

SWI/SNF Complex in Vascular Smooth Muscle Cells and Its Implications in Cardiovascular Pathologies

 ,
,
Abstract
:1. Introduction
2. Roles of VSMCs from Physiology to Pathology
3. Epigenetic and Transcriptional Regulation of VSMC Plasticity in Health and Diseases
4. Chromatin Remodeling and SWI/SNF Complexes
5. SWI/SNF Complex in Cardiovascular Development
6. SMARCA4 and SMARCA2 in VSMC Biology and Cardiovascular Diseases
7. Roles of the SMARCD Family in VSMC
8. Concluding Remarks and Future Directions
Funding
Conflicts of Interest
References
- Pease, D.C.; Paule, W.J. Electron microscopy of elastic arteries; the thoracic aorta of the rat. J. Ultrastruct. Res. 1960, 3, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Geer, J.C.; McGill, H.C.; Strong, J.P. The fine structure of human atherosclerotic lesions. Am. J. Pathol. 1961, 38, 263–287. [Google Scholar] [PubMed]
- Still, W.J.; O’Neal, R.M. Electron microscopic study of experimental atherosclerosis in the rat. Am. J. Pathol. 1962, 40, 21–35. [Google Scholar] [PubMed]
- Parker, F. An Electron Microscopic Study of Experimental Atherosclerosis. Am. J. Pathol. 1960, 36, 19–53. [Google Scholar]
- Basatemur, G.L.; Jorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Landerholm, T.E.; Dong, X.R.; Lu, J.; Belaguli, N.S.; Schwartz, R.J.; Majesky, M.W. A role for serum response factor in coronary smooth muscle differentiation from proepicardial cells. Development 1999, 126, 2053–2062. [Google Scholar] [CrossRef]
- Wang, D.; Chang, P.S.; Wang, Z.; Sutherland, L.; Richardson, J.A.; Small, E.; Krieg, P.A.; Olson, E.N. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 2001, 105, 851–862. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [PubMed]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef]
- Sibinga, N.E.; Foster, L.C.; Hsieh, C.M.; Perrella, M.A.; Lee, W.S.; Endege, W.O.; Sage, E.H.; Lee, M.E.; Haber, E. Collagen VIII is expressed by vascular smooth muscle cells in response to vascular injury. Circ. Res. 1997, 80, 532–541. [Google Scholar] [CrossRef]
- Niklason, L.E.; Lawson, J.H. Bioengineered human blood vessels. Science 2020, 370, eaaw8682. [Google Scholar] [CrossRef]
- Cook, C.L.; Weiser, M.C.; Schwartz, P.E.; Jones, C.L.; Majack, R.A. Developmentally timed expression of an embryonic growth phenotype in vascular smooth muscle cells. Circ. Res. 1994, 74, 189–196. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jørgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef]
- Mizrak, D.; Zhao, Y.; Feng, H.; Macaulay, J.; Tang, Y.; Sultan, Z.; Zhao, G.; Guo, Y.; Zhang, J.; Yang, B.; et al. Single-Molecule Spatial Transcriptomics of Human Thoracic Aortic Aneurysms Uncovers Calcification-Related CARTPT-Expressing Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Majesky, M.W.; Horita, H.; Ostriker, A.; Lu, S.; Regan, J.N.; Bagchi, A.; Dong, X.R.; Poczobutt, J.; Nemenoff, R.A.; Weiser-Evans, M.C. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4. Circ. Res. 2017, 120, 296–311. [Google Scholar] [CrossRef]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Davies, J.D.; Carpenter, K.L.H.; Challis, I.R.; Figg, N.L.; McNair, R.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Adipocytic differentiation and liver x receptor pathways regulate the accumulation of triacylglycerols in human vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 3911–3919. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Shanahan, C.M.; Weissberg, P.L. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 379–388. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef]
- Miano, J.M.; Fisher, E.A.; Majesky, M.W. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021, 143, 2110–2116. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Chen, Y.E. Vascular cell lineage determination and differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1467–1468. [Google Scholar] [CrossRef]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Ritchie, R.P.; Huang, H.; Zhang, J.; Chen, Y.E. Smooth muscle cell differentiation in vitro: Models and underlying molecular mechanisms. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Minty, A.; Kedes, L. Upstream regions of the human cardiac actin gene that modulate its transcription in muscle cells: Presence of an evolutionarily conserved repeated motif. Mol. Cell. Biol. 1986, 6, 2125–2136. [Google Scholar] [CrossRef]
- Sartorelli, V.; Kurabayashi, M.; Kedes, L. Muscle-specific gene expression. A comparison of cardiac and skeletal muscle transcription strategies. Circ. Res. 1993, 72, 925–931. [Google Scholar] [CrossRef]
