patients. | [188] |
| Rifampin/sertraline PXR/SSRIs | Phase | 36 patients with PSC and PBC. Treatments: Group 1: sertraline 100 mg/d (n = 18); Group 2: rifampin 300 mg/d (n = 18). | End points: pruritus severity, ALT, AST, ALP and total bilirubin at baseline and after 4 weeks of treatment. | No difference between sertraline and rifampin on pruritus improvement and total bilirubin. | [189] |
8. Ongoing Clinical Trial PBC 2018–2023
9. Conclusions
Funding
Conflicts of Interest
References
- Lv, T.; Chen, S.; Li, M.; Zhang, D.; Kong, Y.; Jia, J. Regional Variation and Temporal Trend of Primary Biliary Cholangitis Epidemiology: A Systematic Review and Meta-Analysis. J. Gastroenterol. Hepatol. 2021, 36, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Bowlus, C.L.; Levy, C.; Akarca, U.S.; Alvares-da-Silva, M.R.; Andreone, P.; Arrese, M.; Corpechot, C.; Francque, S.M.; Heneghan, M.A.; et al. Efficacy and Safety of Elafibranor in Primary Biliary Cholangitis. N. Engl. J. Med. 2024, 390, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Achufusi, T.G.O.; Safadi, A.O.; Mahabadi, N. Ursodeoxycholic Acid; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Ishizaki, K.; Imada, T.; Tsurufuji, M. Hepatoprotective Bile Acid “ursodeoxycholic Acid (UDCA)” Property and Difference as Bile Acids. Hepatol. Res. 2005, 33, 174–177. [Google Scholar] [CrossRef]
- Zukowski, T.H.; Jorgensen, R.A.; Dickson, E.R.; Lindor, K.D. Autoimmune Conditions Associated with Primary Biliary Cirrhosis: Response to Ursodeoxycholic Acid Therapy. Am. J. Gastroenterol. 1998, 93, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Jepsen, P.; Morenghi, E.; Carbone, M.; Moroni, L.; Battezzati, P.M.; Podda, M.; Mackay, I.R.; Gershwin, M.E.; Invernizzi, P. Evolving Trends in Female to Male Incidence and Male Mortality of Primary Biliary Cholangitis. Sci. Rep. 2016, 6, 25906. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, S.; Maillette de Buy Wenniger, L.; Paulusma, C.C.; van Vliet, S.J.; Jefferson, D.M.; Oude Elferink, R.P.; Beuers, U. A Biliary HCO3− Umbrella Constitutes a Protective Mechanism against Bile Acid-Induced Injury in Human Cholangiocytes. Hepatology 2012, 55, 173–183. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Bowlus, C.L.; Yimam, K.K.; Razavi, H.; Estes, C. Epidemiology, Natural History, and Outcomes of Primary Sclerosing Cholangitis: A Systematic Review of Population-Based Studies. Clin. Gastroenterol. Hepatol. 2022, 20, 1687–1700.e4. [Google Scholar] [CrossRef]
- Harms, M.H.; van Buuren, H.R.; Corpechot, C.; Thorburn, D.; Janssen, H.L.A.; Lindor, K.D.; Hirschfield, G.M.; Parés, A.; Floreani, A.; Mayo, M.J.; et al. Ursodeoxycholic Acid Therapy and Liver Transplant-Free Survival in Patients with Primary Biliary Cholangitis. J. Hepatol. 2019, 71, 357–365. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef]
- Tanaka, A.; Leung, P.S.; Gershwin, M.E. Environmental Basis of Primary Biliary Cholangitis. Exp. Biol. Med. 2018, 243, 184–189. [Google Scholar] [CrossRef]
- Joshita, S.; Umemura, T.; Tanaka, E.; Ota, M. Genetics and Epigenetics in the Pathogenesis of Primary Biliary Cholangitis. Clin. J. Gastroenterol. 2018, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.G.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; et al. Primary Biliary Cirrhosis in Monozygotic and Dizygotic Twins: Genetics, Epigenetics, and Environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, P.; Selmi, C.; MacKay, I.R.; Podda, M.; Gershwin, M.E. From Bases to Basis: Linking Genetics to Causation in Primary Biliary Cirrhosis. Clin. Gastroenterol. Hepatol. 2005, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Bremm, A.; Schneider, P.M.; Notghi, A.; Gerken, G.; Prager-Eberle, M.; Stradmann-Bellinghausen, B.; Zum Büschenfelde, K.H.M.; Rittner, C. HLA DRw8 and Complement C4 Deficiency as Risk Factors in Primary Biliary Cirrhosis. Gastroenterology 1991, 101, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Begovich, A.B.; Klitz, W.; Moonsamy, P.V.; Van de Water, J.; Peltz, G.; Gershwin, M.E. Genes within the HLA Class II Region Confer Both Predisposition and Resistance to Primary Biliary Cirrhosis. Tissue Antigens 1994, 43, 71–77. [Google Scholar] [CrossRef]
- Agarwal, K.; Jones, D.E.J.; Daly, A.K.; James, O.F.W.; Vaidya, B.; Pearce, S.; Bassendine, M.F. CTLA-4 Gene Polymorphism Confers Susceptibility to Primary Biliary Cirrhosis. J. Hepatol. 2000, 32, 538–541. [Google Scholar] [CrossRef]
- Donaldson, P.; Agarwal, K.; Craggs, A.; Craig, W.; James, O.; Jones, D. HLA and Interleukin 1 Gene Polymorphisms in Primary Biliary Cirrhosis: Associations with Disease Progression and Disease Susceptibility. Gut 2001, 48, 397–402. [Google Scholar] [CrossRef]
- Matsushita, M.; Tanaka, A.; Kikuchi, K.; Kitazawa, E.; Kawaguchi, N.; Kawashima, Y.; Kato, T.; Fujikawa, H.; Quaranta, S.; Rosina, F. Association of Single Nucleotide Polymorphisms of the Interleukin-10 Promoter Gene and Susceptibility to Primary Biliary Cirrhosis: Immunogenetic Differences in Italian and Japanese Patients. Autoimmunity 2002, 35, 531–536. [Google Scholar] [CrossRef]
- Mulinacci, G.; Palermo, A.; Gerussi, A.; Asselta, R.; Gershwin, M.E.; Invernizzi, P. New Insights on the Role of Human Leukocyte Antigen Complex in Primary Biliary Cholangitis. Front. Immunol. 2022, 13, 975115. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Lu, Y.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; et al. Primary Biliary Cirrhosis Associated with HLA, IL12A, and IL12RB2 Variants. N. Engl. J. Med. 2009, 360, 2544–2555. [Google Scholar] [CrossRef]
- Nakamura, M.; Nishida, N.; Kawashima, M.; Aiba, Y.; Tanaka, A.; Yasunami, M.; Nakamura, H.; Komori, A.; Nakamuta, M.; Zeniya, M.; et al. Genome-Wide Association Study Identifies TNFSF15 and POU2AF1 as Susceptibility Loci for Primary Biliary Cirrhosis in the Japanese Population. Am. J. Hum. Genet. 2012, 91, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Leung, P.S.C.; Gershwin, M.E. Evolution of Our Understanding of PBC. Best Pract. Res. Clin. Gastroenterol. 2018, 34–35, 3–9. [Google Scholar] [CrossRef]
- Smyk, D.S.; Bogdanos, D.P.; Kriese, S.; Billinis, C.; Burroughs, A.K.; Rigopoulou, E.I. Urinary Tract Infection as a Risk Factor for Autoimmune Liver Disease: From Bench to Bedside. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Shamriz, O.; Nussinovitch, U.; Rose, N.R. Chapter 1—Pathophysiology of Autoimmunity and Immune-Mediated Mechanisms in Cardiovascular Diseases. In The Heart in Rheumatic, Autoimmune and Inflammatory Diseases; Academic Press: Cambridge, MA, USA, 2017; pp. 3–23. ISBN 978-0-12-803267-1. [Google Scholar]
- Mao, T.K.; Davis, P.A.; Odin, J.A.; Coppel, R.L.; Gershwin, M.E. Sidechain Biology and the Immunogenicity of PDC-E2, the Major Autoantigen of Primary Biliary Cirrhosis. Hepatology 2004, 40, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.-P.; Baum, H.; Grasso, A.; Okamoto, M.; Butler, P.; Ma, Y.; Rigopoulou, E.; Montalto, P.; Davies, E.T.; Burroughs, A.K.; et al. Microbial Mimics Are Major Targets of Crossreactivity with Human Pyruvate Dehydrogenase in Primary Biliary Cirrhosis. J. Hepatol. 2004, 40, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.M. Novosphingobium Aromaticivorans: A Potential Initiator of Primary Biliary Cirrhosis. Off. J. Am. Coll. Gastroenterol.|ACG 2004, 99, 2147–2149. [Google Scholar] [CrossRef]
- Selmi, C.; Balkwill, D.L.; Invernizzi, P.; Ansari, A.A.; Coppel, R.L.; Podda, M.; Leung, P.S.; Kenny, T.P.; Van De Water, J.; Nantz, M.H.; et al. Patients with Primary Biliary Cirrhosis React against a Ubiquitous Xenobiotic-Metabolizing Bacterium. Hepatology 2003, 38, 1250–1257. [Google Scholar] [CrossRef]
- Gershwin, M.E.; Selmi, C.; Worman, H.J.; Gold, E.B.; Watnik, M.; Utts, J.; Lindor, K.D.; Kaplan, M.M.; Vierling, J.M.; USA PBC Epidemiology Group. Risk Factors and Comorbidities in Primary Biliary Cirrhosis: A Controlled Interview-Based Study of 1032 Patients. Hepatology 2005, 42, 1194–1202. [Google Scholar] [CrossRef]
