
Kisspeptin Alleviates Human Hepatic Fibrogenesis by Inhibiting TGFβ Signaling in Hepatic Stellate Cells
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Precision-Cut Liver Slices
2.2. Human LX-2 Stellate Cell Culture and Treatment
2.3. Primary Mouse Hepatic Stellate Cell (HSC) Culture and Treatment
2.4. Real-Time PCR (RT-qPCR) Quantification of Gene Expression
2.5. RNA-Seq Data Analysis
2.6. Immunoblot Analysis
2.7. LX-2 Cell Proliferation Assay
2.8. LX-2 Cell Transformation Assay
2.9. Kisspeptin Secretion
2.10. Immunohistochemistry of Human Liver
2.11. Statistical Analysis
3. Results
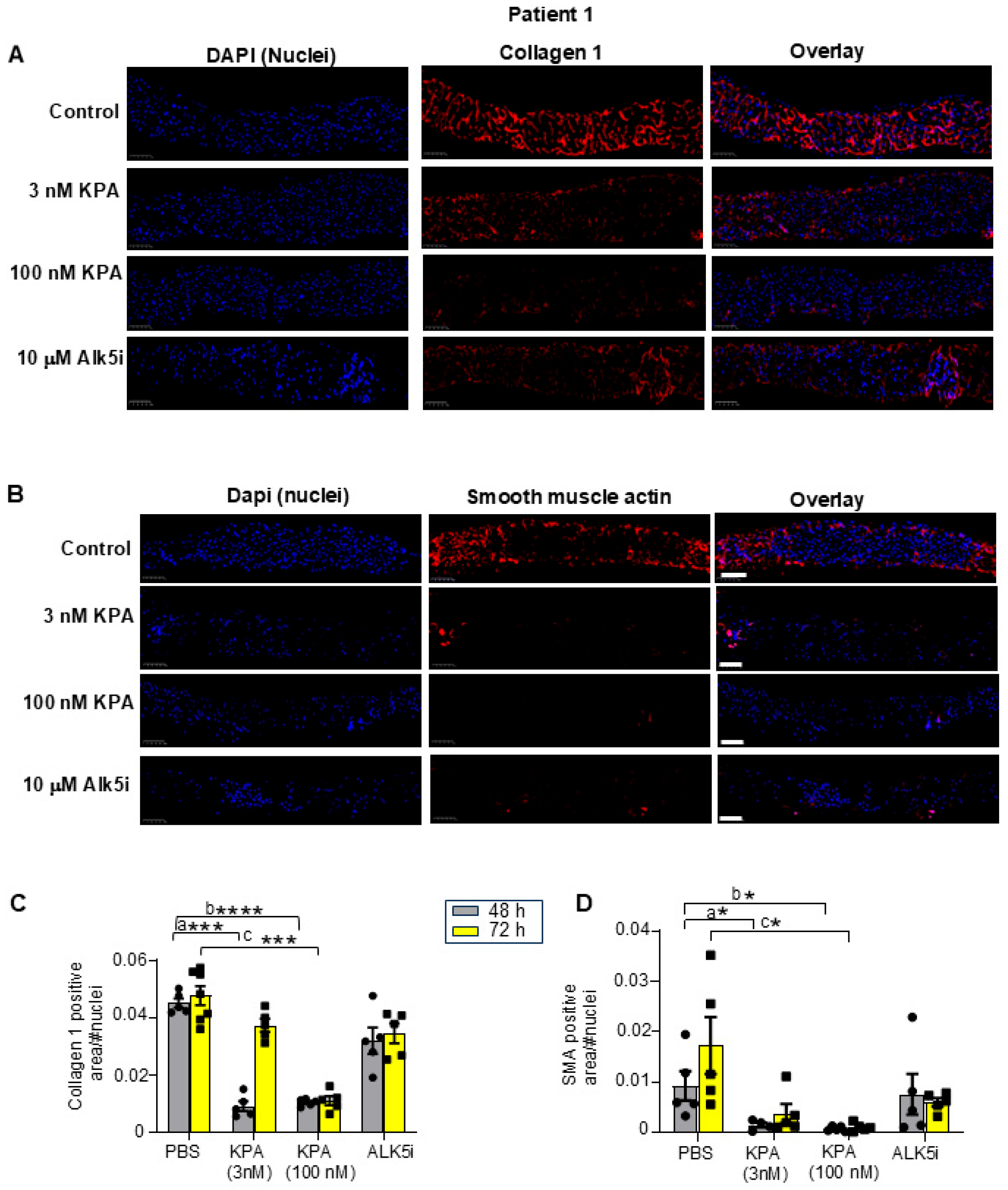
3.1. Kisspeptin Analog, TAK-448 (KPA), Reduces Fibrogenic and Inflammatory Markers as Well as Collagen Secretion in Diseased Human Patient-Derived Precision-Cut Liver Slices (hPCLS)
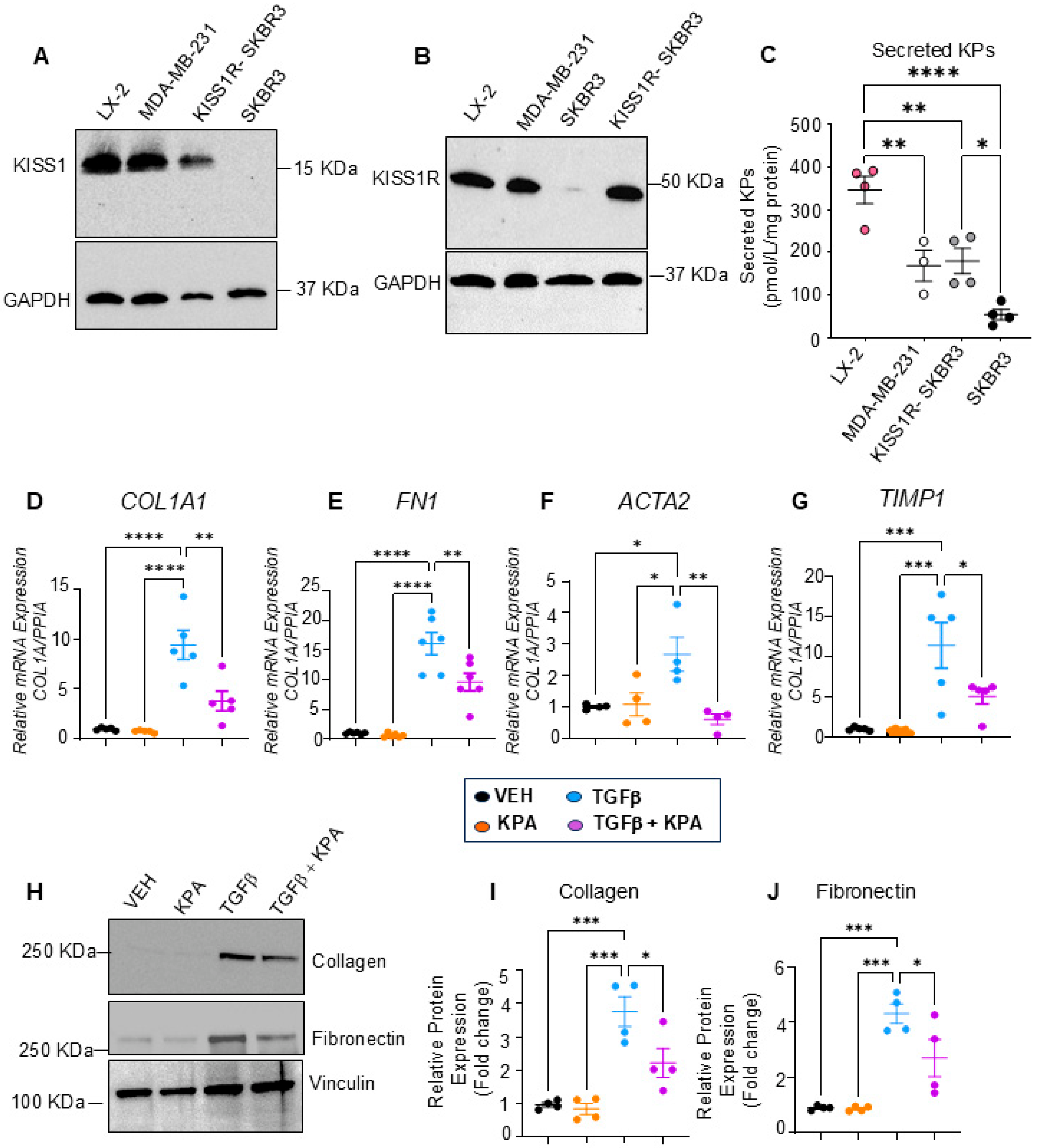
3.2. KPA Treatment Upregulates KISS1 and KISS1R in hPCLS, and KISS1R Is Strongly Expressed in Human HSCs
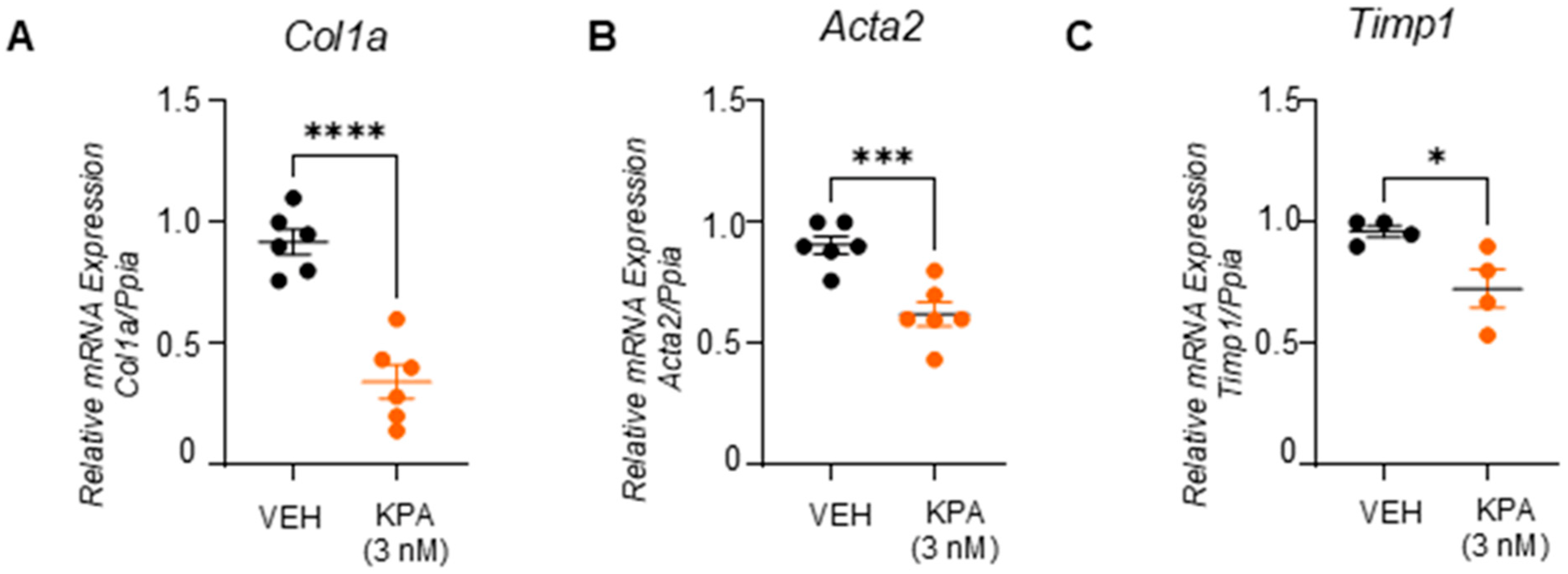
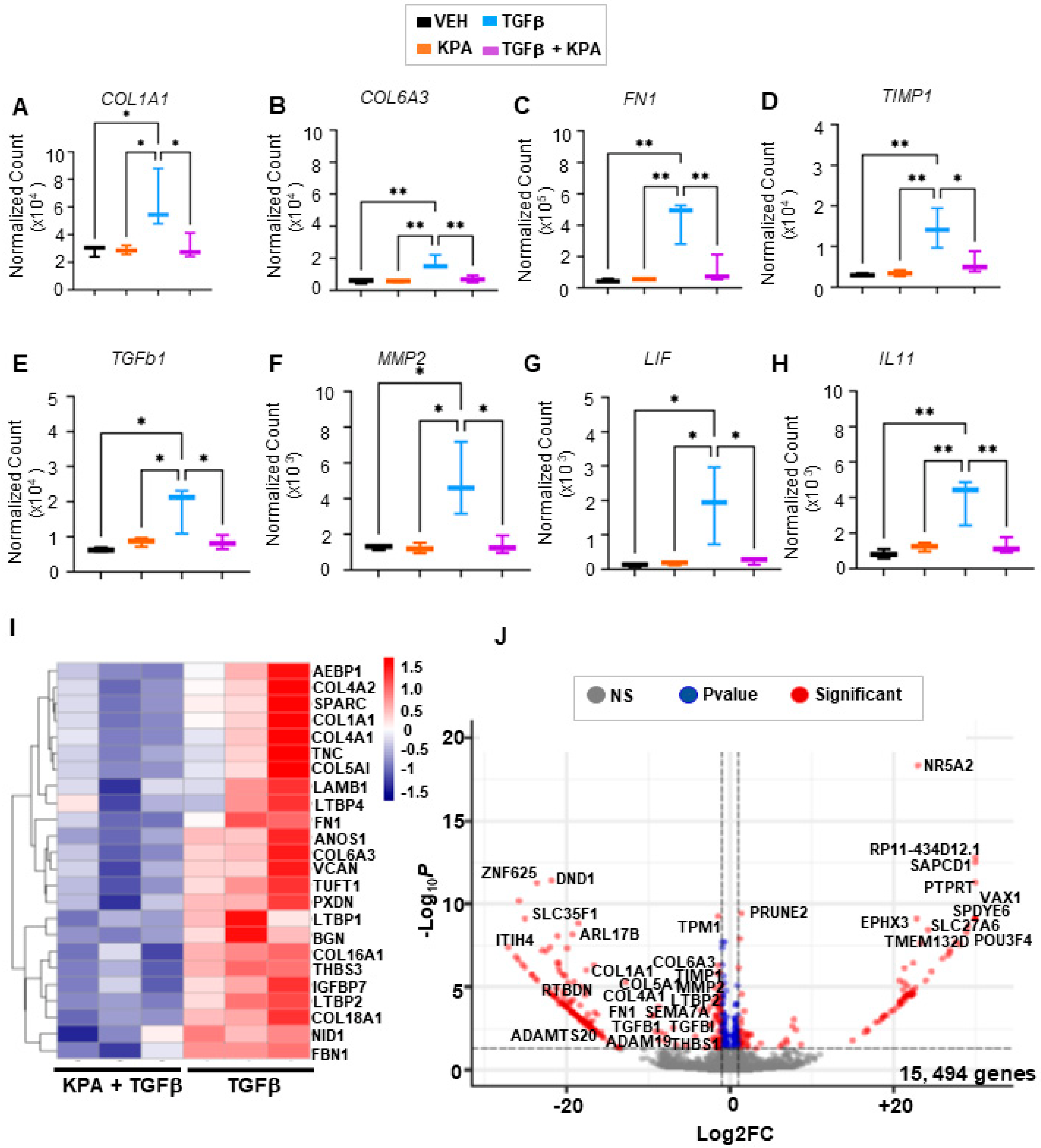
3.3. KPA Treatment Attenuates Fibrogenesis in Hepatic Stellate Cells (HSCs)
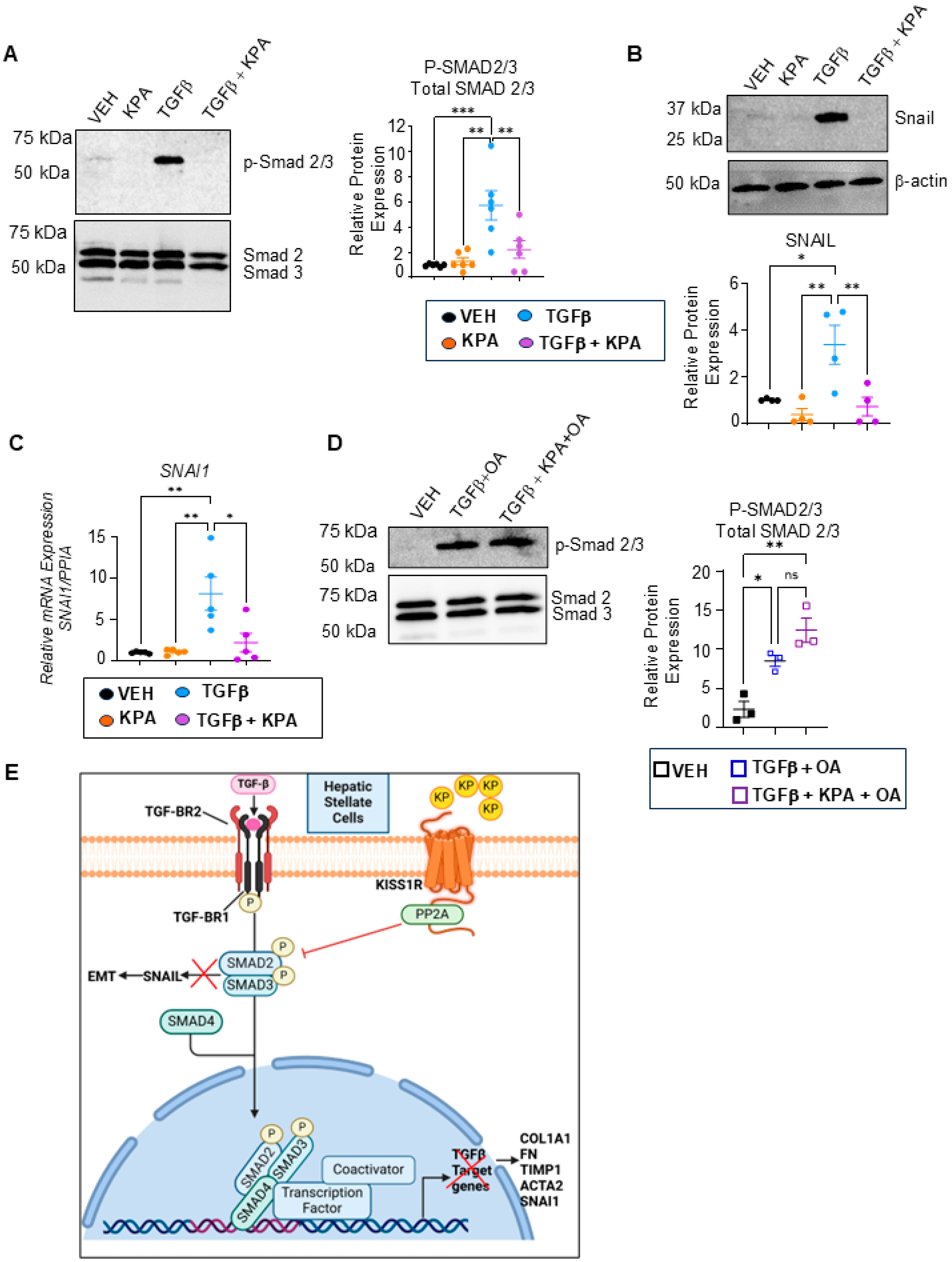
3.4. KPA Inhibits TGFβ Signaling in Hepatic Stellate Cells via Activation of Protein Phosphatase PP2A
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akkiz, H.; Gieseler, R.K.; Canbay, A. Liver Fibrosis: From Basic Science towards Clinical Progress, Focusing on the Central Role of Hepatic Stellate Cells. Int. J. Mol. Sci. 2024, 25, 7873. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, B.; Yashaswini, C.N.; Wang, S.; Sia, D.; Friedman, S.L. Friend or foe? The elusive role of hepatic stellate cells in liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Willoughby, C.E.; Singal, A.G.; Greten, T.F.; Heikenwalder, M.; El-Serag, H.B.; Finn, R.S.; Friedman, S.L. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: Pathogenesis and treatment. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic Fibrosis and Cancer: The Silent Threats of Metabolic Syndrome. Diabetes Metab. J. 2024, 48, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Tapper, E.B.; Parikh, N.D. Diagnosis and Management of Cirrhosis and Its Complications: A Review. JAMA 2023, 329, 1589–1602. [Google Scholar] [CrossRef]
- Hagstrom, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stal, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef]
- Noureddin, M.; Vipani, A.; Bresee, C.; Todo, T.; Kim, I.K.; Alkhouri, N.; Setiawan, V.W.; Tran, T.; Ayoub, W.S.; Lu, S.C.; et al. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. Am. J. Gastroenterol. 2018, 113, 1649–1659. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Babwah, A.V. Kisspeptin: Beyond the brain. Endocrinology 2015, 156, 1218–1227. [Google Scholar] [CrossRef]
- Kirby, H.R.; Maguire, J.J.; Colledge, W.H.; Davenport, A.P. International Union of Basic and Clinical Pharmacology. LXXVII. Kisspeptin receptor nomenclature, distribution, and function. Pharmacol. Rev. 2010, 62, 565–578. [Google Scholar] [CrossRef]
- Li, X.; Liang, C.; Yan, Y. Novel Insight into the Role of the Kiss1/GPR54 System in Energy Metabolism in Major Metabolic Organs. Cells 2022, 11, 3148. [Google Scholar] [CrossRef]
- Wolfe, A.; Hussain, M.A. The Emerging Role(s) for Kisspeptin in Metabolism in Mammals. Front. Endocrinol. 2018, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Shoji, I.; Hirose, T.; Mori, N.; Hiraishi, K.; Kato, I.; Shibasaki, A.; Yamamoto, H.; Ohba, K.; Kaneko, K.; Morimoto, R.; et al. Expression of kisspeptins and kisspeptin receptor in the kidney of chronic renal failure rats. Peptides 2010, 31, 1920–1925. [Google Scholar] [CrossRef] [PubMed]