- Boxer, L.M.; Prywes, R.; Roeder, R.G.; Kedes, L. The sarcomeric actin CArG-binding factor is indistinguishable from the c-fos serum response factor. Mol. Cell. Biol. 1989, 9, 515–522. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004, 428, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fang, H.; Zhou, J.; Herring, B.P. A novel role of Brg1 in the regulation of SRF/MRTFA-dependent smooth muscle-specific gene expression. J. Biol. Chem. 2007, 282, 25708–25716. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, M.; Fang, H.; El-Mounayri, O.; Rodenberg, J.M.; Imbalzano, A.N.; Herring, B.P. The SWI/SNF chromatin remodeling complex regulates myocardin-induced smooth muscle-specific gene expression. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zhao, Y.; Lu, H.; Chang, Z.; Liu, H.; Wang, H.; Liang, W.; Liu, Y.; Zhu, T.; Rom, O.; et al. BAF60c prevents abdominal aortic aneurysm formation through epigenetic control of vascular smooth muscle cell homeostasis. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Herring, B.P. Mechanisms responsible for the promoter-specific effects of myocardin. J. Biol. Chem. 2005, 280, 10861–10869. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Li, S.; Hockemeyer, D.; Sutherland, L.; Wang, Z.; Schratt, G.; Richardson, J.A.; Nordheim, A.; Olson, E.N. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. USA 2002, 99, 14855–14860. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Gan, Q.; Shang, Y.; Owens, G.K. Platelet-derived growth factor-BB represses smooth muscle cell marker genes via changes in binding of MKL factors and histone deacetylases to their promoters. Am. J. Physiol. Cell Physiol. 2007, 292, C886–C895. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ritchie, R.P.; Fu, Z.; Cao, D.; Cumming, J.; Miano, J.M.; Wang, D.-Z.; Li, H.J.; Li, L. Myocardin enhances Smad3-mediated transforming growth factor-beta1 signaling in a CArG box-independent manner: Smad-binding element is an important cis element for SM22alpha transcription in vivo. Circ. Res. 2005, 97, 983–991. [Google Scholar] [CrossRef]
- Charron, F.; Paradis, P.; Bronchain, O.; Nemer, G.; Nemer, M. Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol. Cell. Biol. 1999, 19, 4355–4365. [Google Scholar] [CrossRef]
- Mano, T.; Luo, Z.; Malendowicz, S.L.; Evans, T.; Walsh, K. Reversal of GATA-6 downregulation promotes smooth muscle differentiation and inhibits intimal hyperplasia in balloon-injured rat carotid artery. Circ. Res. 1999, 84, 647–654. [Google Scholar] [CrossRef]
- Wada, H.; Hasegawa, K.; Morimoto, T.; Kakita, T.; Yanazume, T.; Sasayama, S. A p300 protein as a coactivator of GATA-6 in the transcription of the smooth muscle-myosin heavy chain gene. J. Biol. Chem. 2000, 275, 25330–25335. [Google Scholar] [CrossRef] [PubMed]
- Nishida, W.; Nakamura, M.; Mori, S.; Takahashi, M.; Ohkawa, Y.; Tadokoro, S.; Yoshida, K.; Hiwada, K.; Hayashi, K.; Sobue, K. A triad of serum response factor and the GATA and NK families governs the transcription of smooth and cardiac muscle genes. J. Biol. Chem. 2002, 277, 7308–7317. [Google Scholar] [CrossRef] [PubMed]
- Kurz, J.; Weiss, A.C.; Lüdtke, T.H.; Deuper, L.; Trowe, M.O.; Thiesler, H.; Hildebrandt, H.; Heineke, J.; Duncan, S.A.; Kispert, A. GATA6 is a crucial factor for Myocd expression in the visceral smooth muscle cell differentiation program of the murine ureter. Development 2022, 149. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.F.; Belaguli, N.S.; Iyer, D.; Roberts, W.B.; Wu, S.P.; Dong, X.R.; Marx, J.G.; Moore, M.S.; Beckerle, M.C.; Majesky, M.W.; et al. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev. Cell 2003, 4, 107–118. [Google Scholar] [CrossRef]
- Chang, D.F.; Belaguli, N.S.; Chang, J.; Schwartz, R.J. LIM-only protein, CRP2, switched on smooth muscle gene activity in adult cardiac myocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 157–162. [Google Scholar] [CrossRef]
- Yoshida, T.; Hoofnagle, M.H.; Owens, G.K. Myocardin and Prx1 contribute to angiotensin II-induced expression of smooth muscle alpha-actin. Circ. Res. 2004, 94, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yoshida, T.; Amendt, B.A.; Martin, J.F.; Owens, G.K. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. J. Cell Biol. 2008, 181, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Kawai-Kowase, K.; Kumar, M.S.; Hoofnagle, M.H.; Yoshida, T.; Owens, G.K. PIAS1 activates the expression of smooth muscle cell differentiation marker genes by interacting with serum response factor and class I basic helix-loop-helix proteins. Mol. Cell. Biol. 2005, 25, 8009–8023. [Google Scholar] [CrossRef]
- Katoh, Y.; Molkentin, J.D.; Dave, V.; Olson, E.N.; Periasamy, M. MEF2B is a component of a smooth muscle-specific complex that binds an A/T-rich element important for smooth muscle myosin heavy chain gene expression. J. Biol. Chem. 1998, 273, 1511–1518. [Google Scholar] [CrossRef]
- Noseda, M.; Fu, Y.; Niessen, K.; Wong, F.; Chang, L.; McLean, G.; Karsan, A. Smooth Muscle alpha-actin is a direct target of Notch/CSL. Circ. Res. 2006, 98, 1468–1470. [Google Scholar] [CrossRef]
- Tang, Y.; Urs, S.; Boucher, J.; Bernaiche, T.; Venkatesh, D.; Spicer, D.B.; Vary, C.P.; Liaw, L. Notch and transforming growth factor-beta (TGFbeta) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J. Biol. Chem. 2010, 285, 17556–17563. [Google Scholar] [CrossRef]
