
EF24, a Curcumin Analog, Reverses Interleukin-18-Induced miR-30a or miR-342-Dependent TRAF3IP2 Expression, RECK Suppression, and the Proinflammatory Phenotype of Human Aortic Smooth Muscle Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Adenoviral Vectors
2.4. mRNA Expression
2.5. Micro RNA Expression, Mimics, and Transfections
2.6. Western Blot Analysis and Quantification of Cleaved Caspase-3 Levels
2.7. Cell Proliferation, Migration and Phenotypic Modulation
2.8. Cell Migration
2.9. Phenotypic Modulation
Cell Viability
2.10. Statistical Analysis
3. Results
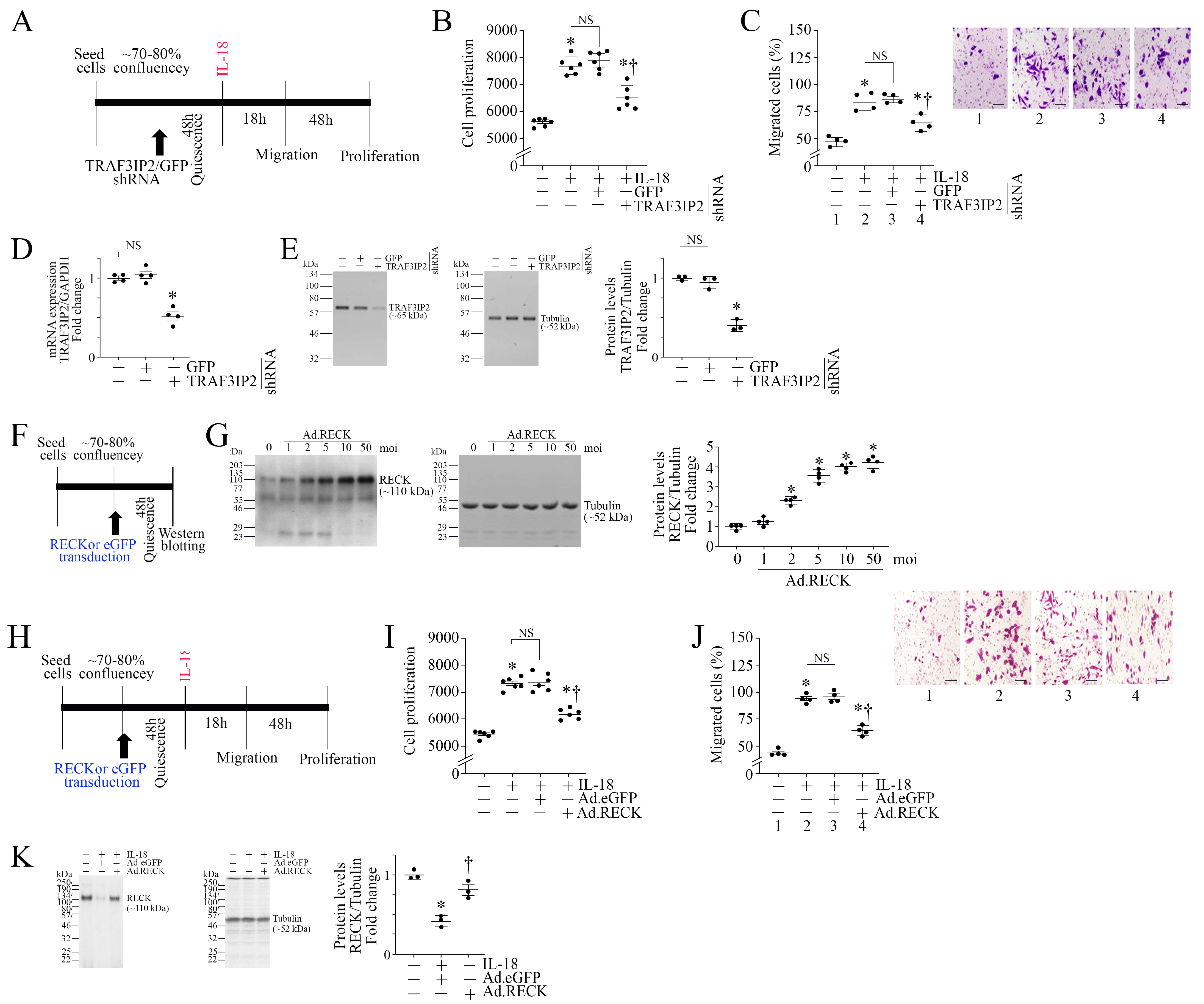
3.1. IL-18 Differentially Regulates TRAF3IP2 and RECK Expression in ASMC
3.2. TRAF3IP2 Knockdown or Ectopic Expression of RECK Reverses IL-18-Induced ASMC Proliferation and Migration
3.3. Silencing TRAF3IP2 Reverses IL-18-Induced Suppression in SMC Markers and Inhibits the Induction of Proinflammatory Phenotype Markers
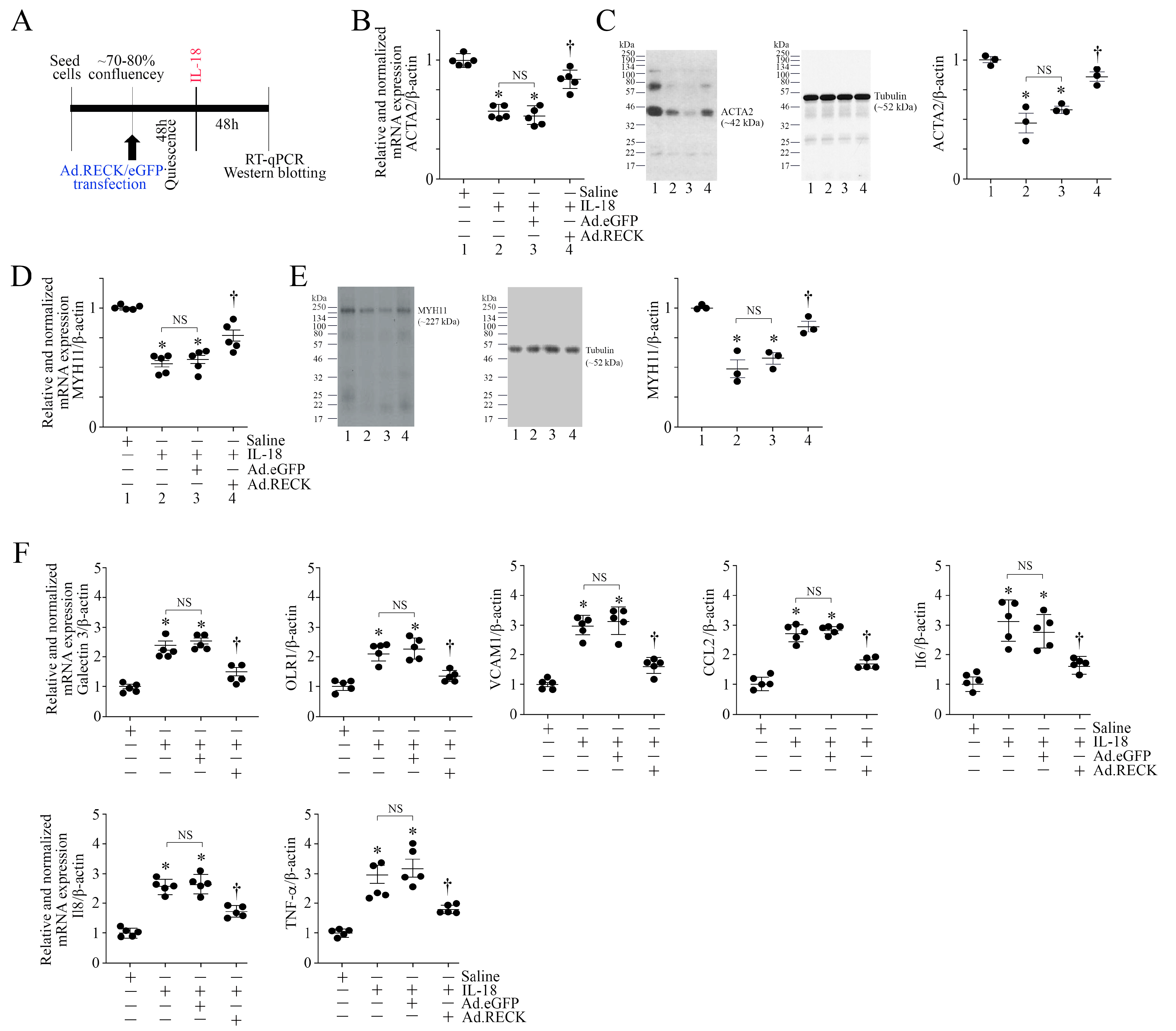
3.4. Ectopic Expression of RECK Reverses IL-18-Induced Suppression in SMC Markers and Inhibits the Induction of Proinflammatory Phenotype Markers
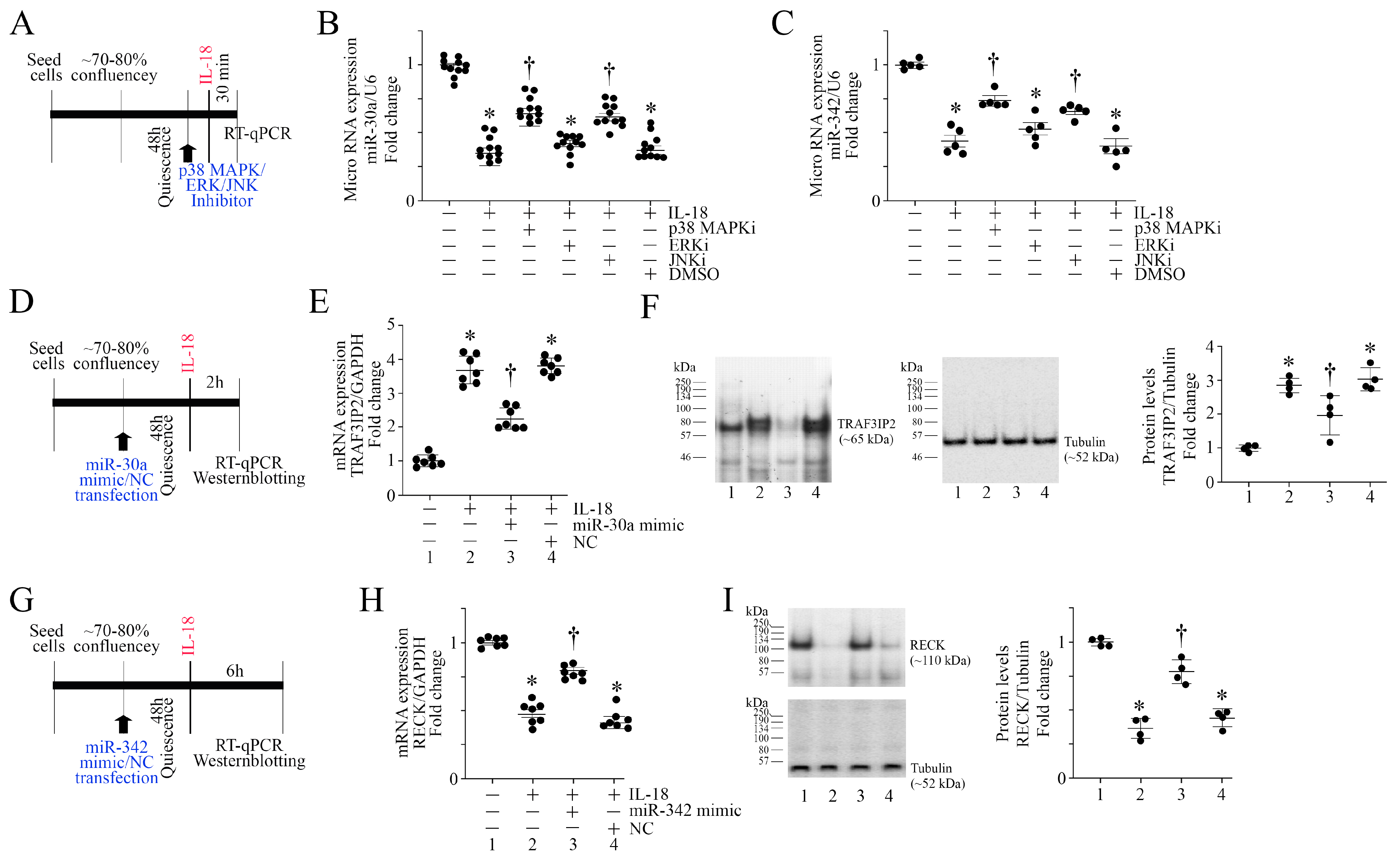
3.5. IL-18 Inhibits miR-30a and miR-342 Expression via Activation of Stress Activated Kinases
3.6. miR-30a and miR-342 Mimics Reverse IL-18-Induced TRAF3IP2 Expression and RECK Suppression
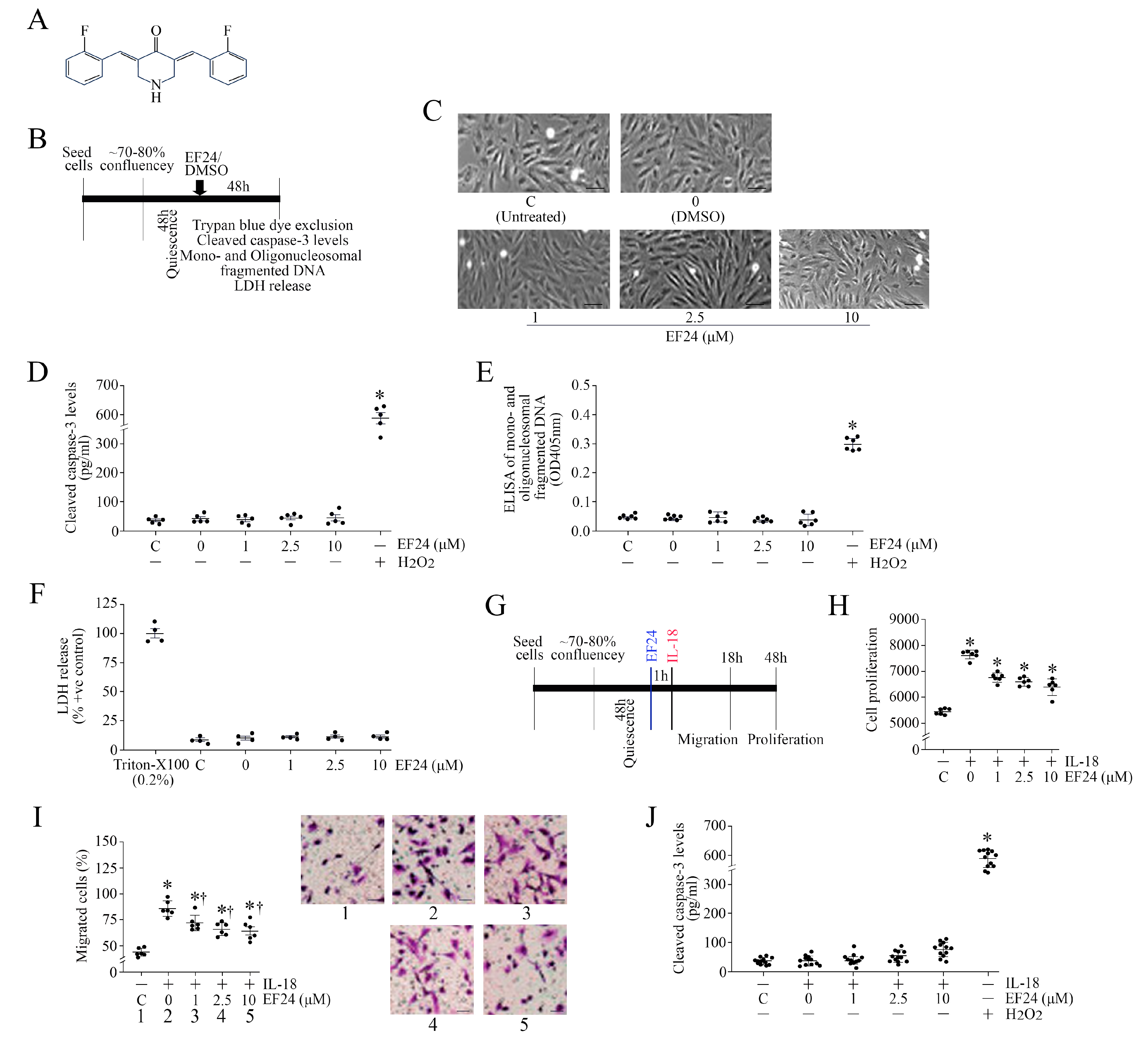
3.7. EF24, a Curcumin Analog, Inhibits IL-18-Induced ASMC Proliferation and Migration without Affecting Cell Viability
3.8. EF24 Inhibits IL-18-Induced TRAF3IP2 Expression and RECK Suppression
3.9. EF24 Inhibits IL-18-Induced Proinflammatory Phenotype Changes in ASMC
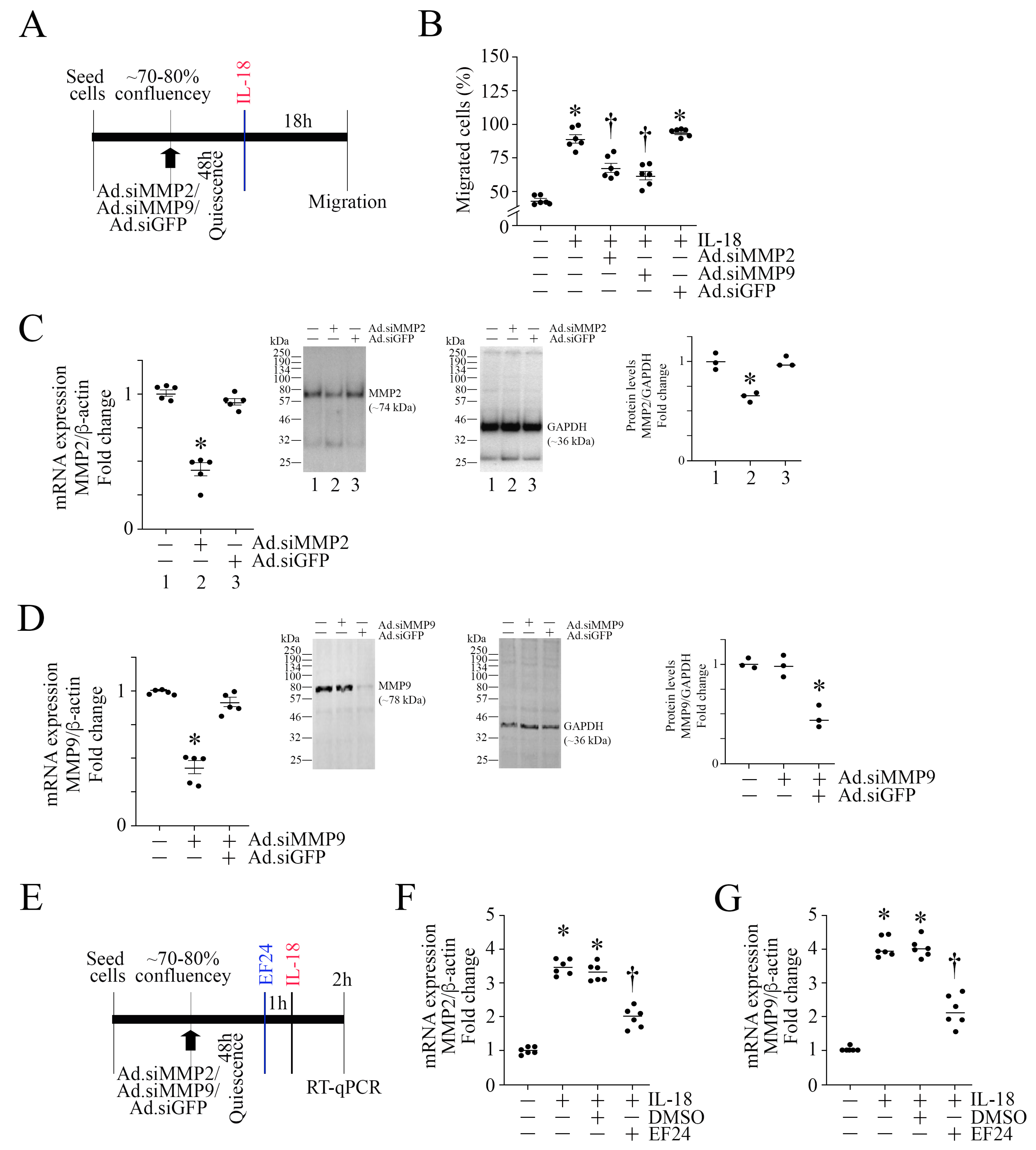
3.10. EF24 Inhibits MMP Expression in ASMC
4. Discussion
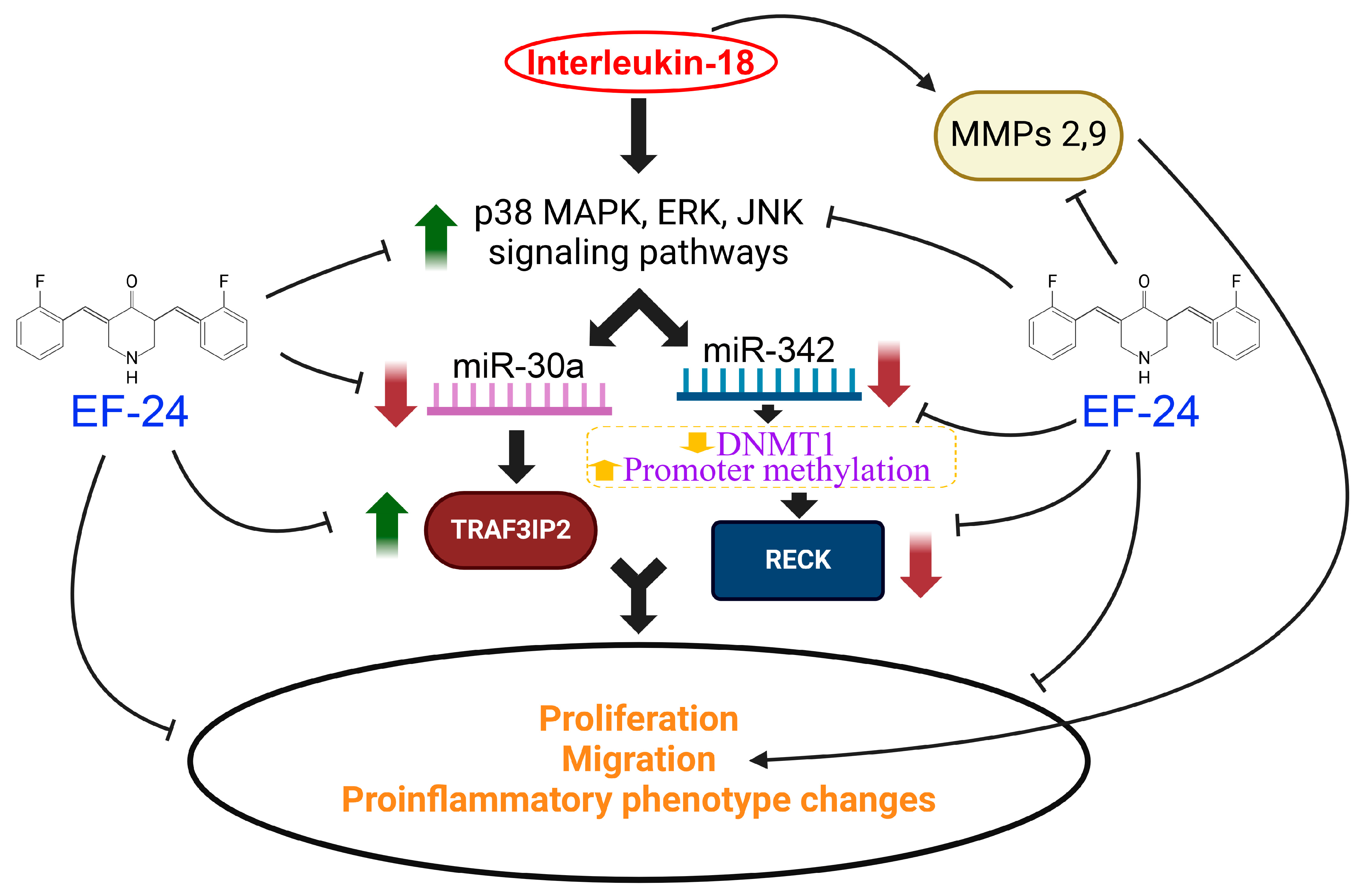
5. Conclusions
Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Martin, S.S.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Barone Gibbs, B.; Beaton, A.Z.; Boehme, A.K.; et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2024, 149, e347–e913. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Argulian, E.; Leipsic, J.; Newby, D.E.; Narula, J. From Subclinical Atherosclerosis to Plaque Progression and Acute Coronary Events: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, A.; Da Dalt, L.; Norata, G.D. New insights into the therapeutic options to lower lipoprotein(a). Eur. J. Clin. Investig. 2024, 54, e14254. [Google Scholar] [CrossRef]
- Nielsen, R.V.; Fuster, V.; Bundgaard, H.; Fuster, J.J.; Johri, A.M.; Kofoed, K.F.; Douglas, P.S.; Diederichsen, A.; Shapiro, M.D.; Nicholls, S.J.; et al. Personalized Intervention Based on Early Detection of Atherosclerosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2024, 83, 2112–2127. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Le, A.; Peng, H.; Golinsky, D.; Di Scipio, M.; Lali, R.; Pare, G. What Causes Premature Coronary Artery Disease? Curr. Atheroscler. Rep. 2024, 26, 189–203. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; Rupprecht, H.J.; AtheroGene, I. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation 2002, 106, 24–30. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2020, 41, 2153–2163. [Google Scholar] [CrossRef]
- Okamura, H.; Tsutsui, H.; Kashiwamura, S.; Yoshimoto, T.; Nakanishi, K. Interleukin-18: A novel cytokine that augments both innate and acquired immunity. Adv. Immunol. 1998, 70, 281–312. [Google Scholar] [CrossRef] [PubMed]
- Puren, A.J.; Fantuzzi, G.; Gu, Y.; Su, M.S.; Dinarello, C.A. Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J. Clin. Investig. 