- Guzman, S.; Dragan, M.; Kwon, H.; de Oliveira, V.; Rao, S.; Bhatt, V.; Kalemba, K.M.; Shah, A.; Rustgi, V.K.; Wang, H.; et al. Targeting hepatic kisspeptin receptor ameliorates nonalcoholic fatty liver disease in a mouse model. J. Clin. Investig. 2022, 132, e145889. [Google Scholar] [CrossRef]
- Song, W.J.; Mondal, P.; Wolfe, A.; Alonso, L.C.; Stamateris, R.; Ong, B.W.; Lim, O.C.; Yang, K.S.; Radovick, S.; Novaira, H.J.; et al. Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab. 2014, 19, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; She, Y.; Zhao, J.; Yan, J.; Yu, X.; Jin, Y.; Wei, Q.; Zhang, Z.; Shang, W. Emerging roles of kisspeptin/galanin in age-related metabolic disease. Mech. Ageing Dev. 2021, 199, 111571. [Google Scholar] [CrossRef] [PubMed]
- Izzi-Engbeaya, C.; Dhillo, W.S. Emerging roles for kisspeptin in metabolism. J. Physiol. 2021, 600, 1079–1088. [Google Scholar] [CrossRef]
- Musa, E.; Matjila, M.; Levitt, N.S. Kisspeptins and Glucose Homeostasis in Pregnancy: Implications for Gestational Diabetes Mellitus-a Review Article. Reprod. Sci. 2022, 29, 321–327. [Google Scholar] [CrossRef]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.-K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Paish, H.L.; Reed, L.H.; Brown, H.; Bryan, M.C.; Govaere, O.; Leslie, J.; Barksby, B.S.; Macia, M.G.; Watson, A.; Xu, X.; et al. A Bioreactor Technology for Modeling Fibrosis in Human and Rodent Precision-Cut Liver Slices. Hepatology 2019, 70, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-cut liver slices: A versatile tool to advance liver research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Becker, C.; Readhead, B.; Goossens, N.; Novik, J.; Fiel, M.I.; Cousens, L.P.; Magnusson, B.; Backmark, A. Repositioning of a novel GABA-B receptor agonist, AZD3355 (Lesogaberan), for the treatment of non-alcoholic steatohepatitis. Sci. Rep. 2021, 11, 20827. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2, New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Izzi-Engbeaya, C.; Comninos, A.N.; Clarke, S.A.; Jomard, A.; Yang, L.; Jones, S.; Abbara, A.; Narayanaswamy, S.; Chia, P.; Papadopoulou, D.; et al. The effects of kisspeptin on beta-cell function, serum metabolites and appetite in humans. Diabetes Obes. Metab. 2018, 20, 2800–2810. [Google Scholar] [CrossRef]
- Zhu, C.; Kim, K.; Wang, X.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018, 10, eaat0344. [Google Scholar] [CrossRef]
- Mederacke, I.; Dapito, D.H.; Affo, S.; Uchinami, H.; Schwabe, R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. [Google Scholar] [CrossRef]
- Blake, A.; Dragan, M.; Tirona, R.G.; Hardy, D.B.; Brackstone, M.; Tuck, A.B.; Babwah, A.V.; Bhattacharya, M. G protein-coupled KISS1 receptor is overexpressed in triple negative breast cancer and promotes drug resistance. Sci. Rep. 2017, 7, 46525. [Google Scholar] [CrossRef]
- Dragan, M.; Nguyen, M.U.; Guzman, S.; Goertzen, C.; Brackstone, M.; Dhillo, W.S.; Bech, P.R.; Clarke, S.; Abbara, A.; Tuck, A.B.; et al. G protein-coupled kisspeptin receptor induces metabolic reprograming and tumorigenesis in estrogen receptor-negative breast cancer. Cell Death Dis. 2020, 11, 106. [Google Scholar] [CrossRef]
- Cvetkovic, D.; Dragan, M.; Leith, S.J.; Mir, Z.M.; Leong, H.S.; Pampillo, M.; Lewis, J.D.; Babwah, A.V.; Bhattacharya, M. KISS1R induces invasiveness of estrogen receptor-negative human mammary epithelial and breast cancer cells. Endocrinology 2013, 154, 1999–2014. [Google Scholar] [CrossRef]