- Leung, P.S.; Coppel, R.L.; Ansari, A.; Munoz, S.; Gershwin, M.E. Antimitochondrial Antibodies in Primary Biliary Cirrhosis. Semin. Liver Dis. 1997, 17, 61–69. [Google Scholar] [CrossRef]
- Palmer, J.M.; Yeaman, S.J.; Jones, D.E.J. Epitope Specificity of Anti-PDC E1 Alpha Antibodies in Primary Biliary Cirrhosis (PBC). J. Hepatol. 2001, 34, 214. [Google Scholar] [CrossRef]
- Palmer, J.M.; Doshi, M.; Kirby, J.A.; Yeaman, S.J.; Bassendine, M.F.; Jones, D.E. Secretory Autoantibodies in Primary Biliary Cirrhosis (PBC). Clin. Exp. Immunol. 2000, 122, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Courvalin, J.C. Antinuclear Antibodies Specific for Primary Biliary Cirrhosis. Autoimmun. Rev. 2003, 2, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, P.; Podda, M.; Battezzati, P.M.; Crosignani, A.; Zuin, M.; Hitchman, E.; Maggioni, M.; Meroni, P.L.; Penner, E.; Wesierska-Gadek, J. Autoantibodies against Nuclear Pore Complexes Are Associated with More Active and Severe Liver Disease in Primary Biliary Cirrhosis. J. Hepatol. 2001, 34, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Tsuneyama, K.; Baba, H.; Morimoto, Y.; Tsunematsu, T.; Ogawa, H. Primary Biliary Cholangitis: Its Pathological Characteristics and Immunopathological Mechanisms. J. Med. Investig. 2017, 64, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Van de Water, J.; Ansari, A.; Prindiville, T.; Coppel, R.L.; Ricalton, N.; Kotzin, B.L.; Liu, S.; Roche, T.E.; Krams, S.M.; Munoz, S.; et al. Heterogeneity of Autoreactive T Cell Clones Specific for the E2 Component of the Pyruvate Dehydrogenase Complex in Primary Biliary Cirrhosis. J. Exp. Med. 1995, 181, 723–733. [Google Scholar] [CrossRef]
- Bernuzzi, F.; Fenoglio, D.; Battaglia, F.; Fravega, M.; Gershwin, M.E.; Indiveri, F.; Ansari, A.A.; Podda, M.; Invernizzi, P.; Filaci, G. Phenotypical and Functional Alterations of CD8 Regulatory T Cells in Primary Biliary Cirrhosis. J. Autoimmun. 2010, 35, 176–180. [Google Scholar] [CrossRef]
- Harada, K.; Nakanuma, Y. Molecular Mechanisms of Cholangiopathy in Primary Biliary Cirrhosis. Med. Mol. Morphol. 2006, 39, 55–61. [Google Scholar] [CrossRef]
- Harada, K.; Shimoda, S.; Sato, Y.; Isse, K.; Ikeda, H.; Nakanuma, Y. Periductal Interleukin-17 Production in Association with Biliary Innate Immunity Contributes to the Pathogenesis of Cholangiopathy in Primary Biliary Cirrhosis. Clin. Exp. Immunol. 2009, 157, 261–270. [Google Scholar] [CrossRef]
- Lin, C.-I.; Wang, Y.-W.; Liu, C.-Y.; Chen, H.-W.; Liang, P.-H.; Chuang, Y.-H. Regulatory T Cells in Inflamed Liver Are Dysfunctional in Murine Primary Biliary Cholangitis. Clin. Exp. Immunol. 2024, 215, 225–239. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Masyuk, A.I.; Masyuk, T.V.; O’Hara, S.P.; LaRusso, N.F. Physiology of Cholangiocytes. Compr. Physiol. 2013, 3, 541–565. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fabris, L. Functional Anatomy of Normal Bile Ducts. Anat. Rec. 2008, 291, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Masyuk, T.V.; LaRusso, N.F. Chapter 44—Physiology of Cholngiocytes. In Physiology of the Gastrointestinal Tract, 6th ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 1003–1023. ISBN 978-0-12-809954-4. [Google Scholar]
- Chen, X.-M.; O’Hara, S.P.; Nelson, J.B.; Splinter, P.L.; Small, A.J.; Tietz, P.S.; Limper, A.H.; LaRusso, N.F. Multiple TLRs Are Expressed in Human Cholangiocytes and Mediate Host Epithelial Defense Responses to Cryptosporidium Parvum via Activation of NF-KappaB. J. Immunol. 2005, 175, 7447–7456. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Haibo, B.; Ray, D.; Zhou, T.; Wan, Y.; Meng, F.; Marzioni, M.; Alpini, G. Functional and Structural Features of Cholangiocytes in Health and Disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Ronca, V.; Mancuso, C.; Milani, C.; Carbone, M.; Oo, Y.H.; Invernizzi, P. Immune System and Cholangiocytes: A Puzzling Affair in Primary Biliary Cholangitis. J. Leukoc. Biol. 2020, 108, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Milani, S.; Herbst, H.; Schuppan, D.; Stein, H.; Surrenti, C. Transforming Growth Factors Beta 1 and Beta 2 Are Differentially Expressed in Fibrotic Liver Disease. Am. J. Pathol. 1991, 139, 1221. [Google Scholar]
- Kruglov, E.A.; Nathanson, R.A.; Nguyen, T.; Dranoff, J.A. Secretion of MCP-1/CCL2 by Bile Duct Epithelia Induces Myofibroblastic Transdifferentiation of Portal Fibroblasts. Am. J. Physiol. Liver Physiol. 2006, 290, G765–G771. [Google Scholar] [CrossRef]
- Pinto, C.; Giordano, D.M.; Maroni, L.; Marzioni, M. Role of Inflammation and Proinflammatory Cytokines in Cholangiocyte Pathophysiology. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 1270–1278. [Google Scholar] [CrossRef]
- Syal, G.; Fausther, M.; Dranoff, J.A. Advances in Cholangiocyte Immunobiology. Am. J. Physiol. Liver Physiol. 2012, 303, G1077–G1086. [Google Scholar] [CrossRef]
- Matsumoto, K.; Fujii, H.; Michalopoulos, G.; Fung, J.J.; Demetris, A.J. Human Biliary Epithelial Cells Secrete and Respond to Cytokines and Hepatocyte Growth Factors in Vitro: Interleukin-6, Hepatocyte Growth Factor and Epidermal Growth Factor Promote DNA Synthesis in Vitro. Hepatology 1994, 20, 376–382. [Google Scholar] [CrossRef]
- Wu, C.-T.; Davis, P.A.; Luketic, V.A.; Gershwin, M.E. A Review of the Physiological and Immunological Functions of Biliary Epithelial Cells: Targets for Primary Biliary Cirrhosis, Primary Sclerosing Cholangitis and Drug-Induced Ductopenias. Clin. Dev. Immunol. 2004, 11, 403720. [Google Scholar] [CrossRef]
- Ayres, R.C.; Neuberger, J.M.; Shaw, J.; Joplin, R.; Adams, D.H. Intercellular Adhesion Molecule-1 and MHC Antigens on Human Intrahepatic Bile Duct Cells: Effect of pro-Inflammatory Cytokines. Gut 1993, 34, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Bour, E.S.; Ward, L.K.; Cornman, G.A.; Isom, H.C. Tumor Necrosis Factor-α-Induced Apoptosis in Hepatocytes in Long-Term Culture. Am. J. Pathol. 1996, 148, 485–495. [Google Scholar] [PubMed]
- Harada, K.; Ohba, K.; Ozaki, S.; Isse, K.; Hirayama, T.; Wada, A.; Nakanuma, Y. Peptide Antibiotic Human Beta-Defensin-1 and -2 Contribute to Antimicrobial Defense of the Intrahepatic Biliary Tree. Hepatology 2004, 40, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ohira, S.; Isse, K.; Ozaki, S.; Zen, Y.; Sato, Y.; Nakanuma, Y. Lipopolysaccharide Activates Nuclear Factor-KappaB through Toll-like Receptors and Related Molecules in Cultured Biliary Epithelial Cells. Lab. Investig. 2003, 83, 1657–1667. [Google Scholar] [CrossRef]
- Hazlett, L.; Wu, M. Defensins in Innate Immunity. Cell Tissue Res. 2011, 343, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dranoff, J.A.; Chan, E.P.; Uemura, M.; Sévigny, J.; Wells, R.G. Transforming Growth Factor-β and Substrate Stiffness Regulate Portal Fibroblast Activation in Culture. Hepatology 2007, 46, 1246–1256. [Google Scholar] [CrossRef]
- Cichon, M.A.; Radisky, D.C. Extracellular Matrix as a Contextual Determinant of Transforming Growth Factor-β Signaling in Epithelial-Mesenchymal Transition and in Cancer. Cell Adh. Migr. 2014, 8, 588–594. [Google Scholar] [CrossRef]
- Banales, J.M.; Sáez, E.; Uriz, M.; Sarvide, S.; Urribarri, A.D.; Splinter, P.; Tietz Bogert, P.S.; Bujanda, L.; Prieto, J.; Medina, J.F.; et al. Up-Regulation of MicroRNA 506 Leads to Decreased Cl-/HCO3- Anion Exchanger 2 Expression in Biliary Epithelium of Patients with Primary Biliary Cirrhosis. Hepatology 2012, 56, 687–697. [Google Scholar] [CrossRef]
- Kita, H.; Matsumura, S.; He, X.-S.; Ansari, A.A.; Lian, Z.-X.; Van de Water, J.; Coppel, R.L.; Kaplan, M.M.; Gershwin, M.E. Quantitative and Functional Analysis of PDC-E2-Specific Autoreactive Cytotoxic T Lymphocytes in Primary Biliary Cirrhosis. J. Clin. Investig. 2002, 109, 1231–1240. [Google Scholar] [CrossRef]
- Salas-Silva, S.; Simoni-Nieves, A.; Chávez-Rodríguez, L.; Gutiérrez-Ruiz, M.C.; Bucio, L.; Quiroz, L.E.G. Mechanism of Cholangiocellular Damage and Repair during Cholestasis. Ann. Hepatol. 2021, 26, 100530. [Google Scholar] [CrossRef]