1998, 101, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Vanaclocha, F.; Fantuzzi, G.; Mendoza, L.; Fuentes, A.M.; Anasagasti, M.J.; Martin, J.; Carrascal, T.; Walsh, P.; Reznikov, L.L.; Kim, S.H.; et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc. Natl. Acad. Sci. USA 2000, 97, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Bhat, O.M.; Kumar, P.U.; Giridharan, N.V.; Kaul, D.; Kumar, M.J.; Dhawan, V. Interleukin-18-induced atherosclerosis involves CD36 and NF-kappaB crosstalk in Apo E-/- mice. J. Cardiol. 2015, 66, 28–35. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Graber, P.; Alouani, S.; Esposito, B.; Humbert, Y.; Chvatchko, Y.; Tedgui, A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 2001, 89, E41–E45. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, N.; Sukhova, G.K.; Libby, P.; Reynolds, R.S.; Young, J.L.; Schonbeck, U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: Implications for atherogenesis. J. Exp. Med. 2002, 195, 245–257. [Google Scholar] [CrossRef]
- Leonardi, A.; Chariot, A.; Claudio, E.; Cunningham, K.; Siebenlist, U. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc. Natl. Acad. Sci. USA 2000, 97, 10494–10499. [Google Scholar] [CrossRef]
- Li, X.; Commane, M.; Nie, H.; Hua, X.; Chatterjee-Kishore, M.; Wald, D.; Haag, M.; Stark, G.R. Act1, an NF-kappa B-activating protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10489–10493. [Google Scholar] [CrossRef]
- Valente, A.J.; Sakamuri, S.S.; Siddesha, J.M.; Yoshida, T.; Gardner, J.D.; Prabhu, R.; Siebenlist, U.; Chandrasekar, B. TRAF3IP2 mediates interleukin-18-induced cardiac fibroblast migration and differentiation. Cell. Signal. 2013, 25, 2176–2184. [Google Scholar] [CrossRef]
- Takahashi, C.; Sheng, Z.; Horan, T.P.; Kitayama, H.; Maki, M.; Hitomi, K.; Kitaura, Y.; Takai, S.; Sasahara, R.M.; Horimoto, A.; et al. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc. Natl. Acad. Sci. USA 1998, 95, 13221–13226. [Google Scholar] [CrossRef] [PubMed]
