
Oncological Aspects of Lysosomal Storage Diseases


Abstract
1. Introduction
- -
- chronic, progressive nature of the disease;
- -
- in the neonatal period, often symptoms not very pronounced;
- -
- sometimes hydrops fetalis;
- -
- dysmorphic facial features;
- -
- skeletal changes (especially dysostosis multiplex, Erlenmayer’s beaker type deformity of long bones);
- -
- skin lesions (e.g., angiokeratoma);
- -
- muscular hypotonia;
- -
- delayed motor and then mental development;
- -
- progressive organomegaly (liver, spleen, heart);
- -
- features of leukodystrophy (ataxia, hyperactivity, spasticity, paresis);
- -
- cortical and subcortical lesions;
- -
- pyramidal and extrapyramidal symptoms;
- -
- polyneuropathy;
- -
- cerebellar ataxia;
- -
- epilepsy of unclear etiology;
- -
- hearing loss, vision loss;
- -
- cherry red spot on the fundus, corneal opacity, lens subluxation [29].
2. Gaucher Disease
- -
- Type 1—non-neuronopathic, bone involvement (bone pains, osteopenia, pathological fractures, deformity of long bones known as ‘Erlenmayer flask’ bones).
- -
- Type 2—neuronopathic, severe lung involvement.
- -
- Type 3—neuronopathic, slowly progressive, supranuclear horizontal gaze palsy.
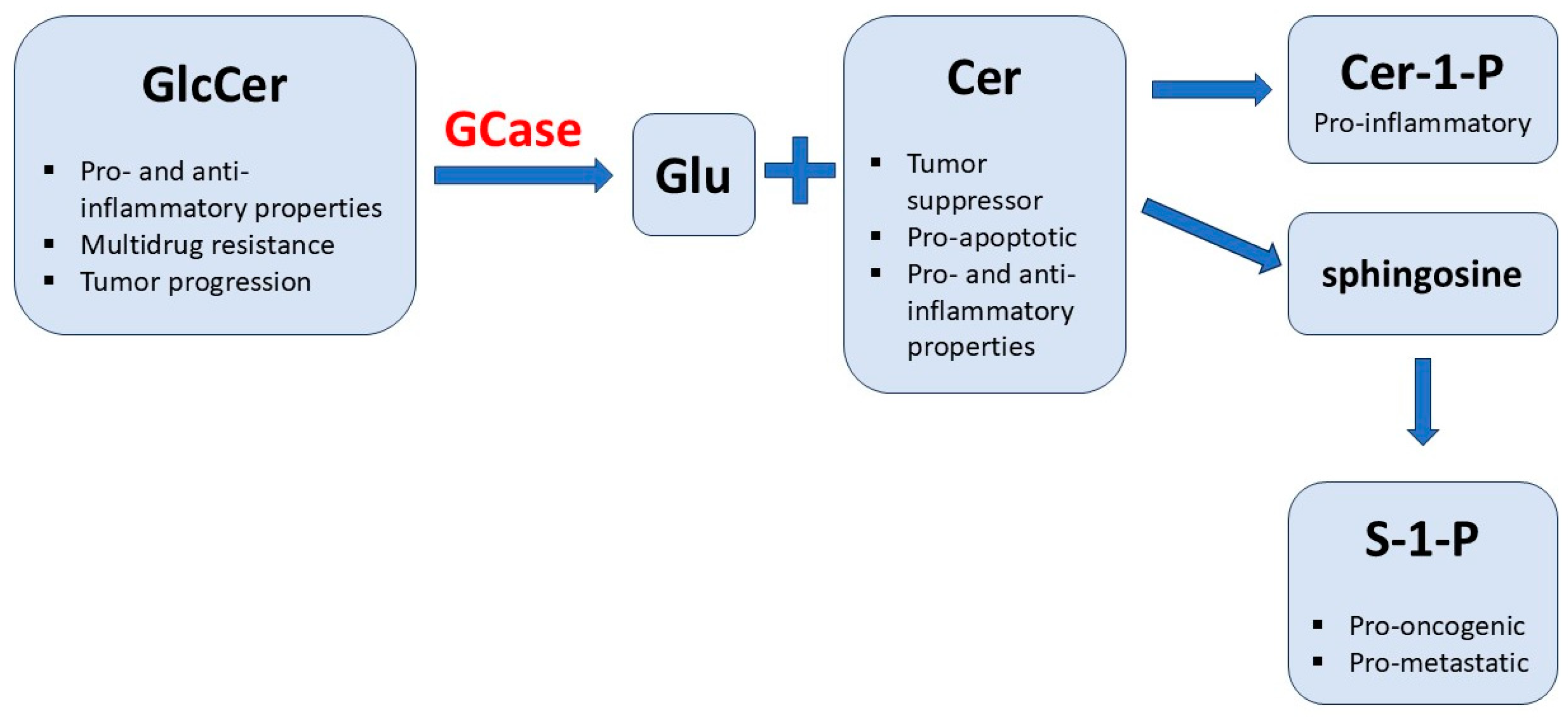
Theoretical Relationship between GD and Cancers
3. Alfa-Mannosidosis
- -
- facial features (e.g., coarse facial features, macrocephaly, prominent forehead, highly arched brows, depressed nasal bridge, widely spaced teeth, macroglossia, prognathism);
- -
- skeletal abnormalities;
- -
- hearing loss;
- -
- frequent infections;
- -
- developmental delay;
- -
- intellectual disability;
- -
- ataxia [59].