- Fejfar, T.; Vaňásek, T.; Hůlek, P. Chronic Cholestatic Liver Diseases—Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. Vnitr. Lek. 2020, 66, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wei, Y.; Xiong, A.; Li, Y.; Guan, H.; Wang, Q.; Miao, Q.; Bian, Z.; Xiao, X.; Lian, M.; et al. Comprehensive Analysis of Serum and Fecal Bile Acid Profiles and Interaction with Gut Microbiota in Primary Biliary Cholangitis. Clin. Rev. Allergy Immunol. 2020, 58, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Marchianò, S.; Urbani, G.; Di Giorgio, C.; Distrutti, E.; Zampella, A.; Biagioli, M. Immunology of Bile Acids Regulated Receptors. Prog. Lipid Res. 2024, 95, 101291. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Di Giorgio, C.; Massa, C.; Marchianò, S.; Bellini, R.; Bordoni, M.; Urbani, G.; Roselli, R.; Lachi, G.; Morretta, E.; et al. A Microbial Derived Bile Acid Acts as GPBAR1 Agonist and RORγt Inverse Agonist and Reverses Inflammation in Inflammatory Bowel Disease. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Strazzabosco, M.; Larusso, N.F. The Cholangiopathies: Disorders of Biliary Epithelia. Gastroenterology 2004, 127, 1565–1577. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. The Cholangiopathies. Mayo Clin. Proc. 2015, 90, 791–800. [Google Scholar] [CrossRef]
- Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Interplay of Autophagy, Apoptosis, and Senescence in Primary Biliary Cholangitis. Explor. Dig. Dis. 2023, 2, 223–245. [Google Scholar] [CrossRef]
- Obeng, E. Apoptosis (Programmed Cell Death) and Its Signals—A Review. Braz. J. Biol. 2021, 81, 1133–1143. [Google Scholar] [CrossRef]
- Xu, G.; Shi, Y. Apoptosis Signaling Pathways and Lymphocyte Homeostasis. Cell Res. 2007, 17, 759–771. [Google Scholar] [CrossRef]
- Bhosale, P.B.; Ha, S.E.; Vetrivel, P.; Kim, H.H.; Kim, J.-A.; Park, K.-I.; Kim, S.M.; Kim, G.S. Flavonoid-Induced Apoptotic Cell Death in Human Cancer Cells and Its Mechanisms. J. Biomed. Transl. Res. 2020, 21, 50–58. [Google Scholar] [CrossRef]
- Lemke, G. How Macrophages Deal with Death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ozaki, S.; Gershwin, M.E.; Nakanuma, Y. Enhanced Apoptosis Relates to Bile Duct Loss in Primary Biliary Cirrhosis. Hepatology 1997, 26, 1399–1405. [Google Scholar] [CrossRef]
- Koga, H.; Sakisaka, S.; Ohishi, M.; Sata, M.; Tanikawa, K. Nuclear DNA Fragmentation and Expression of Bcl-2 in Primary Biliary Cirrhosis. Hepatology 1997, 25, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, P.; Mizuochi, T.; Mourya, R.; Gutta, S.; Yang, L.; Luo, Z.; Bezerra, J.A. Preferential TNFα Signaling via TNFR2 Regulates Epithelial Injury and Duct Obstruction in Experimental Biliary Atresia. JCI Insight 2017, 2, e88747. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Peng, J.; Liao, L.; Su, H.; Zhao, G.; Zoubek, M.E.; Macías-Rodríguez, R.; Ruiz-Margain, A.; Reißing, J.; Zimmermann, H.W.; et al. Inactivation of Caspase 8 in Liver Parenchymal Cells Confers Protection against Murine Obstructive Cholestasis. J. Hepatol. 2018, 69, 1326–1334. [Google Scholar] [CrossRef]
- Manousou, P.; Kolios, G.; Drygiannakis, I.; Koulentaki, M.; Pyrovolaki, K.; Voumvouraki, A.; Notas, G.; Bourikas, L.; Papadaki, H.A.; Kouroumalis, E. CXCR3 Axis in Patients with Primary Biliary Cirrhosis: A Possible Novel Mechanism of the Effect of Ursodeoxycholic Acid. Clin. Exp. Immunol. 2013, 172, 9–15. [Google Scholar] [CrossRef]
- Schoemaker, M.H.; Conde de la Rosa, L.; Buist-Homan, M.; Vrenken, T.E.; Havinga, R.; Poelstra, K.; Haisma, H.J.; Jansen, P.L.M.; Moshage, H. Tauroursodeoxycholic Acid Protects Rat Hepatocytes from Bile Acid-Induced Apoptosis via Activation of Survival Pathways. Hepatology 2004, 39, 1563–1573. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L.; Chu, H. Targeting Gut Microbiota for the Treatment of Primary Biliary Cholangitis: From Bench to Bedside. J. Clin. Transl. Hepatol. 2023, 11, 958–966. [Google Scholar] [CrossRef]
- Wang, Y.-W.; Lin, C.-I.; Chen, H.-W.; Wu, J.-C.; Chuang, Y.-H. Apoptotic Biliary Epithelial Cells and Gut Dysbiosis in the Induction of Murine Primary Biliary Cholangitis. J. Transl. Autoimmun. 2023, 6, 100182. [Google Scholar] [CrossRef]
- Amaral, J.D.; Castro, R.E.; Steer, C.J.; Rodrigues, C.M.P. P53 and the Regulation of Hepatocyte Apoptosis: Implications for Disease Pathogenesis. Trends Mol. Med. 2009, 15, 531–541. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Guicciardi, M.E.; Splinter, N.P.; Al Suraih, M.S.; Nasser-Ghodsi, N.; Stollenwerk, D.; Gores, G.J.; LaRusso, N.F. The Transcription Factor ETS1 Promotes Apoptosis Resistance of Senescent Cholangiocytes by Epigenetically Up-Regulating the Apoptosis Suppressor BCL2L1. J. Biol. Chem. 2019, 294, 18698–18713. [Google Scholar] [CrossRef] [PubMed]
- Meadows, V.; Baiocchi, L.; Kundu, D.; Sato, K.; Fuentes, Y.; Wu, C.; Chakraborty, S.; Glaser, S.; Alpini, G.; Kennedy, L.; et al. Biliary Epithelial Senescence in Liver Disease: There Will Be SASP. Front. Mol. Biosci. 2021, 8, 803098. [Google Scholar] [CrossRef] [PubMed]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Sasaki, M.; Yoshimura-Miyakoshi, M.; Sato, Y.; Nakanuma, Y. A Possible Involvement of Endoplasmic Reticulum Stress in Biliary Epithelial Autophagy and Senescence in Primary Biliary Cirrhosis. J. Gastroenterol. 2015, 50, 984–995. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Yamaguchi, J.; Nakada, S.; Nakanuma, Y. Telomere Shortening in the Damaged Small Bile Ducts in Primary Biliary Cirrhosis Reflects Ongoing Cellular Senescence. Hepatology 2008, 48, 186–195. [Google Scholar] [CrossRef]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. Increased P16(INK4a)-Expressing Senescent Bile Ductular Cells Are Associated with Inadequate Response to Ursodeoxycholic Acid in Primary Biliary Cholangitis. J. Autoimmun. 2020, 107, 102377. [Google Scholar] [CrossRef]
- Carino, A.; Biagioli, M.; Marchianò, S.; Fiorucci, C.; Zampella, A.; Monti, M.C.C.; Scarpelli, P.; Ricci, P.; Distrutti, E.; Fiorucci, S. Ursodeoxycholic Acid Is a GPBAR1 Agonist and Resets Liver/Intestinal FXR Signaling in a Model of Diet-Induced Dysbiosis and NASH. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1422–1437. [Google Scholar] [CrossRef]
- Marchianò, S.; Biagioli, M.; Morretta, E.; Di Giorgio, C.; Roselli, R.; Bordoni, M.; Bellini, R.; Urbani, G.; Massa, C.; Monti, M.C.; et al. Combinatorial Therapy with BAR502 and UDCA Resets FXR and GPBAR1 Signaling and Reverses Liver Histopathology in a Model of NASH. Sci. Rep. 2023, 13, 1602. [Google Scholar] [CrossRef]
- Maillette de Buy Wenniger, L.J.; Hohenester, S.; Maroni, L.; van Vliet, S.J.; Oude Elferink, R.P.; Beuers, U. The Cholangiocyte Glycocalyx Stabilizes the “Biliary HCO3 Umbrella”: An Integrated Line of Defense against Toxic Bile Acids. Dig. Dis. 2015, 33, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Merlen, G.; Kahale, N.; Ursic-Bedoya, J.; Bidault-Jourdainne, V.; Simerabet, H.; Doignon, I.; Tanfin, Z.; Garcin, I.; Péan, N.; Gautherot, J.; et al. TGR5-Dependent Hepatoprotection through the Regulation of Biliary Epithelium Barrier Function. Gut 2020, 69, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Renga, B.; Bucci, M.; Cipriani, S.; Carino, A.; Monti, M.C.C.; Zampella, A.; Gargiulo, A.; d’Emmanuele di Villa Bianca, R.; Distrutti, E.; Fiorucci, S.; et al. Cystathionine γ-Lyase, a H2S-Generating Enzyme, Is a GPBAR1-Regulated Gene and Contributes to Vasodilation Caused by Secondary Bile Acids. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H114-26. [Google Scholar] [CrossRef] [PubMed]
- Renga, B.; Cipriani, S.; Carino, A.; Simonetti, M.; Zampella, A.; Fiorucci, S. Reversal of Endothelial Dysfunction by GPBAR1 Agonism in Portal Hypertension Involves a AKT/FOXOA1 Dependent Regulation of H2S Generation and Endothelin-1. PLoS ONE 2015, 10, e0141082. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Yu, D.; Forman, B.M.; Huang, W. The G-Protein-Coupled Bile Acid Receptor, Gpbar1 (TGR5), Negatively Regulates Hepatic Inflammatory Response through Antagonizing Nuclear Factor κ Light-Chain Enhancer of Activated B Cells (NF-ΚB) in Mice. Hepatology 2011, 54, 1421–1432. [Google Scholar] [CrossRef]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and Function of the Bile Acid Receptor TGR5 in Kupffer Cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef]