- Niki, T.; De Bleser, P.J.; Xu, G.; Van Den Berg, K.; Wisse, E.; Geerts, A. Comparison of glial fibrillary acidic protein and desmin staining in normal and CCl4-induced fibrotic rat livers. Hepatology 1996, 23, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Kim, M.J.; Park, S.Y.; Kim, J.S.; Lim, W.; Nam, J.S.; Sheen, Y.Y. TIMP-1 mediates TGF-beta-dependent crosstalk between hepatic stellate and cancer cells via FAK signaling. Sci. Rep. 2015, 5, 16492. [Google Scholar] [CrossRef]
- Yoshida, K.; Matsuzaki, K.; Murata, M.; Yamaguchi, T.; Suwa, K.; Okazaki, K. Clinico-Pathological Importance of TGF-beta/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis. Cancers 2018, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ju, H.L.; Chung, S.I.; Cho, K.J.; Eun, J.W.; Nam, S.W.; Han, K.W.; Calvisi, D.F.; Ro, S.W. Transforming Growth Factor-beta Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017, 153, 1378–1391.e1376. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Grillo, A.R.; Brun, P.; Macchi, V.; Stefani, A.; Signori, S.; Buda, A.; Fabris, P.; Giordani, M.T.; De Caro, R.; et al. Snail1 transcription factor is a critical mediator of hepatic stellate cell activation following hepatic injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G316–G326. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.J.; Wang, Z.; Mobley, L.; Khosravi, D.; Fujii, N.; Navenot, J.M.; Peiper, S.C. Physical association of GPR54 C-terminal with protein phosphatase 2A. Biochem. Biophys. Res. Commun. 2008, 377, 1067–1071. [Google Scholar] [CrossRef]
- Rizvi, F.; Siddiqui, R.; DeFranco, A.; Homar, P.; Emelyanova, L.; Holmuhamedov, E.; Ross, G.; Tajik, A.J.; Jahangir, A. Simvastatin reduces TGF-beta1-induced SMAD2/3-dependent human ventricular fibroblasts differentiation: Role of protein phosphatase activation. Int. J. Cardiol. 2018, 270, 228–236. [Google Scholar] [CrossRef]