- -
- Type 1 is a benign form, with onset after the age of 10 years and very slow disease progression; skeletal abnormalities are absent.
- -
- Type 2 is a moderate, slowly progressing form, with onset before the age of 10 years; skeletal abnormalities are present, and ataxia can be revealed by age 20 to 30 years.
- -
- Type 3 is the severe form, with onset in early infancy, skeletal abnormalities are present, and early death may occur due to primary central nervous system involvement or myopathy [http://www.omim.org/entry/248500 (accessed on 1 August 2024)].
4. Fabry Disease
- -
- 7 cases of breast cancer in females;
- -
- renal cell carcinoma in 2 males;
- -
- 4 cases with melanoma; incidence rate ratio of 3.1;
- -
- 5 cases with urological malignancies (1 with bladder cancer, 1 with ureteric cancer; incidence rate ratio of 2.7 and 3 cases with renal cancer; incidence rate ratio of 4.3).
- -
- 5 cases with growths in neurological tissues (including two benign neoplasms of the meninges; the incidence rate ratio was 12);
- -
- 5 cases with colon polyps;
- -
- 4 cases with benign breast lesions;
- -
- 3 cases with atypical moles;
- -
- 2 cases with benign renal lesions;
- -
- 2 cases with cervical intraepithelial neoplasm.
5. Niemann-Pick Disease Type A/B (Acid Sphingomyelinase Deficiency, ASMD)
6. Sialidosis
7. Carriers of Lysosomal Diseases
- (1)
- CLN3-osteosarcoma (prevalence ratio 52.0);
- (2)
- CLN3-myeloproliferative neoplasm (32.5);
- (3)
- GUSB-pancreas neuroendocrine carcinoma (61.8);
- (4)
- NEU1-uterine cervix squamous cell carcinoma (46.4);
- (5)
- NEU1-urinary bladder transitional cell carcinoma (36.3);
- (6)
- SGSH-myeloproliferative neoplasm (51.1);
- (7)
- SGSH-pancreas neuroendocrine carcinoma (30.9);
- (8)
- SGSH-osteosarcoma (30.7);
- (9)
- SGSH-breast invasive ductal carcinoma (16.5);
- (10)
- SGSH-pancreas adenocarcinoma (15.4).
7.1. Gene CLN3 and Neuronal Ceroid Lipofuscinosis Type 3 (CLN3)
- -
- motor dysfunction due to extrapyramidal, slight pyramidal, and cerebellar disturbances;
- -
- slowly developing mental retardation;
- -
- dementia becomes profound later in the course of the disease;
- -
- disordered speech leading to dysarthria;
- -
7.2. Gene GUSB and Mucopolysaccharidosis Type VII (Sly Disease)
7.3. Gene SGSH and Mucopolysaccharidosis Type IIIA (Sanfilippo Syndrome Type A)
8. Conclusions
- -
- Most often, cancer is found in individuals affected with Gaucher, Fabry, Niemann-Pick type A and B diseases, alfa-mannosidosis, and sialidosis.
- -
- Carriers of pathogenic variants in LSD genes and patients affected with some lysosomal diseases are at an increased risk for cancer, especially carriers of variants in CLN3, SGSH, GUSB, NEU1 genes;
- -
- There are controversial results from studies on the frequency of oncological diseases in LSD patients.