- Zhang, F.; Xiao, X.; Li, Y.; Wu, H.; Deng, X.; Jiang, Y.; Zhang, W.; Wang, J.; Ma, X.; Zhao, Y. Therapeutic Opportunities of GPBAR1 in Cholestatic Diseases. Front. Pharmacol. 2022, 12, 805269. [Google Scholar] [CrossRef]
- Fiorucci, S.; Carino, A.; Baldoni, M.; Santucci, L.; Costanzi, E.; Graziosi, L.; Distrutti, E.; Biagioli, M. Bile Acid Signaling in Inflammatory Bowel Diseases. Dig. Dis. Sci. 2021, 66, 674–693. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef]
- Carey, E.J.; Ali, A.H.; Lindor, K.D. Primary Biliary Cirrhosis. Lancet 2015, 386, 1565–1575. [Google Scholar] [CrossRef]
- Floreani, A.; Gabbia, D.; De Martin, S. Update on the Pharmacological Treatment of Primary Biliary Cholangitis. Biomedicines 2022, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Jazrawi, R.P.; Ahmed, H.A.; Davis, T.; Bland, J.M.; Benson, M.; Orchard, R.T.; Theodossi, A.; Maxwell, J.D.; Northfield, T.C. Optimum Dose of Ursodeoxycholic Acid in Primary Biliary Cirrhosis. Eur. J. Gastroenterol. Hepatol. 1999, 11, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Rudic, J.S.; Poropat, G.; Krstic, M.N.; Bjelakovic, G.; Gluud, C. Ursodeoxycholic Acid for Primary Biliary Cirrhosis. Cochrane Database Syst. Rev. 2012, 12, CD000551. [Google Scholar] [CrossRef] [PubMed]
- Marchianò, S.; Biagioli, M.; Roselli, R.; Zampella, A.; Di Giorgio, C.; Bordoni, M.; Bellini, R.; Urbani, G.; Morretta, E.; Monti, M.C.; et al. Beneficial Effects of UDCA and NorUDCA in a Rodent Model of Steatosis Are Linked to Modulation of GPBAR1/FXR Signaling. Biochim. Biophys. Mol. Cell Biol. Lipids 2022, 1867, 159218. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Carino, A.; Fiorucci, C.; Marchianò, S.; Di Giorgio, C.; Bordoni, M.; Roselli, R.; Baldoni, M.; Distrutti, E.; Zampella, A.; et al. The Bile Acid Receptor GPBAR1 Modulates CCL2/CCR2 Signaling at the Liver Sinusoidal/Macrophage Interface and Reverses Acetaminophen-Induced Liver Toxicity. J. Immunol. 2020, 204, 2535–2551. [Google Scholar] [CrossRef]
- Brevini, T.; Maes, M.; Webb, G.J.; John, B.V.; Fuchs, C.D.; Buescher, G.; Wang, L.; Griffiths, C.; Brown, M.L.; Scott, W.E., 3rd; et al. FXR Inhibition May Protect from SARS-CoV-2 Infection by Reducing ACE2. Nature 2023, 615, 134–142. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Regulation of Gene Expression: Roles of Nuclear Hormone Receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef]
- Panzitt, K.; Wagner, M. FXR in Liver Physiology: Multiple Faces to Regulate Liver Metabolism. Biochim. Biophys. Acta—Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef]
- Renga, B.; Migliorati, M.; Mencarelli, A.; Fiorucci, S. Reciprocal Regulation of the Bile Acid-Activated Receptor FXR and the Interferon-Gamma-STAT-1 Pathway in Macrophages. Biochim. Biophys. Acta 2009, 1792, 564–573. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Chen, W.-D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X Receptor Antagonizes Nuclear Factor ΚB in Hepatic Inflammatory Response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous Hepatocarcinogenesis in Farnesoid X Receptor-Null Mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous Development of Liver Tumors in the Absence of the Bile Acid Receptor Farnesoid X Receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Renga, B.; Mencarelli, A.; Migliorati, M.; Cipriani, S.; D’Amore, C.; Distrutti, E.; Fiorucci, S. SHP-Dependent and -Independent Induction of Peroxisome Proliferator-Activated Receptor-γ by the Bile Acid Sensor Farnesoid X Receptor Counter-Regulates the pro-Inflammatory Phenotype of Liver Myofibroblasts. Inflamm. Res. 2011, 60, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The Nuclear Receptor SHP Mediates Inhibition of Hepatic Stellate Cells by FXR and Protects against Liver Fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Da Costa, K.-A.A.; Lee, S.; Renga, B.; Jaeschke, H.; Yang, Z.; Orena, S.J.J.; Goedken, M.J.J.; Zhang, Y.; Kong, B.; et al. Interactions Between Nuclear Receptor SHP and FOXA1 Maintain Oscillatory Homocysteine Homeostasis in Mice. Gastroenterology 2015, 148, 1012–1023.e14. [Google Scholar] [CrossRef]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867.e5. [Google Scholar] [CrossRef]
- Sepe, V.; Distrutti, E.; Fiorucci, S.; Zampella, A. Farnesoid X Receptor Modulators 2014-Present: A Patent Review. Expert Opin. Ther. Pat. 2018, 28, 351–364. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Sepe, V.; Zampella, A.; Distrutti, E. Bile Acid Modulators for the Treatment of Nonalcoholic Steatohepatitis (NASH). Expert. Opin. Investig. Drugs 2020, 29, 623–632. [Google Scholar] [CrossRef]
- Stedman, C.; Liddle, C.; Coulter, S.; Sonoda, J.; Alvarez, J.G.; Evans, R.M.; Downes, M. Benefit of Farnesoid X Receptor Inhibition in Obstructive Cholestasis. Proc. Natl. Acad. Sci. USA 2006, 103, 11323–11328. [Google Scholar] [CrossRef]
- Renga, B.; Migliorati, M.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Distrutti, E.; Fiorucci, S. Farnesoid X Receptor Suppresses Constitutive Androstane Receptor Activity at the Multidrug Resistance Protein-4 Promoter. Biochim. Biophys. Acta 2011, 1809, 157–165. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; D’Amore, C.; Cipriani, S.; D’Auria, M.V.; Sepe, V.; Chini, M.G.; Monti, M.C.; Bifulco, G.; Zampella, A.; et al. Discovery That Theonellasterol a Marine Sponge Sterol Is a Highly Selective FXR Antagonist That Protects against Liver Injury in Cholestasis. PLoS ONE 2012, 7, e30443. [Google Scholar] [CrossRef] [PubMed]
- Sepe, V.; Bifulco, G.; Renga, B.; D’Amore, C.; Fiorucci, S.; Zampella, A. Discovery of Sulfated Sterols from Marine Invertebrates as a New Class of Marine Natural Antagonists of Farnesoid-X-Receptor. J. Med. Chem. 2011, 54, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.R.; Morelli, A.; Parks, D.J.J.; Willson, T.M.M. 6alpha-Ethyl-Chenodeoxycholic Acid (6-ECDCA), a Potent and Selective FXR Agonist Endowed with Anticholestatic Activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Murillo Perez, C.F.; Fisher, H.; Hiu, S.; Kareithi, D.; Adekunle, F.; Mayne, T.; Malecha, E.; Ness, E.; van der Meer, A.J.; Lammers, W.J.; et al. Greater Transplant-Free Survival in Patients Receiving Obeticholic Acid for Primary Biliary Cholangitis in a Clinical Trial Setting Compared to Real-World External Controls. Gastroenterology 2022, 163, 1630–1642.e3. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Parés, A.; Mason, A.; et al. A Randomized Trial of Obeticholic Acid Monotherapy in Patients with Primary Biliary Cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Pockros, P.J.; Kremer, A.E.; Parés, A.; Forman, L.M.; Drenth, J.P.H.; Ryder, S.D.; Terracciano, L.; Jin, Y.; Liberman, A.; et al. Long-Term Obeticholic Acid Therapy Improves Histological Endpoints in Patients with Primary Biliary Cholangitis. Clin. Gastroenterol. Hepatol. 2020, 18, 1170–1178.e6. [Google Scholar] [CrossRef]
- Fiorucci, S.; Di Giorgio, C.; Distrutti, E. Obeticholic Acid: An Update of Its Pharmacological Activities in Liver Disorders. Handb. Exp. Pharmacol. 2019, 256, 283–295. [Google Scholar] [CrossRef]
- Han, B.; Kim, B.-K.; Kim, K.; Fang, S. Essential Roles of Bile Acids and Their Nuclear Receptors, FXR and PXR, in the Cholestatic Liver Disease. Animal Cells Syst. 2016, 20, 175–178. [Google Scholar] [CrossRef]