- Siddesha, J.M.; Valente, A.J.; Sakamuri, S.S.; Yoshida, T.; Gardner, J.D.; Somanna, N.; Takahashi, C.; Noda, M.; Chandrasekar, B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J. Mol. Cell. Cardiol. 2013, 65, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Siddesha, J.M.; Valente, A.J.; Sakamuri, S.S.; Gardner, J.D.; Delafontaine, P.; Noda, M.; Chandrasekar, B. Acetylsalicylic acid inhibits IL-18-induced cardiac fibroblast migration through the induction of RECK. J. Cell. Physiol. 2014, 229, 845–855. [Google Scholar] [CrossRef]
- Mummidi, S.; Das, N.A.; Carpenter, A.J.; Yoshida, T.; Yariswamy, M.; Mostany, R.; Izadpanah, R.; Higashi, Y.; Sukhanov, S.; Noda, M.; et al. RECK suppresses interleukin-17/TRAF3IP2-mediated MMP-13 activation and human aortic smooth muscle cell migration and proliferation. J. Cell. Physiol. 2019, 234, 22242–22259. [Google Scholar] [CrossRef]
- Sukhanov, S.; Higashi, Y.; Yoshida, T.; Mummidi, S.; Aroor, A.R.; Jeffrey Russell, J.; Bender, S.B.; DeMarco, V.G.; Chandrasekar, B. The SGLT2 inhibitor Empagliflozin attenuates interleukin-17A-induced human aortic smooth muscle cell proliferation and migration by targeting TRAF3IP2/ROS/NLRP3/Caspase-1-dependent IL-1beta and IL-18 secretion. Cell. Signal. 2021, 77, 109825. [Google Scholar] [CrossRef]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar] [CrossRef]
- Wongcharoen, W.; Phrommintikul, A. The protective role of curcumin in cardiovascular diseases. Int. J. Cardiol. 2009, 133, 145–151. [Google Scholar] [CrossRef]
- Jurenka, J.S. Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: A review of preclinical and clinical research. Altern. Med. Rev. 2009, 14, 141–153. [Google Scholar] [PubMed]
- Banez, M.J.; Geluz, M.I.; Chandra, A.; Hamdan, T.; Biswas, O.S.; Bryan, N.S.; Von Schwarz, E.R. A systemic review on the antioxidant and anti-inflammatory effects of resveratrol, curcumin, and dietary nitric oxide supplementation on human cardiovascular health. Nutr. Res. 2020, 78, 11–26. [Google Scholar] [CrossRef]