- Millar, R.P.; Babwah, A.V. KISS1R: Hallmarks of an Effective Regulator of the Neuroendocrine Axis. Neuroendocrinology 2015, 101, 193–210. [Google Scholar] [CrossRef]
- Patel, B.; Koysombat, K.; Mills, E.G.; Tsoutsouki, J.; Comninos, A.N.; Abbara, A.; Dhillo, W.S. The Emerging Therapeutic Potential of Kisspeptin and Neurokinin B. Endocr. Rev. 2024, 45, 30–68. [Google Scholar] [CrossRef]
- Radovick, S.; Babwah, A.V. Regulation of Pregnancy: Evidence for Major Roles by the Uterine and Placental Kisspeptin/KISS1R Signaling Systems. Semin. Reprod. Med. 2019, 37, 182–190. [Google Scholar] [CrossRef]
- Hauge-Evans, A.C.; Richardson, C.C.; Milne, H.M.; Christie, M.R.; Persaud, S.J.; Jones, P.M. A role for kisspeptin in islet function. Diabetologia 2006, 49, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Bowe, J.E.; Foot, V.L.; Amiel, S.A.; Huang, G.C.; Lamb, M.W.; Lakey, J.; Jones, P.M.; Persaud, S.J. GPR54 peptide agonists stimulate insulin secretion from murine, porcine and human islets. Islets 2012, 4, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Dewyse, L.; Reynaert, H.; van Grunsven, L.A. Best Practices and Progress in Precision-Cut Liver Slice Cultures. Int. J. Mol. Sci. 2021, 22, 7137. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Asami, T.; Nishizawa, N.; Matsui, H.; Nishibori, K.; Ishibashi, Y.; Horikoshi, Y.; Nakayama, M.; Matsumoto, S.-I.; Tarui, N.; Yamaguchi, M.; et al. Design, synthesis, and biological evaluation of novel investigational nonapeptide KISS1R agonists with testosterone-suppressive activity. J. Med. Chem. 2013, 56, 8298–8307. [Google Scholar] [CrossRef]