- Soret, P.-A.; Lam, L.; Carrat, F.; Smets, L.; Berg, T.; Carbone, M.; Invernizzi, P.; Leroy, V.; Trivedi, P.; Cazzagon, N.; et al. Combination of Fibrates with Obeticholic Acid Is Able to Normalise Biochemical Liver Tests in Patients with Difficult-to-Treat Primary Biliary Cholangitis. Aliment. Pharmacol. Ther. 2021, 53, 1138–1146. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; DeLuca, H.F. Where Is the Vitamin D Receptor? Arch. Biochem. Biophys. 2012, 523, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D. Nonclassic Actions of Vitamin D. J. Clin. Endocrinol. Metab. 2009, 94, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Gascon-Barré, M.; Demers, C.; Mirshahi, A.; Néron, S.; Zalzal, S.; Nanci, A. The Normal Liver Harbors the Vitamin D Nuclear Receptor in Nonparenchymal and Biliary Epithelial Cells. Hepatology 2003, 37, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D Receptor as an Intestinal Bile Acid Sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Lipsky, P.E. Modulatory Effects of 1,25-Dihydroxyvitamin D3 on Human B Cell Differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef]
- Tang, J.; Zhou, R.; Luger, D.; Zhu, W.; Silver, P.B.; Grajewski, R.S.; Su, S.B.; Chan, C.C.; Adorini, L.; Caspi, R.R. Calcitriol Suppresses Antiretinal Autoimmunity through Inhibitory Effects on the Th17 Effector Response. J. Immunol. 2009, 182, 4624–4632. [Google Scholar] [CrossRef]
- Penna, G.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Berti, E.; Colonna, M.; Adorini, L. Expression of the Inhibitory Receptor ILT3 on Dendritic Cells Is Dispensable for Induction of CD4+Foxp3+ Regulatory T Cells by 1,25-Dihydroxyvitamin D3. Blood 2005, 106, 3490–3497. [Google Scholar] [CrossRef]
- Almerighi, C.; Sinistro, A.; Cavazza, A.; Ciaprini, C.; Rocchi, G.; Bergamini, A. 1Alpha,25-Dihydroxyvitamin D3 Inhibits CD40L-Induced pro-Inflammatory and Immunomodulatory Activity in Human Monocytes. Cytokine 2009, 45, 190–197. [Google Scholar] [CrossRef]
- Ding, N.; Yu, R.T.; Subramaniam, N.; Sherman, M.H.; Wilson, C.; Rao, R.; Leblanc, M.; Coulter, S.; He, M.; Scott, C.; et al. A Vitamin D Receptor/SMAD Genomic Circuit Gates Hepatic Fibrotic Response. Cell 2013, 153, 601–613. [Google Scholar] [CrossRef]
- Kempinska-Podhorodecka, A.; Milkiewicz, M.; Wasik, U.; Ligocka, J.; Zawadzki, M.; Krawczyk, M.; Milkiewicz, P. Decreased Expression of Vitamin D Receptor Affects an Immune Response in Primary Biliary Cholangitis via the VDR-MiRNA155-SOCS1 Pathway. Int. J. Mol. Sci. 2017, 18, 289. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hang, S.; Fang, Y.; Bae, S.; Zhang, Y.; Zhang, M.; Wang, G.; McCurry, M.D.; Bae, M.; Paik, D.; et al. A Bacterial Bile Acid Metabolite Modulates T(Reg) Activity through the Nuclear Hormone Receptor NR4A1. Cell Host Microbe 2021, 29, 1366–1377.e9. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile Acid Metabolites Control T(H)17 and T(Reg) Cell Differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Oladimeji, P.O.; Chen, T. PXR: More Than Just a Master Xenobiotic Receptor. Mol. Pharmacol. 2018, 93, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Lou, X.; Zhang, F.; Xu, G. Nuclear Receptors in the Pathogenesis and Management of Inflammatory Bowel Disease. Mediat. Inflamm. 2019, 2019, 2624941. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Migliorati, M.; Barbanti, M.; Cipriani, S.; Palladino, G.; Distrutti, E.; Renga, B.; Fiorucci, S. Pregnane-X-Receptor Mediates the Anti-Inflammatory Activities of Rifaximin on Detoxification Pathways in Intestinal Epithelial Cells. Biochem. Pharmacol. 2010, 80, 1700–1707. [Google Scholar] [CrossRef]
- Mencarelli, A.; Renga, B.; Palladino, G.; Claudio, D.; Ricci, P.; Distrutti, E.; Barbanti, M.; Baldelli, F.; Fiorucci, S. Inhibition of NF-ΚB by a PXR-Dependent Pathway Mediates Counter-Regulatory Activities of Rifaximin on Innate Immunity in Intestinal Epithelial Cells. Eur. J. Pharmacol. 2011, 668, 317–324. [Google Scholar] [CrossRef]
- Stedman, C.A.M.; Liddle, C.; Coulter, S.A.; Sonoda, J.; Alvarez, J.G.A.; Moore, D.D.; Evans, R.M.; Downes, M. Nuclear Receptors Constitutive Androstane Receptor and Pregnane X Receptor Ameliorate Cholestatic Liver Injury. Proc. Natl. Acad. Sci. USA 2005, 102, 2063–2068. [Google Scholar] [CrossRef]
- Teng, S.; Piquette-Miller, M. Hepatoprotective Role of PXR Activation and MRP3 in Cholic Acid-Induced Cholestasis. Br. J. Pharmacol. 2007, 151, 367–376. [Google Scholar] [CrossRef]
- Bachs, L.; Parés, A.; Elena, M.; Piera, C.; Rodés, J. Effects of Long-Term Rifampicin Administration in Primary Biliary Cirrhosis. Gastroenterology 1992, 102, 2077–2080. [Google Scholar] [CrossRef]
- De Marino, S.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; Renga, B.; D’Amore, C.; Fiorucci, S.; Debitus, C.; Zampella, A. Theonellasterols and Conicasterols from Theonella Swinhoei. Novel Marine Natural Ligands for Human Nuclear Receptors. J. Med. Chem. 2011, 54, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Shizu, R.; Abe, T.; Kodama, S.; Hosaka, T.; Sasaki, T.; Yoshinari, K. PXR Functionally Interacts with NF-ΚB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice. Cells 2020, 9, 2296. [Google Scholar] [CrossRef] [PubMed]
- Özdirik, B.; Schnabl, B. Microbial Players in Primary Sclerosing Cholangitis: Current Evidence and Concepts. Cell. Mol. Gastroenterol. Hepatol. 2024, 17, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, G.; Tang, Y.; Liu, H.; Ge, X.; Peng, R.; Cao, J.; Tu, D.; Su, B.; Jin, S.; et al. Causal Associations between Gut Microbiota and Primary Biliary Cholangitis: A Bidirectional Two-Sample Mendelian Randomization Study. Front. Microbiol. 2023, 14, 1273024. [Google Scholar] [CrossRef]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut Microbial Profile Is Altered in Primary Biliary Cholangitis and Partially Restored after UDCA Therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Wang, R.; Li, B.; Huang, B.; Li, Y.; Liu, Q.; Lyu, Z.; Chen, R.; Qian, Q.; Liang, X.; Pu, X.; et al. Gut Microbiota-Derived Butyrate Induces Epigenetic and Metabolic Reprogramming in Myeloid-Derived Suppressor Cells to Alleviate Primary Biliary Cholangitis. Gastroenterology 2024, 167, 733–749.e3. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Beuers, U.; Corpechot, C.; Invernizzi, P.; Jones, D.; Marzioni, M.; Schramm, C. EASL Clinical Practice Guidelines: The Diagnosis and Management of Patients with Primary Biliary Cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Lammers, W.J.; Hirschfield, G.M.; Corpechot, C.; Nevens, F.; Lindor, K.D.; Janssen, H.L.A.; Floreani, A.; Ponsioen, C.Y.; Mayo, M.J.; Invernizzi, P.; et al. Development and Validation of a Scoring System to Predict Outcomes of Patients with Primary Biliary Cirrhosis Receiving Ursodeoxycholic Acid Therapy. Gastroenterology 2015, 149, 1804–1812.e4. [Google Scholar] [CrossRef]
- Corpechot, C.; Abenavoli, L.; Rabahi, N.; Chrétien, Y.; Andréani, T.; Johanet, C.; Chazouillères, O.; Poupon, R. Biochemical Response to Ursodeoxycholic Acid and Long-Term Prognosis in Primary Biliary Cirrhosis. Hepatology 2008, 48, 871–877. [Google Scholar] [CrossRef]
- Corpechot, C.; Chazouillères, O.; Poupon, R. Early Primary Biliary Cirrhosis: Biochemical Response to Treatment and Prediction of Long-Term Outcome. J. Hepatol. 2011, 55, 1361–1367. [Google Scholar] [CrossRef]
- Kuiper, E.M.M.; Hansen, B.E.; de Vries, R.A.; den Ouden-Muller, J.W.; van Ditzhuijsen, T.J.M.; Haagsma, E.B.; Houben, M.H.M.G.; Witteman, B.J.M.; van Erpecum, K.J.; van Buuren, H.R. Improved Prognosis of Patients with Primary Biliary Cirrhosis That Have a Biochemical Response to Ursodeoxycholic Acid. Gastroenterology 2009, 136, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Parés, A.; Caballería, L.; Rodés, J. Excellent Long-Term Survival in Patients with Primary Biliary Cirrhosis and Biochemical Response to Ursodeoxycholic Acid. Gastroenterology 2006, 130, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Kumagi, T.; Guindi, M.; Fischer, S.E.; Arenovich, T.; Abdalian, R.; Coltescu, C.; Heathcote, E.J.; Hirschfield, G.M. Baseline Ductopenia and Treatment Response Predict Long-Term Histological Progression in Primary Biliary Cirrhosis. Am. J. Gastroenterol. 2010, 105, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Lindor, K.D.; Therneau, T.M.; Jorgensen, R.A.; Malinchoc, M.; Kamath, P.S.; Dickson, E.R. Utilization of the Mayo Risk Score in Patients with Primary Biliary Cirrhosis Receiving Ursodeoxycholic Acid. Liver 1999, 19, 115–121. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver; Clinical Practice Guideline Panel. EASL Clinical Practice Guidelines on Non-Invasive Tests for Evaluation of Liver Disease Severity and Prognosis—2021 Update. J. Hepatol. 2021, 75, 659–689. [Google Scholar] [CrossRef]