- Surma, S.; Sahebkar, A.; Urbanski, J.; Penson, P.E.; Banach, M. Curcumin—The Nutraceutical with Pleiotropic Effects? Which Cardiometabolic Subjects Might Benefit the Most? Front. Nutr. 2022, 9, 865497. [Google Scholar] [CrossRef]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.L.; Zhong, D.; Zhou, W.; Malik, S.; Liotta, D.; Snyder, J.P.; Hamel, E.; Giannakakou, P. EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF-1. Cell Cycle 2008, 7, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Lagisetty, P.; Subramaniam, D.; Sahoo, K.; Anant, S.; Awasthi, V. Anticancer activity of an imageable curcuminoid 1-[2-aminoethyl-(6-hydrazinopyridine-3-carbamidyl)-3,5-bis-(2-fluorobenzylidene)-4-piperidone (EFAH). Chem. Biol. Drug Des. 2012, 79, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, S.; Shi, L.; Zhang, S.; Ren, J.; Yao, S.; Yun, D.; Huang, L.; Wang, J.; Li, W.; et al. Design, synthesis, and evaluation of asymmetric EF24 analogues as potential anti-cancer agents for lung cancer. Eur. J. Med. Chem. 2017, 125, 1321–1331. [Google Scholar] [CrossRef]
- Xie, X.; Tu, J.; You, H.; Hu, B. Design, synthesis, and biological evaluation of novel EF24 and EF31 analogs as potential IkappaB kinase beta inhibitors for the treatment of pancreatic cancer. Drug Des. Dev. Ther. 2017, 11, 1439–1451. [Google Scholar] [CrossRef]
- Schmitt, F.; Gold, M.; Begemann, G.; Andronache, I.; Biersack, B.; Schobert, R. Fluoro and pentafluorothio analogs of the antitumoral curcuminoid EF24 with superior antiangiogenic and vascular-disruptive effects. Bioorg Med. Chem. 2017, 25, 4894–4903. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Zheng, R.; Hou, K.; Zhang, Y.; Jia, T.; Lu, X.; Samarawickrama, P.N.; Jia, S.; He, Y.; et al. Curcumin analogue EF24 prevents alveolar epithelial cell senescence to ameliorate idiopathic pulmonary fibrosis via activation of PTEN. Phytomedicine 2024, 133, 155882. [Google Scholar] [CrossRef]