- Liu, Y.; Sinha, S.; McDonald, O.G.; Shang, Y.; Hoofnagle, M.H.; Owens, G.K. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 2005, 280, 9719–9727. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Gan, Q.; Owens, G.K. Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am. J. Physiol. Cell Physiol. 2008, 295, C1175–C1182. [Google Scholar] [CrossRef] [PubMed]
- Salmon, M.; Gomez, D.; Greene, E.; Shankman, L.; Owens, G.K. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22alpha promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ. Res. 2012, 111, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sinha, S.; Owens, G. A transforming growth factor-beta control element required for SM alpha-actin expression in vivo also partially mediates GKLF-dependent transcriptional repression. J. Biol. Chem. 2003, 278, 48004–48011. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Wang, Z.; Zhang, C.L.; Oh, J.; Xing, W.; Li, S.; Richardson, J.A.; Wang, D.Z.; Olson, E.N. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol. Cell. Biol. 2005, 25, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Ostriker, A.C.; Xie, Y.; Dave, J.M.; Gamez-Mendez, A.; Chatterjee, P.; Abu, Y.; Valentine, J.; Lezon-Geyda, K.; Greif, D.M.; et al. Histone Acetyltransferases p300 and CBP Coordinate Distinct Chromatin Remodeling Programs in Vascular Smooth Muscle Plasticity. Circulation 2022, 145, 1720–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sharma, S.; Sun, X.; Guan, X.; Hou, Y.; Yang, Z.; Shi, H.; Zou, M.H.; Song, P.; Zhou, J.; et al. SMYD2 regulates vascular smooth muscle cell phenotypic switching and intimal hyperplasia via interaction with myocardin. Cell. Mol. Life Sci. 2023, 80, 264. [Google Scholar] [CrossRef]
- Lockman, K.; Taylor, J.M.; Mack, C.P. The histone demethylase, Jmjd1a, interacts with the myocardin factors to regulate SMC differentiation marker gene expression. Circ. Res. 2007, 101, e115–e123. [Google Scholar] [CrossRef]
- Gan, Q.; Thiébaud, P.; Thézé, N.; Jin, L.; Xu, G.; Grant, P.; Owens, G.K. WD repeat-containing protein 5, a ubiquitously expressed histone methyltransferase adaptor protein, regulates smooth muscle cell-selective gene activation through interaction with pituitary homeobox 2. J. Biol. Chem. 2011, 286, 21853–21864. [Google Scholar] [CrossRef]
- Li, N.; Subrahmanyan, L.; Smith, E.; Yu, X.; Zaidi, S.; Choi, M.; Mane, S.; Nelson-Williams, C.; Behjati, M.; Kazemi, M.; et al. Mutations in the Histone Modifier PRDM6 Are Associated with Isolated Nonsyndromic Patent Ductus Arteriosus. Am. J. Hum. Genet. 2016, 98, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Haberland, M.; Arnold, M.A.; Sutherland, L.B.; McDonald, O.G.; Richardson, J.A.; Childs, G.; Harris, S.; Owens, G.K.; Olson, E.N. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol. Cell. Biol. 2006, 26, 2626–2636. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Chakraborty, R.; Xie, Y.; Sizer, A.; Hwa, J.; Martin, K.A. The Histone Methyl Transferase (SUV39H1) Promotes Smooth Muscle Cell Dedifferentiation. In Arteriosclerosis, Thrombosis, and Vascular Biology; 2022. [Google Scholar]
- Liang, J.; Li, Q.; Cai, W.; Zhang, X.; Yang, B.; Li, X.; Jiang, S.; Tian, S.; Zhang, K.; Song, H.; et al. Inhibition of polycomb repressor complex 2 ameliorates neointimal hyperplasia by suppressing trimethylation of H3K27 in vascular smooth muscle cells. Br. J. Pharmacol. 2019, 176, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Lino Cardenas, C.L.; Kessinger, C.W.; MacDonald, C.; Jassar, A.S.; Isselbacher, E.M.; Jaffer, F.A.; Lindsay, M.E. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Ostriker, A.C.; Xie, Y.; Chakraborty, R.; Sizer, A.J.; Bai, Y.; Ding, M.; Song, W.L.; Huttner, A.; Hwa, J.; Martin, K.A. TET2 Protects Against Vascular Smooth Muscle Cell Apoptosis and Intimal Thickening in Transplant Vasculopathy. Circulation 2021, 144, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jin, Y.; Tang, W.H.; Qin, L.; Zhang, X.; Tellides, G.; Hwa, J.; Yu, J.; Martin, K.A. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 2013, 128, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Guo, Z.-F.; Kazama, K.; Yi, B.; Tongmuang, N.; Yao, H.; Yang, R.; Zhang, C.; Qin, Y.; Han, L.; et al. Epigenetic regulation of vascular smooth muscle cell phenotypic switch and neointimal formation by PRMT5. Cardiovasc. Res. 2023, 119, 2244–2255. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.M.; Groves, A.K.; Anderson, D.J. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell 1996, 85, 331–343. [Google Scholar] [CrossRef]
- Hautmann, M.B.; Madsen, C.S.; Owens, G.K. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J. Biol. Chem. 1997, 272, 10948–10956. [Google Scholar] [CrossRef]
- Janknecht, R.; Wells, N.J.; Hunter, T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 1998, 12, 2114–2119. [Google Scholar] [CrossRef]