- Corpechot, C.; Carrat, F.; Gaouar, F.; Chau, F.; Hirschfield, G.; Gulamhusein, A.; Montano-Loza, A.J.; Lytvyak, E.; Schramm, C.; Pares, A.; et al. Liver Stiffness Measurement by Vibration-Controlled Transient Elastography Improves Outcome Prediction in Primary Biliary Cholangitis. J. Hepatol. 2022, 77, 1545–1553. [Google Scholar] [CrossRef]
- Cipriani, S.; Renga, B.; D’Amore, C.; Simonetti, M.; De Tursi, A.A.; Carino, A.; Monti, M.C.; Sepe, V.; Zampella, A.; Fiorucci, S. Impaired Itching Perception in Murine Models of Cholestasis Is Supported by Dysregulation of GPBAR1 Signaling. PLoS ONE 2015, 10, e0129866. [Google Scholar] [CrossRef]
- Schnabl, B. PPAR Agonists in Primary Biliary Cholangitis. N. Engl. J. Med. 2024, 390, 855–858. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective Effects of the Dual Peroxisome Proliferator-Activated Receptor Alpha/Delta Agonist, GFT505, in Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Fiorucci, S.; Zampella, A.; Ricci, P.; Distrutti, E.; Biagioli, M. Immunomodulatory Functions of FXR. Mol. Cell. Endocrinol. 2022, 551, 111650. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A Multi-Society Delphi Consensus Statement on New Fatty Liver Disease Nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Bowlus, C.L.; Mayo, M.J.; Kremer, A.E.; Vierling, J.M.; Kowdley, K.V.; Levy, C.; Villamil, A.; Ladrón de Guevara Cetina, A.L.; Janczewska, E.; et al. A Phase 3 Trial of Seladelpar in Primary Biliary Cholangitis. N. Engl. J. Med. 2024, 390, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Assis, D.N. Advancing Second-Line Treatment for Primary Biliary Cholangitis. N. Engl. J. Med. 2024, 390, 853–854. [Google Scholar] [CrossRef]
- Levy, C.; Kendrick, S.; Bowlus, C.L.; Tanaka, A.; Jones, D.; Kremer, A.E.; Mayo, M.J.; Haque, N.; von Maltzahn, R.; Allinder, M.; et al. GLIMMER: A Randomized Phase 2b Dose-Ranging Trial of Linerixibat in Primary Biliary Cholangitis Patients with Pruritus. Clin. Gastroenterol. Hepatol. 2023, 21, 1902–1912.e13. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Caldwell, S.H.; Pyrsopoulos, N.; deLemos, A.S.; Rossi, S.; Levy, C.; Goldberg, D.S.; Mena, E.A.; Sheikh, A.; Ravinuthala, R.; et al. Proof-of-Concept Study to Evaluate the Safety and Efficacy of Saroglitazar in Patients with Primary Biliary Cholangitis. J. Hepatol. 2022, 76, 75–85. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, G.; Zheng, L.; Sun, R.; Wang, X.; Deng, J.; Jia, G.; Yang, C.; Cui, L.; Guo, C.; et al. Effectiveness of Fenofibrate in Treatment-Naive Patients with Primary Biliary Cholangitis: A Randomized Clinical Trial. Am. J. Gastroenterol. 2023, 118, 1973–1979. [Google Scholar] [CrossRef]
- Khanna, A.; Jopson, L.; Howel, D.; Bryant, A.; Blamire, A.; Newton, J.L.; Jones, D.E. Rituximab Is Ineffective for Treatment of Fatigue in Primary Biliary Cholangitis: A Phase 2 Randomized Controlled Trial. Hepatology 2019, 70, 1646–1657. [Google Scholar] [CrossRef]
- Kimura, M.; Ogawa, E.; Harada, K.; Imamura, J.; Saio, M.; Ikura, Y.; Yatsuhashi, H.; Murata, K.; Miura, K.; Ieiri, I.; et al. Feasibility, Safety and Tolerability of the CREB-Binding Protein/β-Catenin Inhibitor OP-724 in Patients with Advanced Primary Biliary Cholangitis: An Investigator-Initiated, Open-Label, Non-Randomised, Two-Centre, Phase 1 Study. BMJ open Gastroenterol. 2022, 9, e001001. [Google Scholar] [CrossRef]
- Invernizzi, P.; Carbone, M.; Jones, D.; Levy, C.; Little, N.; Wiesel, P.; Nevens, F. Setanaxib, a First-in-Class Selective NADPH Oxidase 1/4 Inhibitor for Primary Biliary Cholangitis: A Randomized, Placebo-Controlled, Phase 2 Trial. Liver 2023, 43, 1507–1522. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Yang, X.; Shen, M.; Huang, C.; Liu, Y.; Fan, X.; Yang, L. Ursodeoxycholic Acid at 18-22 Mg/Kg/d Showed a Promising Capacity for Treating Refractory Primary Biliary Cholangitis. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6691425. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Beuers, U.; Kupcinskas, L.; Ott, P.; Bergquist, A.; Färkkilä, M.; Manns, M.P.; Parés, A.; Spengler, U.; Stiess, M.; et al. A Placebo-Controlled Randomised Trial of Budesonide for PBC Following an Insufficient Response to UDCA. J. Hepatol. 2021, 74, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Al-Dury, S.; Wahlström, A.; Wahlin, S.; Langedijk, J.; Elferink, R.O.; Ståhlman, M.; Marschall, H.-U. Pilot Study with IBAT Inhibitor A4250 for the Treatment of Cholestatic Pruritus in Primary Biliary Cholangitis. Sci. Rep. 2018, 8, 6658. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.; Bolier, R.; Goet, J.; Parés, A.; Verbeek, J.; de Vree, M.; Drenth, J.; van Erpecum, K.; van Nieuwkerk, K.; van der Heide, F.; et al. Fibrates for Itch (FITCH) in Fibrosing Cholangiopathies: A Double-Blind, Randomized, Placebo-Controlled Trial. Gastroenterology 2021, 160, 734–743.e6. [Google Scholar] [CrossRef] [PubMed]
- Ataei, S.; Kord, L.; Larki, A.; Yasrebifar, F.; Mehrpooya, M.; Seyedtabib, M.; Hasanzarrini, M. Comparison of Sertraline with Rifampin in the Treatment of Cholestatic Pruritus: A Randomized Clinical Trial. Rev. Recent. Clin. Trials 2019, 14, 217–223. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. The Pharmacology of Bile Acids and Their Receptors. Handb. Exp. Pharmacol. 2019, 256, 3–18. [Google Scholar] [CrossRef]
- Matsumoto, K.; Hirohara, J.; Takeuchi, A.; Miura, R.; Asaoka, Y.; Nakano, T.; Tanaka, A. Determinants of the Effectiveness of Bezafibrate Combined with Ursodeoxycholic Acid in Patients with Primary Biliary Cholangitis. Hepatol. Res. 2023, 53, 989–997. [Google Scholar] [CrossRef]

| Name | Response Criteria |
|---|---|
| GLOBAL-PBC [161] | Bilirubin, ALP, albumin and platelet count after 12 months of UDCA and age at baseline |
| Paris-I [162] | ALP > 3 × upper normal values or AST > 2 × upper normal values or bilirubin > 1.0 mg/dL after 12 months of UDCA |
| Paris-II [163] | All three of the following: ALP > 1.5 × upper normal values, AST 1.5 × upper normal values, bilirubin > 1 mg/dL after 12 months of UDCA |
| Rotterdam [164] | Bilirubin > 1 × upper normal values and/or albumin < 1 × upper normal values afer 12 months UDCA |
| Barcelona [165] | Decrease in ALP < 40% and ALP > 1.0 × upper normal values after 12 months of UDCA |
| Toronto [166] | ALP > 1.67 × upper normal values after 24 months UDCA |
| Rochester [167] | ALP > 2 × upper normal values after 6 months or 12 months of UDCA |
| Intervention/Target | Mechanism(s) | Drug |
|---|---|---|
| Bile acids | Various mechanisms, including increased bile acid flow and immunemodulation | UDCA and norUDCA |
| Antifibrotic agents NOX1/4 inhibitor | Prevents generation of ROS and transformation of hepatic stellate cells in myo-fibroblasts | Setanaxib |
| Apical Sodium BA Transporter (ABST) Inhibitors | Mitigates the reabsorption of conjugated BAs from the ileum | A3907 Volixibat Linerixibat |
| FXR agonist | Various mechanisms | Linafexor, Cilofexor OCA, TQA3526 ASC42 |
| PPAR agonists | Various mechanisms including immune modulation | Elafibranor (α/δ) Seladelpar (δ) Benzafibrate (α) Fenofibrate (α) Saroglitazar (α/γ) |
Anti-pruritogens
| Attenuates itching induced by pruritogens (e.g., BAs, bilirubin) Agosnitsfor the opioid K receptors | EP547 Difelikefalin (CR845) |
| HMG-CoA reductase inhibitors | Lowers cholesterol production and reduces dyslipidemia-associated complications | Atorvastatin Rosuvastatin Simvastatin |
| Gut microbiome-based therapies | Shifts the gut microbiota profile toward a lower abundance of pathological species while promoting taxa responsible for immune tolerance | Probiotics and Fecal microbiota transplantation |
| IMPDH inhibitor | Prodrug of mycophenolic acid (MPA), limits de-novo synthesis of guanosine nucleotides | Mycophenolate |