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- de Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. VI Intracellular distribution patterns of enzymes in rat liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef]
- Hall, J.L.; Flowers, T.J.; Roberts, R.M. Lizosomy. In Struktura i Metabolizm Komórek Roślinnych, 1st ed.; PWN: Warszawa, Poland, 1982; pp. 526–543. [Google Scholar]
- de Duve, C. Lysosomes revisited. Eur. J. Biochem. 1983, 137, 391–397. [Google Scholar] [CrossRef]
- Luebke, T.; Lobel, P.; Sleat, D.E. Proteomics of the lysosome. Biochim. Biophys. Acta 2009, 1793, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.W.; Bagshaw, R.D.; Mahuran, D.J. The integral membrane of lysosomes: Its proteins and their roles in disease. J. Proteom. 2009, 72, 23–33. [Google Scholar] [CrossRef]
- Kundra, R.; Kornfeld, S. Asparagine-linked oligosaccharides protect Lamp-1 and Lamp-2 from intracellular proteolysis. J. Biol. Chem. 1999, 274, 31039–31046. [Google Scholar] [CrossRef] [PubMed]
- Sly, W.S.; Fischer, H.D. The phosphomannosyl recognition system for intracellular and intercellular transport of lysosomal enzymes. J. Cell Biochem. 1982, 18, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Dahms, N.M.; Lobel, P.; Kornfeld, S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J. Biol. Chem. 1989, 264, 12115–12118. [Google Scholar] [CrossRef] [PubMed]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate independent targeting of β-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Zhao, Q.; Gao, S.M.; Wang, M.C. Molecular Mechanisms of Lysosome and Nucleus Communication. Trends Biochem. Sci. 2020, 45, 978–991. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Baixauli, F.; Mittelbrunn, M. Interorganelle Communication between Mitochondria and the Endolysosomal System. Front. Cell Dev. Biol. 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Hers, H.G. Type II glycogenosis—A lysosomal disease. In Proceedings of the 1st Meeting of the Federation of European Biochemical Societes, London, UK, 23–25 March 1964. [Google Scholar]
- Hers, H.G. Inborn lysosomal diseases. Gastroenterology 1965, 48, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Jennings, J.J., Jr.; Rbaibi, Y.; Chu, C.T. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy 2007, 3, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef]
- Kuk, M.U.; Lee, Y.H.; Kim, J.W.; Hwang, S.Y.; Park, J.T.; Park, S.C. Potential Treatment of Lysosomal Storage Disease through Modulation of the Mitochondrial-Lysosomal Axis. Cells 2021, 10, 420. [Google Scholar] [CrossRef]
- Walkley, S.U. Pathogenic mechanisms in lysosomal disease: A reappraisal of the role of the lysosome. Acta Pediatr. 2007, S96, 26–32. [Google Scholar] [CrossRef]
- Amaral, O.; Martins, M.; Oliveira, A.R.; Duarte, A.J.; Mondragão-Rodrigues, I.; Macedo, M.F. The Biology of Lysosomes: From Order to Disorder. Biomedicines 2023, 11, 213. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Settembre, C.; Perera, R.M. Lysosomes as coordinators of cellular catabolism, metabolic signalling and organ physiology. Nat. Rev. Mol. Cell Biol. 2024, 25, 223–245. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef]
- Gerasimenko, J.; Gerasimenko, O.; Petersen, O. Membrane repair: Ca2+-elicited lysosomal exocytosis. Curr. Biol. 2001, 11, R971–R974. [Google Scholar] [CrossRef]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primer 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Meikle, P.J.; Hopwood, J.J. Epidemiology of lysosomal storage diseases: An overview. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Chapter 2. Available online: https://www.ncbi.nlm.nih.gov/books/NBK11603/ (accessed on 1 August 2024).