- Kostadinova, R.; Strobel, S.; Chen, L.; Fiaschetti-Egli, K.; Gadient, J.; Pawlowska, A.; Petitjean, J.; Bieri, M.; Thoma, E.; Petitjean, M. Digital pathology with artificial intelligence analysis provides insight to the efficacy of anti-fibrotic compounds in human 3D MASH model. Sci. Rep. 2024, 14, 5885. [Google Scholar] [CrossRef]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic Reticulum Stress in Hepatic Stellate Cells Promotes Liver Fibrosis via PERK-Mediated Degradation of HNRNPA1 and Up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193.e188. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Q.; Li, Z.; Zhang, R.; Jia, C.; Yang, Z.; Zhao, H.; Ya, S.; Mao, R.; Ailijiang, T.; et al. GP73 promotes epithelial-mesenchymal transition and invasion partly by activating TGF-beta1/Smad2 signaling in hepatocellular carcinoma. Carcinogenesis 2018, 39, 900–910. [Google Scholar] [CrossRef]
- Liu, M.; Peng, P.; Wang, J.; Wang, L.; Duan, F.; Jia, D.; Ruan, Y.; Gu, J. RACK1-mediated translation control promotes liver fibrogenesis. Biochem. Biophys. Res. Commun. 2015, 463, 255–261. [Google Scholar] [CrossRef]
- Song, Y.; Kim, S.; Heo, J.; Shum, D.; Lee, S.Y.; Lee, M.; Kim, A.-R.; Seo, H.R. Identification of hepatic fibrosis inhibitors through morphometry analysis of a hepatic multicellular spheroids model. Sci. Rep. 2021, 11, 10931. [Google Scholar] [CrossRef]
- Schaefer, J.; Chadchan, S.B.; George, A.F.; Roan, N.R.; Bhattacharya, M.; Kommagani, R.; Babwah, A.V. Human endometrial KISS1R inhibits stromal cell decidualization in a manner associated with a reduction in ESR1 levels. bioRxiv 2022, bioRxiv:2011.2020.517219. [Google Scholar]
- Dudek, M.; Kolodziejski, P.A.; Pruszynska-Oszmalek, E.; Sassek, M.; Ziarniak, K.; Nowak, K.W.; Sliwowska, J.H. Effects of high-fat diet-induced obesity and diabetes on Kiss1 and GPR54 expression in the hypothalamic-pituitary-gonadal (HPG) axis and peripheral organs (fat, pancreas and liver) in male rats. Neuropeptides 2016, 56, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age (Years) | Sex | Disease Background | Pathological Diagnosis | Fibrosis Stage |