| Calcineurin inhibitor | Immunosuppressant activity | Cyclosporine A |
| JAK1/2 inhibitor | Reduces cell proliferation, shows immunosuppressant activity and induces cell apoptosis | Baricitinib (LY3009104) |
| Treatment | Clinical Trial | Treatments | End Points |
|---|---|---|---|
| Benzafibrate (BZF) PPARα | NCT04514965 Phase N.A. | PBC patients with inadequate response to UDCA. Group 1: BZF, dosages not given | Primary Assessment of sCD163 macrophages marker and other fibrosis markers levels, liver stiffness and bile acid composition from 4 weeks up to 3 years Secondary assessment of itching degree from 4 weeks up to 3 years |
| Benzafibrate (BZF) PPARα | NCT04751188 Phase III | PBC patients with inadequate response to UDCA. Group 1: Benzafibrate 200 mg/b.d. + UDCA 13–15 mg/kg/d Group 2: Placebo Comparator: Placebo b.d. + UDCA 13–15 mg/kg/d | Primary Evaluation of biochemical response as the reduction of ALP ≤ 1.5-fold the upper limit of normal (ULN), AST ≤ 1.5-fold the ULN and Bilirubin ≤ 1 mg/dL at 6 months Secondary Assessment of quality of life and pruritus intensity using visual analogue scales after 6 months |
| Fenofibrate PPARα | NCT05749822 Phase II/III | PBC with compensated cirrhosis and inadequate biochemical response to UDCA. Group 1: Placebo Comparator: Placebo + UDCA 13–15 mg/kg/d Group 2: Fenofibrate 200 mg/d + UDCA 13–15 mg/kg/d | Primary Evaluation of serum ALP levels normalization at 48 weeks Secondary Evaluation of serum ALP levels normalization; changes in pruritus and fatigue; onset of biological or clinical AEs (increased creatinine, blood urea nitrogen, creatine kinase, AST, ALT) at 4, 12, 24, 36 and 48 weeks |
| Fenofibrate PPARα | NCT05751967 Phase III | PBC patients with inadequate biochemical response to UDCA. Group 1: Placebo Comparator: Placebo + UDCA 13–15 mg/kg/d Group 2: Fenofibrate 200 mg/d + UDCA 13–15 mg/kg/d | Primary Assessment of ALP and total bilirubin normalization at 48 weeks Secondary Assessment of ALP and total bilirubin normalization; changes in fatigue, pruritus and quality of life; drug-related adverse events onset; survival rates without liver transplantation or liver decompensation, pruritus, portal hypertension and others at 4, 12, 24, 36 and 48 weeks |
| Fenofibrate PPARα | NCT06174402 Phase II/III | PBC patients. Group 1: Fenofibrate 200 mg/d + UDCA 13–15 mg/kg/d Group 2: Placebo Comparator: Placebo + UDCA 13–15 mg/kg/d | Primary Assessment of ALP levels normalization at 48 weeks Secondary Assessment of ALP levels normalization; changes in pruritus and fatigue (VAS scale); biological or clinical adverse events onset (creatinine, AST and ALT increase); hepatic impairment development at 4, 12, 24, 36 and 48 weeks |
| Obeticholic Acid (OCA) Benzafibrate (BZF) | NCT05239468 Phase II | PBC patients. Group 1: Double Blind phase treatment A: BZF 100 mg/d + 1 OCA Placebo + 1 BZF Placebo Group 2: Double Blind phase treatment B: BZF 400 mg/d + 1 OCA Group 3: Double Blind phase treatment C: OCA 5 mg/d + BZF 100 mg/d + 1 BZF Placebo Group 4: Double Blind phase treatment D: OCA 5 mg/d + BZF 400 mg/d Group 5: Long Term Safety Extension (LTSE) Phase treatment D: OCA 5 mg/d + BZF 400 mg/d | Primary Evaluation of ALP levels change at 2, 4, 6, 8, 10 and 12 weeks Secondary Assessment of percentage changes in ALP levels; AST, ALT, GGT, total and conjugated bilirubin and lipid pool normalization; changes in bile acids plasma values at 2, 4, 6, 8, 10 and 12 weeks |
| Obeticholic Acid (OCA) Benzafibrate (BZF) | NCT04594694 Phase II | PBC patients. Group 1: Treatment A: BZF 200 mg/d Immediate Release (IR) + 1 OCA Placebo + 1 BZF 400 mg/d Placebo Group 2: Treatment B: BZF 400 mg/d SR + 1 BZF 200 mg/d Placebo + 1 OCA Placebo Group 3: Treatment C: OCA 5 to 10 mg/d + BZF 200 mg/d IR + BZF 400 mg/d Placebo Group 4: Treatment D: OCA 5 mg to 10 mg + BZF 400 mg/d SR + BZF 200 mg/d Placebo Group 5: LTSE phase, OCA + BZF: participants will continue the original treatment assigned but OCA and BZF dose may be optimized based on safety and efficacy | Primary Evaluation of ALP levels at day 1 and 4, 8 and 12 weeks Secondary Percentage assessment of ALP, AST, ALT, GGT normalization at day 1 and 4, 8 and 12 weeks; change in total and conjugated bilirubin, lipid and bile acids pool at day 1 and 4, 8 and 12 weeks |
| Obeticholic Acid (OCA) | NCT05450887 Phase III | PBC patients. Group 1: OCA 5 to 10 mg/d + UDCA 13–15 mg/kg/d if already receiving UDCA; if the subjects could not tolerate UDCA, they were not treated with UDCA Group 2: Placebo Comparator + UDCA 13–15 mg/kg/d if already receiving UDCA; if the subjects could not tolerate UDCA, they were not treated with UDCA | Primary Evaluation of ALP ≤ 1.67-fold the ULN, ALP decrease ≥ 15% from baseline and total bilirubin ≤ ULN up to 12 months Secondary Assessment of absolute and percentage change of ALP, AST, ALT, GGT, total and direct bilirubin; quality of life evaluation via PBC-40 score percentage change at 3, 6, 9 and 12 months |
| Volixibat ASBT inhibitor | NCT05050136 Phase II | Group 1: Volixibat 20 mg/b.d. Group 2: Volixibat 80 mg/b.d. Group 3: Placebo | Primary Assessment of mean change in the daily itch scores using the Adult Itch Reported Outcome (Adult ItchRO) questionnaire up to week 28 Secondary Evaluation of ALP, total bilirubin, serum bile acids levels change; adverse events incidents; assessment of quality of life (PBC-40 score), fatigue and sleep disturbance (PROMIS®) up to 28 weeks |
| Linerixibat IBAT imnhibitor | NCT04950127 Phase III | PBC patients. Group 1: Linerixibat, dosages not given Group 2: Linerixibat followed by Placebo, dosages not given Group 3: Placebo Group 4: Placebo followed by Linerixibat, dosages not given | Primary Assessment of change from baseline in Monthly Itch Scores using Numerical Rating Scale (NRS) over 24 weeks Secondary Evaluation of changes in Mean Worst Daily Itch score at Week 2. Changes in PBC-40 score, PGI-S, PGI-C and Monthly Sleep Score, measured by NRS; reduction in the Monthly Itch Score; changes in ALP and bilirubin levels up to 24 weeks |
| Linerixibat Ileal Bile Acid Transporter Inhibitor (IBAT) | NCT04167358 Phase III | Patients with PBC Group 1: Linerixibat in participant who previously participated in the Phase 2 studies (BAT117213 and 201000 GLIMMER [Group 1]) and Phase 3 study (212620 GLISTEN [Group 2]), dosages not given | Primary Assessment of AEs and SAEs onset up to 66 months Secondary Changes in PBC-40 score, in health-related quality of life (EQ-5D-3L score) and self-related health (EQ VAS score); assessment of depression intensity (BDI-II score); changes in hematology, biochemistry and coagulation parameters up to 65 months. Changes in pruritus (MIS-NRS), fatigue (MFS-NRS) and sleep (MSS-NRS) up to week 52 of continuous treatment |
| Obeticholic Acid (OCA) UDCA | NCT04956328 Phase III | PBC patients with inadequate response to UDCA. Group 1: OCA 5–10 mg/d + UDCA (continue pre-study dose) for 24 weeks and then titrating up to 10 mg based on tolerability and response Group 2: Placebo + UDCA (continue pre-study dose) for 48 weeks | Primary Percentage of patients with ALP < 1.67-fold the ULN, ALP decreased at least 15% and total bilirubin ≤ ULN up to 48 weeks Secondary Percentage of patients with ALP < 1.67-fold the ULN, ALP decreased at least 15% and total bilirubin ≤ ULN at 4, 12, 24 and 36 weeks. Assessment of rate of change of AST, ALT, ALP, GGT, total bile acids and total bilirubin ad liver function indicators up to 48 weeks |
| UDCA Total Glucosides of Peony (TGP) Anti-inflammatory and immune regulatory effects | NCT04618575 Phase IV | PBC patients with Autoimmune Hepatitis (AIH) 1. Group 1: UDCA + TGP, dosages not given Group 2: UDCA only, dosages not given | Primary Percentage of patients in biochemical remission defined as normalization of serum ALT and IgG levels after 24 weeks and up to 12 months Secondary Assessment of patients in partial remission (AST/ALT > 1-fold the ULN and <2-fold the ULN), with minimal response (AST/ALT still > 2-fold the ULN) or with treatment failure; drug-related side-effects and clinical symptoms (jaundice, fatigue, itching) onset; changes in the proportion of blood immune cells (% of T cells, DCs, Treg, NK.) up to 12 months |