- Reid, J.M.; Buhrow, S.A.; Gilbert, J.A.; Jia, L.; Shoji, M.; Snyder, J.P.; Ames, M.M. Mouse pharmacokinetics and metabolism of the curcumin analog, 4-piperidinone,3,5-bis[(2-fluorophenyl)methylene]-acetate(3E,5E) (EF-24; NSC 716993). Cancer Chemother. Pharmacol. 2014, 73, 1137–1146. [Google Scholar] [CrossRef]
- Valente, A.J.; Yoshida, T.; Izadpanah, R.; Delafontaine, P.; Siebenlist, U.; Chandrasekar, B. Interleukin-18 enhances IL-18R/Nox1 binding, and mediates TRAF3IP2-dependent smooth muscle cell migration. Inhibition by simvastatin. Cell. Signal. 2013, 25, 1447–1456. [Google Scholar] [CrossRef]
- Venkatesan, B.; Valente, A.J.; Reddy, V.S.; Siwik, D.A.; Chandrasekar, B. Resveratrol blocks interleukin-18-EMMPRIN cross-regulation and smooth muscle cell migration. Am. J. Physiol. Circ. Physiol. 2009, 297, H874–H886. [Google Scholar] [CrossRef]
- Sharma, N.; Khalyfa, A.; Cai, D.; Morales-Quinones, M.; Soares, R.N.; Higashi, Y.; Chen, S.; Gozal, D.; Padilla, J.; Manrique-Acevedo, C.; et al. Chronic Intermittent Hypoxia Facilitates the Development of Angiotensin II-Induced Abdominal Aortic Aneurysm in Male Mice. J. Appl. Physiol. 2024, 137, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Ralay Ranaivo, H.; Hodge, J.N.; Choi, N.; Wainwright, M.S. Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J. Neuroinflamm. 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Ralay Ranaivo, H.; Patel, F.; Wainwright, M.S. Albumin activates the canonical TGF receptor-smad signaling pathway but this is not required for activation of astrocytes. Exp. Neurol. 2010, 226, 310–319. [Google Scholar] [CrossRef]
- Chen, X.; Cobbs, A.; George, J.; Chima, A.; Tuyishime, F.; Zhao, X. Endocytosis of Albumin Induces Matrix Metalloproteinase-9 by Activating the ERK Signaling Pathway in Renal Tubule Epithelial Cells. Int. J. Mol. Sci. 2017, 18, 1758. [Google Scholar] [CrossRef]