- Narita, N.; Heikinheimo, M.; Bielinska, M.; White, R.A.; Wilson, D.B. The gene for transcription factor GATA-6 resides on mouse chromosome 18 and is expressed in myocardium and vascular smooth muscle. Genomics 1996, 36, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.; Mieremet, A.; de Vries, C.J.M.; Micha, D.; de Waard, V. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Kruppel-Like Factor 4). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2693–2707. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.J.; Regan, C.P.; Hautmann, M.B.; Owens, G.K. Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle cell differentiation marker SM22alpha in vivo. J. Biol. Chem. 2000, 275, 37798–37806. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kaestner, K.H.; Owens, G.K. Conditional deletion of Kruppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ. Res. 2008, 102, 1548–1557. [Google Scholar] [CrossRef]
- McDonald, O.G.; Wamhoff, B.R.; Hoofnagle, M.H.; Owens, G.K. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J. Clin. Investig. 2006, 116, 36–48. [Google Scholar] [CrossRef]
- Manabe, I.; Owens, G.K. Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ. Res. 2001, 88, 1127–1134. [Google Scholar] [CrossRef]
- Yoshida, T.; Sinha, S.; Dandré, F.; Wamhoff, B.R.; Hoofnagle, M.H.; Kremer, B.E.; Wang, D.Z.; Olson, E.N.; Owens, G.K. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 2003, 92, 856–864. [Google Scholar] [CrossRef]
- Pellegrini, L.; Tan, S.; Richmond, T.J. Structure of serum response factor core bound to DNA. Nature 1995, 376, 490–498. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- McDonald, O.G.; Owens, G.K. Programming smooth muscle plasticity with chromatin dynamics. Circ. Res. 2007, 100, 1428–1441. [Google Scholar] [CrossRef]
- Gomez, D.; Shankman, L.S.; Nguyen, A.T.; Owens, G.K. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat. Methods 2013, 10, 171–177. [Google Scholar] [CrossRef]
- Liu, M.; Espinosa-Diez, C.; Mahan, S.; Du, M.; Nguyen, A.T.; Hahn, S.; Chakraborty, R.; Straub, A.C.; Martin, K.A.; Owens, G.K.; et al. H3K4 di-methylation governs smooth muscle lineage identity and promotes vascular homeostasis by restraining plasticity. Dev. Cell 2021, 56, 2765–2782.e10. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef]
- de la Serna, I.L.; Ohkawa, Y.; Imbalzano, A.N. Chromatin remodelling in mammalian differentiation: Lessons from ATP-dependent remodellers. Nat. Rev. Genet. 2006, 7, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.A.; Crabtree, G.R. Chromatin regulatory mechanisms in pluripotency. Annu. Rev. Cell Dev. Biol. 2010, 26, 503–532. [Google Scholar] [CrossRef]
- Singhal, N.; Graumann, J.; Wu, G.; Araúzo-Bravo, M.J.; Han, D.W.; Greber, B.; Gentile, L.; Mann, M.; Schöler, H.R. Chromatin-Remodeling Components of the BAF Complex Facilitate Reprogramming. Cell 2010, 141, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Cairns, B.R. The logic of chromatin architecture and remodelling at promoters. Nature 2009, 461, 193–198. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Stern, M.; Jensen, R.; Herskowitz, I. Five SWI genes are required for expression of the HO gene in yeast. J. Mol. Biol. 1984, 178, 853–868. [Google Scholar] [CrossRef]
- Neigeborn, L.; Carlson, M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 1984, 108, 845–858. [Google Scholar] [CrossRef]
- Winston, F.; Carlson, M. Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet. 1992, 8, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994, 370, 477–481. [Google Scholar] [CrossRef]
- Wang, W.; Côté, J.; Xue, Y.; Zhou, S.; Khavari, P.A.; Biggar, S.R.; Muchardt, C.; Kalpana, G.V.; Goff, S.P.; Yaniv, M.; et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996, 15, 5370–5382. [Google Scholar] [CrossRef] [PubMed]
- Lemon, B.; Inouye, C.; King, D.S.; Tjian, R. Selectivity of chromatin-remodelling cofactors for ligand-activated transcription. Nature 2001, 414, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, A.; Dykhuizen, E.C. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 2018, 293, 3892–3903. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [PubMed]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef]
- Tang, L.; Nogales, E.; Ciferri, C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog. Biophys. Mol. Biol. 2010, 102, 122–128. [Google Scholar] [CrossRef]
- Wu, J.I.; Lessard, J.; Crabtree, G.R. Understanding the words of chromatin regulation. Cell 2009, 136, 200–206. [Google Scholar] [CrossRef]
- Otto, J.E.; Ursu, O.; Wu, A.P.; Winter, E.B.; Cuoco, M.S.; Ma, S.; Qian, K.; Michel, B.C.; Buenrostro, J.D.; Berger, B.; et al. Structural and functional properties of mSWI/SNF chromatin remodeling complexes revealed through single-cell perturbation screens. Mol. Cell 2023, 83, 1350–1367.e7. [Google Scholar] [CrossRef]
- Mashtalir, N.; Suzuki, H.; Farrell, D.P.; Sankar, A.; Luo, J.; Filipovski, M.; D’Avino, A.R.; St Pierre, R.; Valencia, A.M.; Onikubo, T.; et al. A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 2020, 183, 802–817.e24. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wu, Z.; Tian, Y.; Yu, Z.; Yu, J.; Wang, X.; Li, J.; Liu, B.; Xu, Y. Structure of nucleosome-bound human BAF complex. Science 2020, 367, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, K.; Zhang, W.; Chen, Z. Structure of human chromatin-remodelling PBAF complex bound to a nucleosome. Nature 2022, 605, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, J.; Yu, Z.; Wang, Q.; Li, W.; Ren, Y.; Chen, Z.; He, S.; Xu, Y. Structure of nucleosome-bound human PBAF complex. Nat. Commun. 2022, 13, 7644. [Google Scholar] [CrossRef]