- Ferreira, C.R. The burden of rare diseases. Am. J. Med. Genet. 2019, 179, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Herder, M. What is the purpose of the orphan drug act? PLoS Med. 2017, 14, e1002191. [Google Scholar] [CrossRef]
- Wraith, J.E.; Beck, M. Clinical aspects and Clinical diagnosis. In Lysosomal Storage Disorders, A Practical Guide, 1st ed.; Mehta, A., Winchester, B., Eds.; Wiley-Blackwell: Cambridge, UK, 2012. [Google Scholar]
- Kos, J.; Mitrović, A.; Perišić Nanut, M.; Pišlar, A. Lysosomal peptidases-intriguing roles in cancer progression and neurodegeneration. FEBS Open Bio. 2022, 12, 708–738. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Yang, Z.Y.; Wang, D.; Yang, X.Y.; Wang, J.; Li, L.; Wen, Q.; Gao, L.; Bian, X.W.; Yu, S.C. The role of lysosomes in cancer development and progression. Cell Biosci. 2020, 10, 131. [Google Scholar] [CrossRef]
- Machado, E.R.; Annunziata, I.; van de Vlekkert, D.; Grosveld, G.C.; d’Azzo, A. Lysosomes and Cancer Progression: A Malignant Liaison. Front. Cell Dev. Biol. 2021, 9, 642494. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B. Classification of Lysosomal Storage Diseases. In Lysosomal Storage Disorders, A Practical Guide, 1st ed.; Mehta, A., Winchester, B., Eds.; Wiley-Blackwell: Cambridge, UK, 2012; pp. 37–46. [Google Scholar]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef]
- Rosenbloom, B.E.; Cappellini, M.D.; Weinreb, N.J.; Dragosky, M.; Revel-Vilk, S.; Batista, J.L.; Sekulic, D.; Mistry, P.K. Cancer risk and gammopathies in 2123 adults with Gaucher disease type 1 in the International Gaucher Group Gaucher Registry. Am. J. Hematol. 2022, 97, 1337–1347. [Google Scholar] [CrossRef]
- Landgren, O.; Turesson, I.; Gridley, G.; Caporaso, N.E. Risk of Malignant Disease among 1525 Adult Male US Veterans with Gaucher Disease. Arch. Intern. Med. 2007, 167, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Choy, F.Y.; Campbell, T.N. Gaucher disease and cancer: Concept and controversy. Int. J. Cell Biol. 2011, 2011, 150450. [Google Scholar] [CrossRef] [PubMed]
- Taddei, T.H.; Kacena, K.A.; Yang, M.; Yang, R.; Malhotra, A.; Boxer, M.; Aleck, K.A.; Rennert, G.; Pastores, G.M.; Mistry, P.K. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 2009, 84, 208–214. [Google Scholar] [CrossRef]
- Hughes, D. Gaucher Disease: Hematologic and Oncologic Implications. Clin. Adv. Hematol. Oncol. 2011, 9, 771–772. [Google Scholar]
- Bozdag, S.C.; Topcuoglu, P.; Kuzu, I.; Arat, M. Acute lymphoblastic leukemia during enzyme replacement therapy in type 1 Gaucher’s disease. Clin. Adv. Hematol. Oncol. 2013, 11, 251–252. [Google Scholar]
- Lo, S.M.; Stein, P.; Mullaly, S.; Bar, M.; Jain, D.; Pastores, G.M.; Mistry, P.K. Expanding spectrum of the association between Type 1 Gaucher disease and cancers: A series of patients with up to 3 sequential cancers of multiple types—Correlation with genotype and phenotype. Am. J. Hematol. 2010, 85, 340–345. [Google Scholar] [CrossRef]