- Stephan, J.P.; Mao, W.; Filvaroff, E.; Cai, L.; Rabkin, R.; Pan, G. Albumin stimulates the accumulation of extracellular matrix in renal tubular epithelial cells. Am. J. Nephrol. 2004, 24, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.J.; Kargiotis, O.; Chetty, C.; Lakka, S.S.; Gujrati, M.; Spomar, D.G.; Dinh, D.H.; Rao, J.S. Downregulation of matrix metalloproteinase-2 (MMP-2) utilizing adenovirus-mediated transfer of small interfering RNA (siRNA) in a novel spinal metastatic melanoma model. Int. J. Oncol. 2008, 32, 557–564. [Google Scholar] [CrossRef]
- Higashi, Y.; Mummidi, S.; Sukhanov, S.; Yoshida, T.; Noda, M.; Delafontaine, P.; Chandrasekar, B. Minocycline inhibits PDGF-BB-induced human aortic smooth muscle cell proliferation and migration by reversing miR-221- and -222-mediated RECK suppression. Cell. Signal. 2019, 57, 10–20. [Google Scholar] [CrossRef]
- Wan, Q.; Zhou, Z.; Ding, S.; He, J. The miR-30a Negatively Regulates IL-17-Mediated Signal Transduction by Targeting Traf3ip2. J. Interf. Cytokine Res. 2015, 35, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, J.; Meng, X.; Ying, X.; Zuo, Y.; Liu, R.; Pan, Z.; Kang, T.; Huang, W. MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis 2011, 32, 1033–1042. [Google Scholar] [CrossRef]
- Reddy, V.S.; Valente, A.J.; Delafontaine, P.; Chandrasekar, B. Interleukin-18/WNT1-inducible signaling pathway protein-1 signaling mediates human saphenous vein smooth muscle cell proliferation. J. Cell Physiol. 2011, 226, 3303–3315. [Google Scholar] [CrossRef]