|---|---|---|---|---|---|
| 1 | 76 | Male | Patient has underlying HBV diagnosed with a mass in segment 5/6 of the liver (consistent with HCC) | Mild portal and mild interface hepatitis, compatible with the patient’s history of hepatitis B | F1 |
| 2 | 66 | Male | HCC (HBV-infected chronic hepatitis) | Mild portal chronic inflammation and periportal fibrosis with bridging fibrous septa. No interface hepatitis or lobular inflammation and steatosis. | F2 |
| 3 | 53 | Male | HCC (HCV-infected chronic hepatitis) | Nodular liver parenchyma without significant portal or lobular inflammation. No significant steatosis. | F4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, K.; Bhattacharya, D.; Shams, S.G.E.; Izarraras, K.; Hart, T.; Mayfield, B.; Blaszczyk, M.B.; Zhou, Z.; Pajvani, U.B.; Friedman, S.L.; et al. Kisspeptin Alleviates Human Hepatic Fibrogenesis by Inhibiting TGFβ Signaling in Hepatic Stellate Cells. Cells 2024, 13, 1651. https://doi.org/10.3390/cells13191651
Prasad K, Bhattacharya D, Shams SGE, Izarraras K, Hart T, Mayfield B, Blaszczyk MB, Zhou Z, Pajvani UB, Friedman SL, et al. Kisspeptin Alleviates Human Hepatic Fibrogenesis by Inhibiting TGFβ Signaling in Hepatic Stellate Cells. Cells. 2024; 13(19):1651. https://doi.org/10.3390/cells13191651
Chicago/Turabian StylePrasad, Kavita, Dipankar Bhattacharya, Shams Gamal Eldin Shams, Kimberly Izarraras, Tia Hart, Brent Mayfield, Maryjka B. Blaszczyk, Zhongren Zhou, Utpal B. Pajvani, Scott L. Friedman, and et al. 2024. "Kisspeptin Alleviates Human Hepatic Fibrogenesis by Inhibiting TGFβ Signaling in Hepatic Stellate Cells" Cells 13, no. 19: 1651. https://doi.org/10.3390/cells13191651