- Mishra, S.; Hajra, S.; Arathi, K.; Gupta, A.K. Gaucher’s disease with acute lymphoblastic leukemia: A rare co-occurrence. J. Hematol. Allied Sci. 2022, 2, 96–98. [Google Scholar] [CrossRef]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. S1), 72–81. [Google Scholar]
- de Fost, M.; vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Haussinger, D.; Hollak, C.E. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef]
- Monge, J.; Chadburn, A.; Gergis, U. Synchronous multiple myeloma and Gaucher disease. Hematol. Oncol. Stem Cell Ther. 2020, 13, 42–45. [Google Scholar] [CrossRef]
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E. Malignancies and monoclonal gammopathy in Gaucher disease; A systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Ridova, N.; Trajkova, S.; Popova-Labachevska, M.; Stojanovska-Jakimovska, S.; Nikolov, F.; Panovska-Stavridis, I. Early-Onset Colorectal Cancer in a Young Woman with Type 1 Gaucher Disease. Eur. J. Case Rep. Intern. Med. 2022, 9, 003412. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Hughes, D.A. Lysosomal Storage Disorders and Malignancy. Diseases 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Dubot, P.; Astudillo, L.; Therville, N.; Sabourdy, F.; Stirnemann, J.; Levade, T.; Andrieu-Abadie, N. Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses. Cancers 2020, 12, 475. [Google Scholar] [CrossRef]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef]
- Nandakumar, S.; Vijayan, B.; Kishore, A.; Thekkuveettil, A. Autophagy enhancement is rendered ineffective in presence of synuclein in melanoma cells. J. Cell Commun. Signal. 2017, 11, 381–394. [Google Scholar] [CrossRef]
- Wahabi, K.; Perwez, A.; Rizvi, M.A. Parkin in Parkinson’s Disease and Cancer: A Double-Edged Sword. Mol. Neurobiol. 2018, 55, 6788–6800. [Google Scholar] [CrossRef]
- Barth, B.M.; Shanmugavelandy, S.S.; Tacelosky, D.M.; Kester, M.; Morad, S.A.; Cabot, M.C. Gaucher’s disease and cancer: A sphingolipid perspective. Crit. Rev. Oncog. 2013, 18, 221–234. [Google Scholar] [CrossRef]
- Mistry, P.K.; Weinreb, N.J.; Brady, R.O.; Grabowski, G.A. Gaucher disease: Resetting the clinical and scientific agenda. Am. J. Hematol. 2009, 84, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Humble, R.; Collart, M. Oligosaccharides in urines of patients with glycoprotein storage diseases. I. Rapid detection by thin-layer chromatography. Clin. Chim. Acta 1975, 60, 143–145. [Google Scholar] [CrossRef]
- Bruggink, C.; Poorthuis, B.J.; Deelder, A.M.; Wuhrer, M. Analysis of urinary oligosaccharides in lysosomal storage disorders by capillary high-performance anion-exchange chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1671–1683. [Google Scholar] [CrossRef]
- Malm, D.; Nilssen, Ø. Alpha-Mannosidosis. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1396/ (accessed on 1 August 2024).
- Zanetta, J.P.; Bonaly, R.; Maschke, S.; Strecker, G.; Michalski, J.C. Hypothesis: Immunodeficiencies in α-mannosidosis, mycosis, AIDS and cancer—A common mechanism of inhibition of the function of the lectin interleukin 2 by oligomannosides. Glycobiology 1998, 8, 6–10. [Google Scholar] [CrossRef]