- Chen, K.; Yuan, J.; Sia, Y.; Chen, Z. Mechanism of action of the SWI/SNF family complexes. Nucleus 2023, 14, 2165604. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell. Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Valencia, A.M.; Collings, C.K.; Dao, H.T.; St Pierre, R.; Cheng, Y.C.; Huang, J.; Sun, Z.Y.; Seo, H.S.; Mashtalir, N.; Comstock, D.E.; et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 2019, 179, 1342–1356.e23. [Google Scholar] [CrossRef]
- Stros, M.; Launholt, D.; Grasser, K.D. The HMG-box: A versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell. Mol. Life Sci. CMLS 2007, 64, 2590–2606. [Google Scholar] [CrossRef]
- Kim, S.; Zhang, Z.; Upchurch, S.; Isern, N.; Chen, Y. Structure and DNA-binding sites of the SWI1 AT-rich interaction domain (ARID) suggest determinants for sequence-specific DNA recognition. J. Biol. Chem. 2004, 279, 16670–16676. [Google Scholar] [CrossRef]
- Priam, P.; Krasteva, V.; Rousseau, P.; D’Angelo, G.; Gaboury, L.; Sauvageau, G.; Lessard, J.A. SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPvarepsilon dependent mechanism. Nat. Genet. 2017, 49, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, C.; Li, N.; Hao, T.; Han, T.; Hill, D.E.; Vidal, M.; Lin, J.D. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab. 2008, 8, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.K.; Zhao, Y.; McCray, A.; Hawk, W.H.; Deary, L.T.; Sugiarto, N.W.; LaCroix, I.S.; Gerber, S.A.; Cheng, C.; Wang, X. Cooperation of chromatin remodeling SWI/SNF complex and pioneer factor AP-1 shapes 3D enhancer landscapes. Nat. Struct. Mol. Biol. 2023, 30, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.A.; Khorasanizadeh, S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 2002, 295, 2080–2083. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.Y.; Suh, J.H.; Kim, K.; Gong, E.-Y.; Jeon, S.H.; Ko, M.; Seong, R.H.; Kwon, H.B.; Lee, K. Modulation of androgen receptor transactivation by the SWI3-related gene product (SRG3) in multiple ways. Mol. Cell. Biol. 2005, 25, 4841–4852. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.R.; Love, I.M.; Imbalzano, A.N.; Grossman, S.R.; Androphy, E.J. The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene 2009, 28, 2492–2501. [Google Scholar] [CrossRef]
- Hsiao, P.-W.; Fryer, C.J.; Trotter, K.W.; Wang, W.; Archer, T.K. BAF60a mediates critical interactions between nuclear receptors and the BRG1 chromatin-remodeling complex for transactivation. Mol. Cell. Biol. 2003, 23, 6210–6220. [Google Scholar] [CrossRef]
- Debril, M.B.; Gelman, L.; Fayard, E.; Annicotte, J.S.; Rocchi, S.; Auwerx, J. Transcription factors and nuclear receptors interact with the SWI/SNF complex through the BAF60c subunit. J. Biol. Chem. 2004, 279, 16677–16686. [Google Scholar] [CrossRef]
- Ito, T.; Yamauchi, M.; Nishina, M.; Yamamichi, N.; Mizutani, T.; Ui, M.; Murakami, M.; Iba, H. Identification of SWI.SNF complex subunit BAF60a as a determinant of the transactivation potential of Fos/Jun dimers. J. Biol. Chem. 2001, 276, 2852–2857. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Yun, R.; Datta, A.; Lacomis, L.; Erdjument-Bromage, H.; Kumar, J.; Tempst, P.; Sif, S. mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol. Cell. Biol. 2003, 23, 7475–7487. [Google Scholar] [CrossRef]
- Sun, X.; Hota, S.K.; Zhou, Y.Q.; Novak, S.; Miguel-Perez, D.; Christodoulou, D.; Seidman, C.E.; Seidman, J.G.; Gregorio, C.C.; Henkelman, R.M.; et al. Cardiac-enriched BAF chromatin-remodeling complex subunit Baf60c regulates gene expression programs essential for heart development and function. Biol. Open 2018, 7, bio029512. [Google Scholar] [CrossRef] [PubMed]
- Flajollet, S.; Lefebvre, B.; Cudejko, C.; Staels, B.; Lefebvre, P. The core component of the mammalian SWI/SNF complex SMARCD3/BAF60c is a coactivator for the nuclear retinoic acid receptor. Mol. Cell Endocrinol. 2007, 270, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Belandia, B.; Orford, R.L.; Hurst, H.C.; Parker, M.G. Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J. 2002, 21, 4094–4103. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.K.; Lickert, H.; Bisgrove, B.W.; Sun, X.; Yamamoto, M.; Chawengsaksophak, K.; Hamada, H.; Yost, H.J.; Rossant, J.; Bruneau, B.G. Baf60c is a nuclear Notch signaling component required for the establishment of left-right asymmetry. Proc. Natl. Acad. Sci. USA 2007, 104, 846–851. [Google Scholar] [CrossRef]
- Tsurusaki, Y.; Okamoto, N.; Ohashi, H.; Kosho, T.; Imai, Y.; Hibi-Ko, Y.; Kaname, T.; Naritomi, K.; Kawame, H.; Wakui, K.; et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 2012, 44, 376–378. [Google Scholar] [CrossRef]
- van der Sluijs, P.J.; Jansen, S.; Vergano, S.A.; Adachi-Fukuda, M.; Alanay, Y.; AlKindy, A.; Baban, A.; Bayat, A.; Beck-Wödl, S.; Berry, K.; et al. The ARID1B spectrum in 143 patients: From nonsyndromic intellectual disability to Coffin-Siris syndrome. Genet. Med. 2019, 21, 1295–1307. [Google Scholar] [CrossRef]