| UDCA Low-Dose Glucocorticoid (GC) Decrease in symptoms severity | NCT04617561 Phase IV | PBC patients with Autoimmune Hepatitis (AIH) 2. Group 1: UDCA 13–15 mg/kg/d Group 2: UDCA 13–15 mg/kg/d + Methylprednisolone 12 mg/d in induction phase (2–4 mg/d in maintenance phase) | Primary Percentage of patients in biochemical remission defined as normalization of serum ALT and IgG levels up to 12 months Secondary Assessment of patients in partial remission (AST/ALT > 1-fold the ULN and <2-fold the ULN), with minimal response (AST/ALT still > 2-fold the ULN) or with treatment failure up to 12 months. Drug-related side-effects onset and changes in the proportion of blood immune cells (% of T cells, DCs, Treg, NK.) at 12 months. Assessment of AST, ALT and IgG serum levels at 3, 6 and 12 months |
| Saroglitazar Magnesium PPARα/γ | NCT05133336 Phase III | PBC patients. Group 1: Saroglitazar Magnesium 2 mg/d Group 2: Saroglitazar Magnesium 1 mg/d Group 3: Placebo | Primary Assessment of number of subjects with biochemical response as ALP < 1.67-fold the ULN, ALP decrease ≥ 15% from baseline and total bilirubin ≤ ULN (or direct bilirubin ≤ ULN in patients with known Gilbert’s Syndrome) up to 52 weeks Secondary Assessment of number of subjects with biochemical response as ALP < 1.67-fold the ULN, ALP decrease ≥ 15% from baseline and total bilirubin ≤ ULN (or direct bilirubin ≤ ULN in patients with known Gilbert’s Syndrome) at 4, 8, 16 and 24 weeks. Percentage improvement or normalization in ALP values; improvement in liver stiffness measurement of at least 25% via FibroScan®; changes in liver enzyme (AST, ALT, GGT, total bilirubin and albumin) and lipid (TG, LDL-C, HDL-C, total cholesterol) parameters; changes in serum bile acids at 24 and 52 weeks. Assessment of changes in health-related quality of life (PBC-40 score) and itching (5D scale, PGI-C scale, PGT-B scale, PGI-Worst Itch Severity scale) at 4, 8, 16, 24 and 52 weeks. Assessment of treatment-related AEs, SAEs, AEs of special interest (e.g., DILI) onset; significant changes in clinical laboratory test results (hematology, biochemistry, urinalysis), in vital signs, in ECG and in body weight at 52 weeks |
| Setanaxib NADP oxidase (NOX) 1/4 inhibitor | NCT05014672 Phase III | PBC patients. Group 1: Setanaxib 1200 mg/day. Eventual escalation to 1600 mg/day will be determined for the extension period Group 2: Setanaxib 1600 mg/d. Eventual reduction to 1200 mg/day mg/day will be determined for the extension period Group 3: Placebo. During the extension period, participants will switch from placebo to Setanaxib at a dose of either 1200 or 1600 mg/d depending on interim analysis outcome | Primary Assessment of biochemical response as ALP < 1.67-fold the ULN, ALP decrease ≥ 15% from baseline and total bilirubin ≤ ULN up to 52 weeks Secondary Assessment of changes in fatigue (PROMIS®, PBC-40 score, PGI-S, PGI-C), liver stiffness (FibroScan®), itching (WI-NRS, PBC-40, PGI-S, PGI-C); TEAEs and AESIs onset up to 52 weeks |
| HTD1801 (BUDCA) Hypolipidemic agent | NCT04604652 Phase II | PBC patients with inadequate response to standard UDCA therapy. Group 1: HTD1801 (BUDCA) 2000 mg/d | Primary Evaluation of changes in serum ALP at 12 weeks Secondary Assessment of serum bilirubin, GGT, total cholesterol, LDL-C, tryglicerides and inflammatory markers (fibrinogen, CRP, haptoglobin, IgG) changes; itching variations (Pruritus VAS), AEs onset as well as changes in physical examinations, vital signs and clinical laboratory values at 12 weeks |
| TQA3526 FXR | NCT04278820 Phase II | PBC patients. Group 1: Climbing Group: TQ3526 drug or Placebo once daily, dosages not given Group 2: Titration Group: TQ3526 drug or Placebo once daily, dosages not given Group 3: Extension Group: TQ3526 drug or Placebo once daily, dosages not given | Primary Evaluation of ALP levels reduction up to 24 weeks Secondary Assessment of ALP, ALT, AST, GGT, total bilirubin, LDL-C, HDL-C, TG and TC at 2, 4, 8, 12, 14, 16, 20 and 24 weeks. Assessment of Cmax and Tmax. Evaluation of TEAEs and SAEs onset up to 24 weeks |
| ASC42 FXR | NCT05190523 Phase II | PBC patients. Group 1: ASC42 5 mg/d Group 2: ASC42 10 mg/d Group 3: ASC42 15 mg/d Group 4: Placebo | Primary Evaluation of percentage changes in ALP levels at day 85 Secondary Evaluation of percentage and absolute changes of ALP, GGT, ALT, AST; incidence of TEAEs, SAEs and AESI onset at day 15, 29, 57 and 85 |
| EP547 MAS related GPR family member X4 (MrgprX4) | NCT05525520 Phase II | PBC or PSC patients with cholestatic pruritus. Group 1: EP547 100 mg/d Group 2: Placebo | Primary Evaluation of changes in pruritus (WI-NRS) up to 6 weeks Secondary Evaluation of changes and reduction in pruritus (5D-Itch scale, PGI-C, PGI-S); assessment of AEs onset; measurement of Cmax up to 6 weeks |
| Probiotics (Micro V Probiotics) | NCT03521297 Phase II | PBC patients with inadequate response to UDCA. Group 1: Placebo + SOC UDCA 13–15 mg/kg/d Group 2: Oral administration three times per day of Probiotics + SOC UDCA 13–15 mg/kg/d | Assessment of percentage of patients with biochemical response as serum ALP or GGT decreased by 20% from baseline after 6 months |
| Mycophenolate Mofetil IMPDH inhibitor Cyclosporin A Calcineurin inhibitor/immunosuppressive agent | NCT04376528 Phase IV | PBC patients with PBC-AIH overlap syndrome and nonresponsive to UDCA standard therapy. Group 1: Cyclosporin A + UDCA SOC, dosages not given Group 2: Mycophenolate Mofetil + UDCA SOC, dosages not given | Primary Evaluation of percentage of patients in biochemical remission as normalization of serum ALT and IgG levels after 24 weeks and up to 6 months Secondary Evaluation of partial remission (AST or ALT serum levels > ULN and <2-fold ULN), minimal response (AST or ALT still > 2-fold ULN) or treatment failure; assessment of changes in liver stiffness (shear-wave elastography); drug-related side effects onset up to 6 months |
| CNP-104 Immunomodulating agent | NCT05104853 Phase I/II | PBC patients non-responsive to UDCA and/or OCA. Group 1: 200 mL intravenous infusion of CNP-104 4 mg/kg on day 1 and day 8 Group 2: 200 mL intravenous infusion of CNP-104 8 mg/kg on day 1 and day 8 Group 3: Placebo Comparator | Primary Assessment of AEs and SAEs onset; laboratory tests (hematology, serum chemistry, coagulation panel, urinalysis) through study completion, an average of 720 days. Assessment of serum cytokines (TNFα, IL-4, IL-6, IL-10, IL-1β, MCP-1, IFN-γ) for an average of 15 days. Evaluation of ALP changes at day 60 Secondary Evaluation of changes in AMA and liver fibrosis (FibroScan®) at day 90 and 720. Changes in modified PBC-40 score, Weekly Mean Itch Score, liver enzyme levels (albumin, bilirubin, AST, ALT, GGT) and antigen-specific CD4+/CD8+ T cells asset at day 60 and 720. Assessment of ALP levels at day 720. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorucci, S.; Urbani, G.; Di Giorgio, C.; Biagioli, M.; Distrutti, E. Current Landscape and Evolving Therapies for Primary Biliary Cholangitis. Cells 2024, 13, 1580. https://doi.org/10.3390/cells13181580
Fiorucci S, Urbani G, Di Giorgio C, Biagioli M, Distrutti E. Current Landscape and Evolving Therapies for Primary Biliary Cholangitis. Cells. 2024; 13(18):1580. https://doi.org/10.3390/cells13181580
Chicago/Turabian StyleFiorucci, Stefano, Ginevra Urbani, Cristina Di Giorgio, Michele Biagioli, and Eleonora Distrutti. 2024. "Current Landscape and Evolving Therapies for Primary Biliary Cholangitis" Cells 13, no. 18: 1580. https://doi.org/10.3390/cells13181580
APA StyleFiorucci, S., Urbani, G., Di Giorgio, C., Biagioli, M., & Distrutti, E. (2024). Current Landscape and Evolving Therapies for Primary Biliary Cholangitis. Cells, 13(18), 1580. https://doi.org/10.3390/cells13181580