- Maschke, S.; Robert, J.; Coindre, J.M.; Kuchler, S.; Vincendon, G.; Zanetta, J.P. Malignant cells have increased levels of common glycoprotein ligands of the endogenous cerebellar soluble lectin. Eur. J. Cell. Biol. 1993, 62, 163–172. [Google Scholar] [PubMed]
- Hennermann, J.B.; Raebel, E.M.; Donà, F.; Jacquemont, M.L.; Cefalo, G.; Ballabeni, A.; Malm, D. Mortality in patients with alpha-mannosidosis: A review of patients’ data and the literature. Orphanet. J. Rare Dis. 2022, 17, 287. [Google Scholar] [CrossRef]
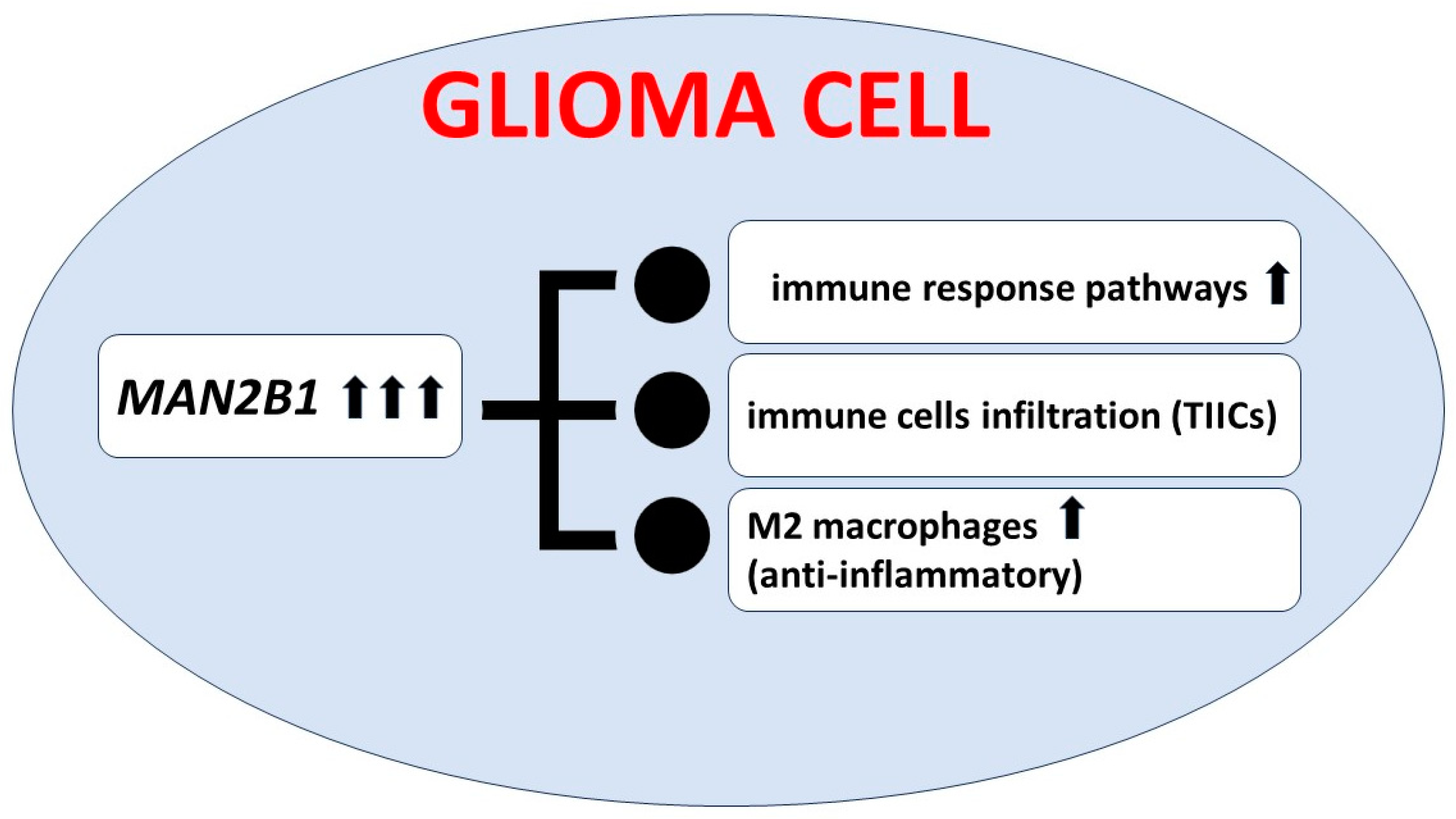
- Lin, X.; Liu, H.; Zhao, H.; Xia, S.; Li, Y.; Wang, C.; Huang, Q.; Wanggou, S.; Li, X. Immune Infiltration Associated MAN2B1 Is a Novel Prognostic Biomarker for Glioma. Front. Oncol. 2022, 12, 842973. [Google Scholar] [CrossRef]
- Bird, S.; Hadjimichael, E.; Mehta, A.; Ramaswami, U.; Hughes, D. Fabry disease and incidence of cancer. Orphanet. J. Rare Dis. 2017, 12, 150. [Google Scholar] [CrossRef]
- Mehta, A.; Hughes, D.A. Fabry Disease. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1292/ (accessed on 1 August 2024).
- Cassiman, D.; Claes, K.; Lerut, E.; Oyen, R.; Joniau, S.; van Damme, B.; Jaeken, J. Bilateral renal cell carcinoma development in long-term Fabry disease. J. Inherit. Metab. Dis. 2007, 30, 830–831. [Google Scholar] [CrossRef]
- Blanco, J.; Herrero, J.; Arias, L.F.; Garcia-Miralles, N.; Gamez, C.; Barrientos, A. Renal variant of Anderson-Fabry disease and bilateral renal cell carcinoma. Pathol. Res. Pract. 2005, 200, 857–860. [Google Scholar] [CrossRef]
- Pagni, F.; Pieruzzi, F.; Zannella, S.; di Giacomo, A.; Bovo, G.; Ferrario, F.; Torti, G.; Rivera, R.; Assi, E.; Viglione, F.; et al. Possible pathogenetic relationship between Fabry disease and renal cell carcinoma. Am. J. Nephrol. 2012, 36, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Tirelli, C.; Rondinone, O.; Italia, M.; Mira, S.; Belmonte, L.A.; De Grassi, M.; Guido, G.; Maggioni, S.; Mondoni, M.; Miozzo, M.R.; et al. The Genetic Basis, Lung Involvement, and Therapeutic Options in Niemann–Pick Disease: A Comprehensive Review. Biomolecules 2024, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Mauhin, W.; Levade, T.; Vanier, M.T.; Froissart, R.; Lidove, O. Prevalence of Cancer in Acid Sphingomyelinase Deficiency. J. Clin. Med. 2021, 10, 5029. [Google Scholar] [CrossRef] [PubMed]
- Sabourdy, F.; Selves, J.; Astudillo, L.; Laurent, C.; Brousset, P.; Delisle, M.-B.; Therville, N.; Andrieu-Abadie, N.; Ségui, B.; Recher, C.; et al. Is active acid sphingomyelinase required for the antiproliferative response to rituximab? Blood 2011, 117, 3695–3696. [Google Scholar] [CrossRef]
- Cassiman, D.; Packman, S.; Bembi, B.; Ben Turkia, H.; Al-Sayed, M.; Schiff, M.; Imrie, J.; Mabe, P.; Takahashi, T.; Mengel, K.E.; et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol. Genet. Metab. 2016, 118, 206–213. [Google Scholar] [CrossRef]