- van der Sluijs, P.J.; Joosten, M.; Alby, C.; Attié-Bitach, T.; Gilmore, K.; Dubourg, C.; Fradin, M.; Wang, T.; Kurtz-Nelson, E.C.; Ahlers, K.P.; et al. Discovering a new part of the phenotypic spectrum of Coffin-Siris syndrome in a fetal cohort. Genet. Med. 2022, 24, 1753–1760. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, S.; Wang, J.; Zhou, X.; Zhang, H.; Yang, H.; He, Y. Expanding the phenotype associated with SMARCC2 variants: A fetus with tetralogy of Fallot. BMC Med. Genom. 2022, 15, 40. [Google Scholar] [CrossRef]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.C.; Barra, J.; Muchardt, C.; Camus, A.; Babinet, C.; Yaniv, M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha). EMBO J. 1998, 17, 6979–6991. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Kaynak, B.; Forster, U.B.; Tönjes, M.; Fischer, J.J.; Grimm, C.; Schlesinger, J.; Just, S.; Dunkel, I.; Krueger, T.; et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 2008, 22, 2370–2384. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Yang, B.; Chen, Y.; Liu, L.; Cheng, X.; Huang, J.; Zhou, K.; Zhang, D.; Xu, E.; Lai, M.; et al. Loss of GLTSCR1 causes congenital heart defects by regulating NPPA transcription. Angiogenesis 2023, 26, 217–232. [Google Scholar] [CrossRef] [PubMed]
- de La Serna, I.L.; Carlson, K.A.; Hill, D.A.; Guidi, C.J.; Stephenson, R.O.; Sif, S.; Kingston, R.E.; Imbalzano, A.N. Mammalian SWI-SNF complexes contribute to activation of the hsp70 gene. Mol. Cell. Biol. 2000, 20, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Herring, B.P. Regulation of microRNAs by Brahma-related gene 1 (Brg1) in smooth muscle cells. J. Biol. Chem. 2013, 288, 6397–6408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, M.; Kim, J.-R.; Zhou, J.; Jones, R.E.; Tune, J.D.; Kassab, G.S.; Metzger, D.; Ahlfeld, S.; Conway, S.J.; et al. SWI/SNF Complexes Containing Brahma or Brahma-Related Gene 1 Play Distinct Roles in Smooth Muscle Development. Mol. Cell. Biol. 2011, 31, 2618–2631. [Google Scholar] [CrossRef]
- Jankowich, M.; Choudhary, G. Endothelin-1 levels and cardiovascular events. Trends Cardiovasc. Med. 2020, 30, 1–8. [Google Scholar] [CrossRef]
- Li, L.; Liu, D.; Bu, D.; Chen, S.; Wu, J.; Tang, C.; Du, J.; Jin, H. Brg1-dependent epigenetic control of vascular smooth muscle cell proliferation by hydrogen sulfide. Biochim. Biophys. Acta 2013, 1833, 1347–1355. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, X.; Tian, W.; Zhou, B.; Wu, X.; Xu, H.; Fang, F.; Fang, M.; Xu, Y. MRTF-A steers an epigenetic complex to activate endothelin-induced pro-inflammatory transcription in vascular smooth muscle cells. Nucleic Acids Res. 2014, 42, 10460–10472. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, C.; Xu, J.; Tao, J.; Xu, Z.; Huang, S. BRG1 overexpression in smooth muscle cells promotes the development of thoracic aortic dissection. BMC Cardiovasc. Disord. 2014, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.L.; Tan, M.W.; Yuan, Y.; Wang, G.K.; Wang, C.; Tang, H.; Xu, Z.Y. Brahma-related gene 1 inhibits proliferation and migration of human aortic smooth muscle cells by directly up-regulating Ras-related associated with diabetes in the pathophysiologic processes of aortic dissection. J. Thorac. Cardiovasc. Surg. 2015, 150, 1292–1301.e2. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, X.; Yuan, Y.; Tan, M.; Zhang, L.; Xue, X.; Yan, Y.; Han, L.; Xu, Z. BRG1 expression is increased in thoracic aortic aneurysms and regulates proliferation and apoptosis of vascular smooth muscle cells through the long non-coding RNA HIF1A-AS1 in vitro. Eur. J. Cardiothorac. Surg. 2015, 47, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Lino Cardenas, C.L.; Kessinger, C.W.; Cheng, Y.; MacDonald, C.; MacGillivray, T.; Ghoshhajra, B.; Huleihel, L.; Nuri, S.; Yeri, A.S.; Jaffer, F.A.; et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018, 9, 1009. [Google Scholar] [CrossRef] [PubMed]
- Jolly, A.J.; Lu, S.; Dubner, A.M.; Strand, K.A.; Mutryn, M.F.; Pilotti-Riley, A.; Danis, E.P.; Nemenoff, R.A.; Moulton, K.S.; Majesky, M.W.; et al. Redistribution of the chromatin remodeler Brg1 directs smooth muscle-derived adventitial progenitor-to-myofibroblast differentiation and vascular fibrosis. JCI Insight 2023, 8, e164862. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.M.; Hafner, A.; Kirkland, J.G.; Braun, S.M.G.; Stanton, B.Z.; Boettiger, A.N.; Crabtree, G.R. mSWI/SNF promotes Polycomb repression both directly and through genome-wide redistribution. Nat. Struct. Mol. Biol. 2021, 28, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xue, Y.; Zhou, S.; Kuo, A.; Cairns, B.R.; Crabtree, G.R. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996, 10, 2117–2130. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.L.; Mercola, M. BAF60 A, B, and Cs of muscle determination and renewal. Genes. Amp Dev. 2012, 26, 2673–2683. [Google Scholar] [CrossRef]