- McGovern, M.M.; Wasserstein, M.P.; Bembi, B.; Giugliani, R.; Mengel, K.E.; Vanier, M.T.; Zhang, Q.; Peterschmitt, M.J. Prospective study of the natural history of chronic acid sphingomyelinase deficiency in children and adults: Eleven years of observation. Orphanet. J. Rare Dis. 2021, 16, 212. [Google Scholar] [CrossRef]
- Yagi, Y.; Machida, A.; Toru, S.; Kobayashi, T.; Uchihara, T. Sialidosis type I with neoplasms in siblings: The first clinical cases. Neurol. Sci. 2011, 32, 737–738. [Google Scholar] [CrossRef]
- Sawada, M.; Moriya, S.; Saito, S.; Shineha, R.; Satomi, S.; Yamori, T.; Tsuruo, T.; Kannagi, R.; Miyagi, T. Reduced sialidase expression in highly metastatic variants of mouse colon adenocarcinoma and retardation of their metastatic ability by sialidase overexpression. Int. J. Cancer 2002, 97, 180–185. [Google Scholar] [CrossRef]
- Yamanami, H.; Shiozaki, K.; Wada, T. Down-regulation of sialidase NEU4 may contribute to invasive properties of human colon cancers. Cancer Sci. 2007, 98, 299–307. [Google Scholar] [CrossRef]
- Shin, J.; Kim, D.; Kim, H.L.; Choi, M.; Koh, Y.; Yoon, S.S. Oncogenic effects of germline variants in lysosomal storage disease genes. Genet. Med. 2019, 21, 2695–2705. [Google Scholar] [CrossRef] [PubMed]
- Cotman, S.L.; Staropoli, J.F. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post Golgi trafficking. Clin. Lipidol. 2012, 7, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.R. Juvenile neuronal ceroid lipofuscinosis (Batten disease): Current insights. Degener Neurol. Neuromuscul. Dis. 2016, 6, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Purzycka-Olewiecka, J.K.; Hetmańczyk-Sawicka, K.; Kmieć, T.; Szczęśniak, D.; Trubicka, J.; Krawczyński, M.; Pronicki, M.; Ługowska, A. Deterioration of visual quality and acuity as the first sign of ceroid lipofuscinosis type 3 (CLN3), a rare neurometabolic disease. Metab. Brain Dis. 2023, 38, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Boustany, R.M.N. Lysosomal storage diseases—The horizon expands. Nat. Rev. Neurol. 2013, 9, 583–598. [Google Scholar] [CrossRef] [PubMed]
- El-Sitt, S.; Soueid, J.; Maalouf, K.; Makhoul, N.; Al Ali, J.; Makoukji, J.; Asser, B.; Daou, D.; Harati, H.; Boustany, R.M. Exogenous Galactosylceramide as Potential Treatment for CLN3 Disease. Ann. Neurol. 2019, 86, 729–742. [Google Scholar] [CrossRef]
- Shematorova, E.K.; Shpakovski, D.G.; Chernysheva, A.D.; Shpakovski, G.V. Molecular mechanisms of the juvenile form of Batten disease: Important role of MAPK signaling pathways (ERK1/ERK2, JNK and p38) in pathogenesis of the malady. Biol. Direct. 2018, 13, 1–9. [Google Scholar] [CrossRef]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Disease | Deficient Enzyme or Protein | Inheritance | Gene |
|---|---|---|---|
| Sphingolipidoses | |||
| Fabry disease | α-Galactosidase A | AR | GLA |
| Gaucher disease -types I, II and III | β-Glucosidase | AR | GBA1 |
| Gaucher disease, atypical | Saposin C defect | AR | PSAP |
| Niemann–Pick A and B | Sphingomyelinase | AR | SMPD1 |
| Mucopolysaccharidoses (MPS) | |||
| MPS IIIA, Sanfilippo A | Heparan N-sulfatase | AR | SGSH |
| MPS VII, Sly | β-Glucuronidase | AR | GUSB |
| Glycoproteinoses (Oligosaccharidoses) | |||
| α-Mannosidosis | α-D-Mannosidase | AR | MAN2B1 |
| Sialidosis I/II (Mucolipidosis I) | Neuraminidase (Sialidase 1) | AR | NEU1 |
| Neuronal Ceroid Lipofuscinoses (CLNS) | |||
| CLN3 disease (Juvenile NCL, JNCL Batten disease) | CLN3, lysosomal, and/or Golgi transmembrane protein | AR | CLN3 |
| Gene Symbol | Lysosomal Storage Disease | Associated Histological Cohort | Relative Prevalence Ratio * |
|---|---|---|---|
| ARSA | Metachromatic leukodystrophy | Kidney-RCC 1 | 4.8 |
| CLN3 | Neuronal ceroid lipofuscinosis type 3 | Bone-Osteosarc | 52.0 |
| Myeloid-MPN | 32.5 | ||
| Breast-AdenoCA | 11.7 | ||
| Ovary-AdenoCA | 4.1 | ||
| Pan-Cancer | 3.1 | ||
| GAA | Pompe disease | Myeloid-MPN | 9.9 |
| Panc-Endocrine | 2.6 | ||
| CNS-Medullo | 1.5 | ||
| GALC | Krabbe disease | Skin-Melanoma | 2.3 |
| GBA1 | Gaucher disease | Pan-Cancer | 1.3 |
| GNPTAB | Mucolipidosis type II/III; pseudo-Hurler polydystrophy; I-cell disease | ColoRect-AdenoCA | 10.1 |
| Panc-Endocrine | 15.5 | ||
| Uterus-AdenoCA | 14.2 | ||
| GNS | Mucopolysaccharidosis type III D | Kidney-RCC | 4.4 |
| GUSB | Mucopolysaccharidosis type VII | Panc-Endocrine | 61.8 |
| HEXA | Gangliosidosis GM2; Tay-Sachs disease; Sandhoff disease | Breast-AdenoCA | 6.8 |
| Pan-Cancer | 2.2 | ||
| HEXB | Liver-HCC | 3.5 | |
| HGSNAT | Mucopolysaccharidosis type III C | CNS-Medullo | 8.1 |
| Head-SCC | 8.1 | ||
| Ovary-AdenoCA | 6.2 | ||
| Kidney-RCC | 4.8 | ||
| IDUA | Mucopolysaccharidosis type I | Bone-Osteosarc | 11.9 |
| Panc-AdenoCA | 7.2 | ||
| IDS | Mucopolysaccharidosis type II; Hunter syndrome | Lymph-BNHL | 4.7 |
| Prost-AdenoCA | 2.5 | ||
| MAN2B1 | Alfa-mannosidosis | Panc-AdenoCA | 3.9 |
| NEU1 | sialidosis | Cervix-SCC | 46.4 |
| Bladder-TCC | 36. | ||
| NPC1 | Niemann-Pick disease type C | Eso-AdenoCA | 11.5 |
| Skin-Melanoma | 6.5 | ||
| CNS-Medullo | 5.9 | ||
| Lung-SCC | 5.9 | ||
| Ovary-AdenoCA | 3.8 | ||
| SGSH | Mucopolysaccharidosis type III A | Myeloid-MPN | 51.1 |
| Panc-Endocrine | 30.9 | ||
| Bone-Osteosarc | 30.7 | ||
| Breast-AdenoCA | 16.5 | ||
| Panc-AdenoCA | 15.4 | ||
| Pan-Cancer | 6.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ługowska, A. Oncological Aspects of Lysosomal Storage Diseases. Cells 2024, 13, 1664. https://doi.org/10.3390/cells13191664
Ługowska A. Oncological Aspects of Lysosomal Storage Diseases. Cells. 2024; 13(19):1664. https://doi.org/10.3390/cells13191664
Chicago/Turabian StyleŁugowska, Agnieszka. 2024. "Oncological Aspects of Lysosomal Storage Diseases" Cells 13, no. 19: 1664. https://doi.org/10.3390/cells13191664
APA StyleŁugowska, A. (2024). Oncological Aspects of Lysosomal Storage Diseases. Cells, 13(19), 1664. https://doi.org/10.3390/cells13191664