- Ochi, H.; Hans, S.; Westerfield, M. Smarcd3 regulates the timing of zebrafish myogenesis onset. J. Biol. Chem. 2008, 283, 3529–3536. [Google Scholar] [CrossRef]
- Forcales, S.V.; Albini, S.; Giordani, L.; Malecova, B.; Cignolo, L.; Chernov, A.; Coutinho, P.; Saccone, V.; Consalvi, S.; Williams, R.; et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 2012, 31, 301–316. [Google Scholar] [CrossRef]
- Lickert, H.; Takeuchi, J.K.; Von Both, I.; Walls, J.R.; McAuliffe, F.; Adamson, S.L.; Henkelman, R.M.; Wrana, J.L.; Rossant, J.; Bruneau, B.G. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 2004, 432, 107–112. [Google Scholar] [CrossRef]
- Lou, X.; Deshwar, A.R.; Crump, J.G.; Scott, I.C. Smarcd3b and Gata5 promote a cardiac progenitor fate in the zebrafish embryo. Development 2011, 138, 3113–3123. [Google Scholar] [CrossRef]
- Takeuchi, J.K.; Bruneau, B.G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 2009, 459, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Sohni, A.; Mulas, F.; Ferrazzi, F.; Luttun, A.; Bellazzi, R.; Huylebroeck, D.; Ekker, S.C.; Verfaillie, C.M. TGFbeta1-induced Baf60c regulates both smooth muscle cell commitment and quiescence. PLoS ONE 2012, 7, e47629. [Google Scholar] [CrossRef]
- Chen, S.; Ding, Y.; Zhang, Z.; Wang, H.; Liu, C. Hyperlipidaemia impairs the circadian clock and physiological homeostasis of vascular smooth muscle cells via the suppression of Smarcd1. J. Pathol. 2014, 233, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Zhao, G.; Zhao, Y.; Lu, H.; Xiong, W.; Liang, W.; Sun, J.; Wang, H.; Zhu, T.; Rom, O.; et al. BAF60a Deficiency in Vascular Smooth Muscle Cells Prevents Abdominal Aortic Aneurysm by Reducing Inflammation and Extracellular Matrix Degradation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2494–2507. [Google Scholar] [CrossRef] [PubMed]
- Hinterseher, I.; Erdman, R.; Elmore, J.R.; Stahl, E.; Pahl, M.C.; Derr, K.; Golden, A.; Lillvis, J.H.; Cindric, M.C.; Jackson, K.; et al. Novel pathways in the pathobiology of human abdominal aortic aneurysms. Pathobiology 2013, 80, 1–10. [Google Scholar] [CrossRef]
- Zhao, G.; Lu, H.; Chang, Z.; Zhao, Y.; Zhu, T.; Chang, L.; Guo, Y.; Garcia-Barrio, M.T.; Chen, Y.E.; Zhang, J. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc. Res. 2021, 117, 1402–1416. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, Y.; Xu, Z.; Gao, H.; Feng, W.; Li, W.; Miao, Y.; Xu, Z.; Zong, Y.; Zhao, J.; et al. KLF5 inhibition overcomes oxaliplatin resistance in patient-derived colorectal cancer organoids by restoring apoptotic response. Cell Death Dis. 2022, 13, 303. [Google Scholar] [CrossRef]
- Li, X.; He, Y.; Xu, Y.; Huang, X.; Liu, J.; Xie, M.; Liu, X. KLF5 mediates vascular remodeling via HIF-1alpha in hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L299–L310. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, B.; Xiang, L.; Xia, S.; Kucuk, O.; Deng, X.; Boise, L.H.; Dong, J.T. TGF-beta causes Docetaxel resistance in Prostate Cancer via the induction of Bcl-2 by acetylated KLF5 and Protein Stabilization. Theranostics 2020, 10, 7656–7670. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.P.; Gatchalian, J.; McDermott, M.L.; Nakamura, M.; Chambers, K.; Rajbhandari, N.; Lytle, N.K.; Rosenthal, S.B.; Hamilton, M.; Albini, S.; et al. Smarcd3 is an epigenetic modulator of the metabolic landscape in pancreatic ductal adenocarcinoma. Nat. Commun. 2023, 14, 292. [Google Scholar] [CrossRef] [PubMed]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 2022, 601, 434–439. [Google Scholar] [CrossRef]
- Wei, Z.; Yoshihara, E.; He, N.; Hah, N.; Fan, W.; Pinto, A.F.M.; Huddy, T.; Wang, Y.; Ross, B.; Estepa, G.; et al. Vitamin D Switches BAF Complexes to Protect beta Cells. Cell 2018, 173, 1135–1149.e15. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Gatchalian, J.; Ho, J.; Burns, M.J.; Hah, N.; Wei, Z.; Downes, M.; Evans, R.M.; Hargreaves, D.C. BRD9 regulates interferon-stimulated genes during macrophage activation via cooperation with BET protein BRD4. Proc. Natl. Acad. Sci. USA 2022, 119, e2110812119. [Google Scholar] [CrossRef]
- Du, J.; Liu, Y.; Wu, X.; Sun, J.; Shi, J.; Zhang, H.; Zheng, A.; Zhou, M.; Jiang, X. BRD9-mediated chromatin remodeling suppresses osteoclastogenesis through negative feedback mechanism. Nat. Commun. 2023, 14, 1413. [Google Scholar] [CrossRef]
- Wang, L.; Oh, T.G.; Magida, J.; Estepa, G.; Obayomi, S.M.B.; Chong, L.W.; Gatchalian, J.; Yu, R.T.; Atkins, A.R.; Hargreaves, D.; et al. Bromodomain containing 9 (BRD9) regulates macrophage inflammatory responses by potentiating glucocorticoid receptor activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2109517118. [Google Scholar] [CrossRef]
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandona, M.; Ryan, T.; Rojas-Munoz, A.; Madaro, L.; Fasanaro, P.; et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 2014, 28, 841–857. [Google Scholar] [CrossRef]
- Zhang, P.; Li, L.; Bao, Z.; Huang, F. Role of BAF60a/BAF60c in chromatin remodeling and hepatic lipid metabolism. Nutr. Metab. 2016, 13, 30. [Google Scholar] [CrossRef]
- Meng, Z.X.; Tao, W.; Sun, J.; Wang, Q.; Mi, L.; Lin, J.D. Uncoupling Exercise Bioenergetics From Systemic Metabolic Homeostasis by Conditional Inactivation of Baf60 in Skeletal Muscle. Diabetes 2018, 67, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Hota, S.K.; Johnson, J.R.; Verschueren, E.; Thomas, R.; Blotnick, A.M.; Zhu, Y.; Sun, X.; Pennacchio, L.A.; Krogan, N.J.; Bruneau, B.G. Dynamic BAF chromatin remodeling complex subunit inclusion promotes temporally distinct gene expression programs in cardiogenesis. Development 2019, 146, dev174086. [Google Scholar] [CrossRef] [PubMed]
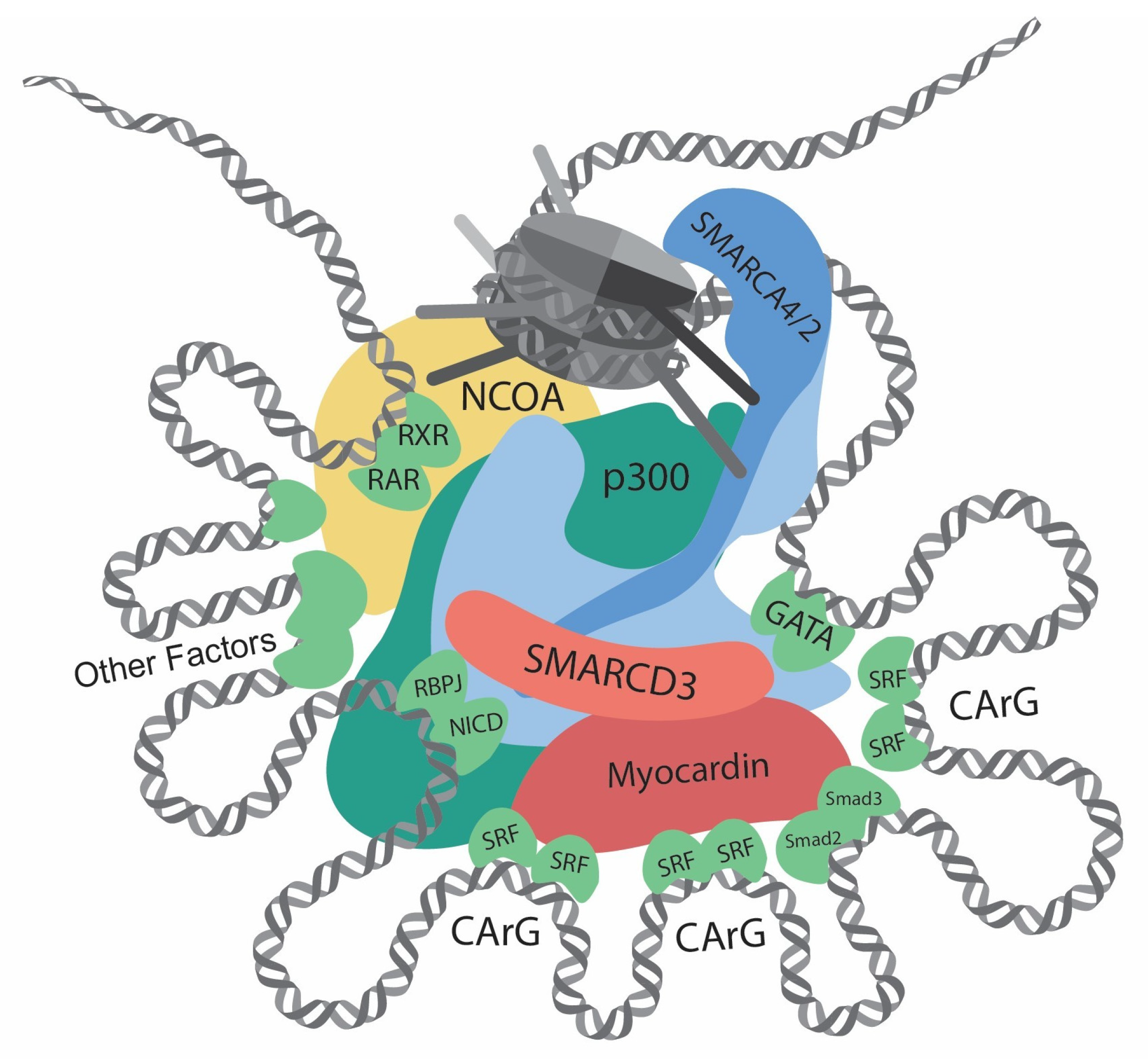



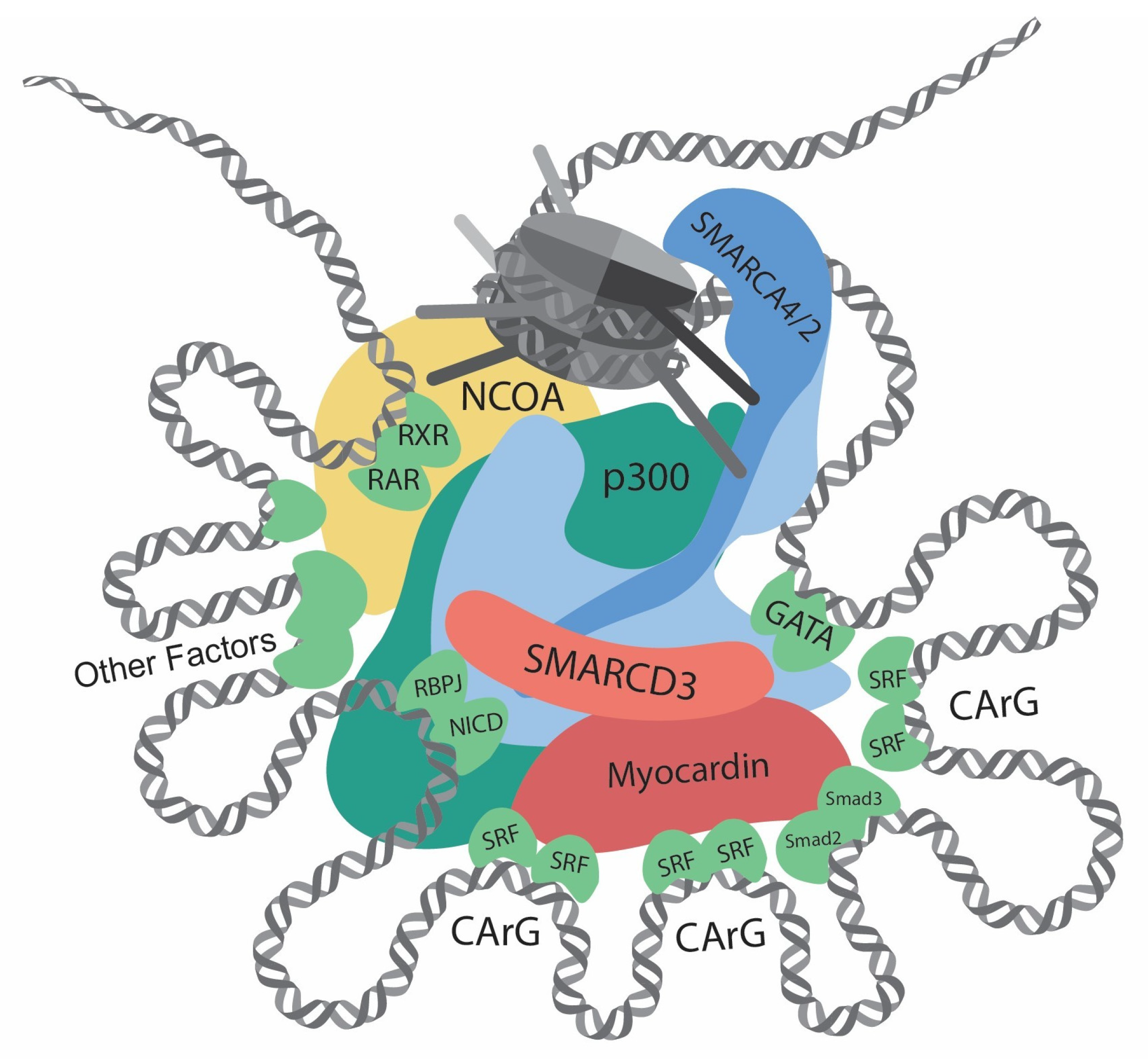{kind=link}
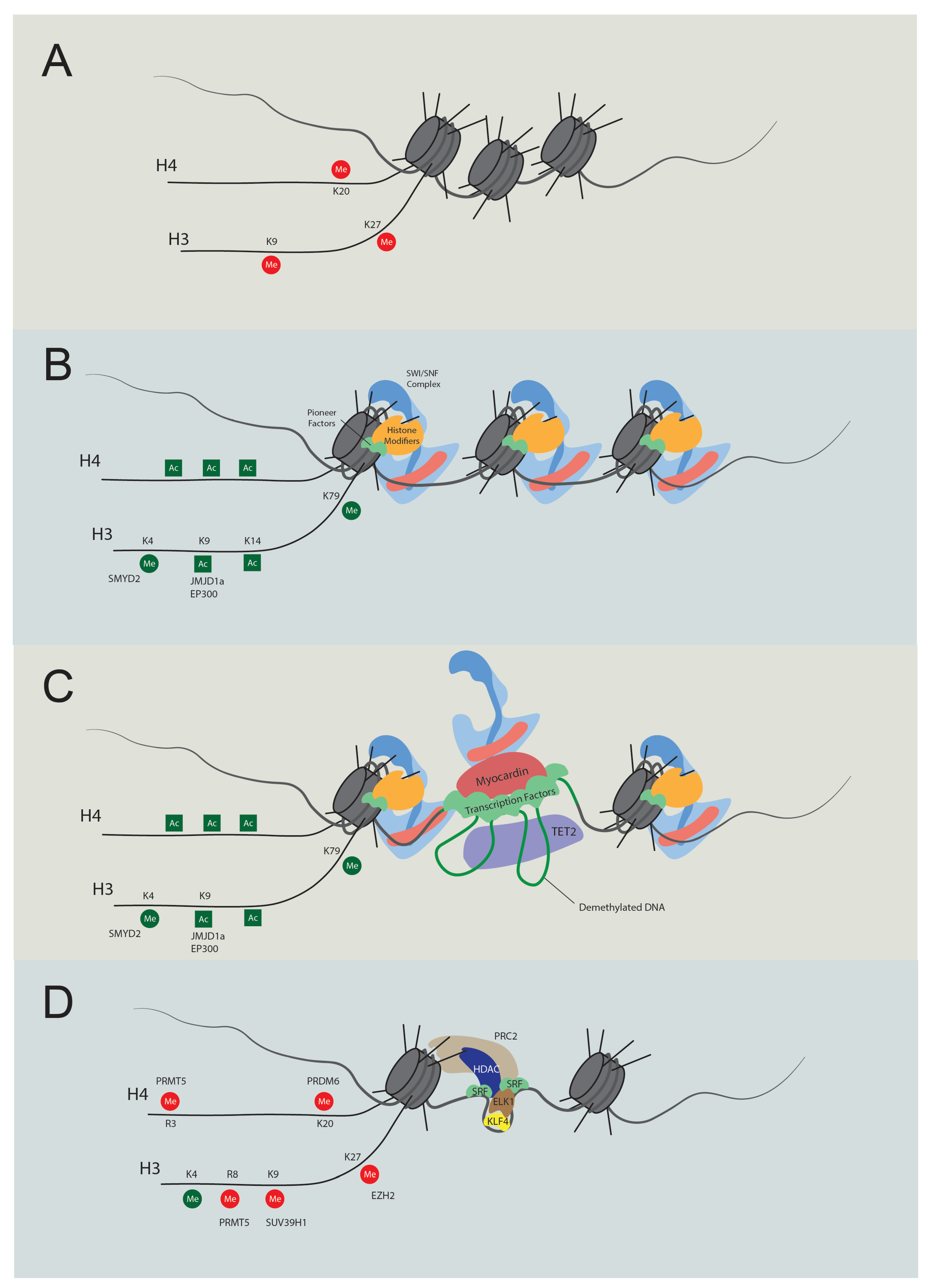{kind=link}
{kind=link}
| Gene | Category | Function | Effect on Contractile Gene Transcription | Reference |
|---|---|---|---|---|
| SMARCA4 | Chromatin remodeler | Mediate chromatin accessibility | Dependent on interactors | [42,43] |
| SMARCD3 | Chromatin remodeler | Mediate chromatin accessibility | Promotion | [44] |
| SRF | Transcription factor | Bind at CArG elements | Dependent on interactors | [8] |
| Myocardin | Cofactor | Interact with SRF | Promotion | [8,41,45] |
| MRTFA/B | Cofactor | Interact with SRF | Promotion | [41,46,47] |
| Smad2/3 | Transcription factor | Bind DNA | Promotion | [48] |
| GATA4/6 | Transcription factor | Bind DNA | Promotion | [49,50,51,52,53] |
| CSRP2 | Cofactor | Interact with SRF, GATA6 | Promotion | [54,55] |
| NKX3-2 | Transcription factor | Bind DNA | Promotion | [52] |
| Prx1 | Transcription factor | Bind DNA | Promotion | [56] |
| PITX2 | Transcription factor | Bind DNA | Promotion | [57] |
| PIAS1 | Transcription factor | Bind DNA | Promotion | [58] |
| MEF2 | Transcription factor | Bind DNA | Promotion | [59] |
| Notch/RBPJ | Transcription factor | Bind DNA | Promotion | [60,61] |
| KLF4 | Transcription factor | Bind G/C repressor element | Repression | [9,62,63,64,65] |
| Elk1 | Cofactor | Interact with SRF | Repression | [47,63,64] |
| p300 | Histone acetyltransferase | Increase histone acetylation | Promotion | [51,66,67] |
| HDAC | Histone deacetylase | Decrease Histone modification | Repression | [47,63,64] |
| SMYD2 | Histone lysine methyltransferase | Increase H3K4me1, H3K4me3 | Promotion | [68] |
| JMJD1A | Histone demethylase | Decrease H3K9me2 | Promotion | [69] |
| WDR5 | Cofactor | Increase H3K4me1, H3K4me3 | Promotion | [70] |
| PRDM6 | Histone lysine methyltransferase | Increase H4K20me2 | Repression | [71,72] |
| SUV39H1 | Histone lysine methyltransferase | Increase H3K9me3 | Repression | [73] |
| EZH2 | Histone lysine methyltransferase | Increase H3K27me3 | Repression | [74,75] |
| TET2 | Methylcytosine dioxygenase | DNA demethylation | Promotion | [76,77] |
| PRMT5 | Histone arginine methyltransferase | H3R8me2, H4R3me2 | Repression | [78] |
| HUGO Name | Common Name |
|---|---|
| SMARCC1 | BAF155, SRG3 |
| SMARCC2 | BAF170 |
| SMARCD1/2/3 | BAF60A/B/C |
| SMARCB1 | BAF47, INI1 |
| SMARCE1 | BAF57 |
| ARID1A/B | BAF250A/B |
| ARID2 | BAF200 |
| PHF10 | BAF45A |
| DPF1/2/3 | BAF45B/D/C |
| BICRA/BICRAL | GLTSCR1/GLTSCR1L |
| SMARCA4 | BRG1 |
| SMARCA2 | BRM |
| ACTL6A/B | BAF53A/B |
| PBRM1 | BAF180 |
| Interactor | SWI/SNF Subunit | Reference |
|---|---|---|
| AR | SMARCC1 | [128] |
| CBP | SMARCA4 | [129] |
| ERα | SMARCD1 | [130] |
| ERα | SMARCD3 | [131] |
| FOS | SMARCD1 | [132] |
| JUN | SMARCD1 | [132] |
| JUN | SMARCD3 | [131] |
| MYC | SMARCA2 | [133] |
| MYC | SMARCA4 | [133] |
| MYC | SMARCB1 | [133] |
| MYC | SMARCE1 | [133] |
| Myocardin | SMARCD3 | [134] |
| NCOA1 | ARID1 | [135] |
| NCOA1 | SMARCC1 | [128] |
| NCOA1 | SMARCE1 | [135,136] |
| Nkx2-5 | SMARCD3 | [134] |
| NR3C1/GR | SMARCD1 | [130] |
| NR3C1/GR | SMARCE1 | [130] |
| PCG1α | SMARCD1 | [123] |
| PPARγ | SMARCD1 | [123] |
| PPARγ | SMARCD3 | [131] |
| PRMT5 | SMARCB1 | [133] |
| PRMT5 | SMARCE1 | [133] |
| RAR | SMARCD3 | [135] |
| RBP-J | SMARCD3 | [137] |
| RORα | SMARCD3 | [131] |
| RXR | SMARCD3 | [131,135] |
| SREBP1α | SMARCD3 | [131] |
| Tbx5 | SMARCD3 | [134,137] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Zhao, Y.; Zhao, G.; Deng, Y.; Chen, Y.E.; Zhang, J. SWI/SNF Complex in Vascular Smooth Muscle Cells and Its Implications in Cardiovascular Pathologies. Cells 2024, 13, 168. https://doi.org/10.3390/cells13020168
Liu H, Zhao Y, Zhao G, Deng Y, Chen YE, Zhang J. SWI/SNF Complex in Vascular Smooth Muscle Cells and Its Implications in Cardiovascular Pathologies. Cells. 2024; 13(2):168. https://doi.org/10.3390/cells13020168
Chicago/Turabian StyleLiu, Hongyu, Yang Zhao, Guizhen Zhao, Yongjie Deng, Y. Eugene Chen, and Jifeng Zhang. 2024. "SWI/SNF Complex in Vascular Smooth Muscle Cells and Its Implications in Cardiovascular Pathologies" Cells 13, no. 2: 168. https://doi.org/10.3390/cells13020168
APA StyleLiu, H., Zhao, Y., Zhao, G., Deng, Y., Chen, Y. E., & Zhang, J. (2024). SWI/SNF Complex in Vascular Smooth Muscle Cells and Its Implications in Cardiovascular Pathologies. Cells, 13(2), 168. https://doi.org/10.3390/cells13020168