- Yamaoka-Tojo, M.; Tojo, T.; Wakaume, K.; Kameda, R.; Nemoto, S.; Takahira, N.; Masuda, T.; Izumi, T. Circulating interleukin-18: A specific biomarker for atherosclerosis-prone patients with metabolic syndrome. Nutr. Metab. 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.C.; Ravisankar, P.; Daugherty, A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(-/-) mice through release of interferon-gamma. Circ. Res. 2002, 90, E34–E38. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-gamma production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Reddy, V.S.; Harskamp, R.E.; van Ginkel, M.W.; Calhoon, J.; Baisden, C.E.; Kim, I.S.; Valente, A.J.; Chandrasekar, B. Interleukin-18 stimulates fibronectin expression in primary human cardiac fibroblasts via PI3K-Akt-dependent NF-kappaB activation. J. Cell. Physiol. 2008, 215, 697–707. [Google Scholar] [CrossRef]
- Reddy, V.S.; Prabhu, S.D.; Mummidi, S.; Valente, A.J.; Venkatesan, B.; Shanmugam, P.; Delafontaine, P.; Chandrasekar, B. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-kappaB activation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1242–H1254. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Valente, A.J.; Freeman, G.L.; Mahimainathan, L.; Mummidi, S. Interleukin-18 induces human cardiac endothelial cell death via a novel signaling pathway involving NF-kappaB-dependent PTEN activation. Biochem. Biophys. Res. Commun. 2006, 339, 956–963. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Mummidi, S.; Mahimainathan, L.; Patel, D.N.; Bailey, S.R.; Imam, S.Z.; Greene, W.C.; Valente, A.J. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J. Biol. Chem. 2006, 281, 15099–15109. [Google Scholar] [CrossRef]
- Ruby, A.J.; Kuttan, G.; Babu, K.D.; Rajasekharan, K.N.; Kuttan, R. Anti-tumour and antioxidant activity of natural curcuminoids. Cancer Lett. 1995, 94, 79–83. [Google Scholar] [CrossRef]
- Dehzad, M.J.; Ghalandari, H.; Nouri, M.; Askarpour, M. Antioxidant and anti-inflammatory effects of curcumin/turmeric supplementation in adults: A GRADE-assessed systematic review and dose-response meta-analysis of randomized controlled trials. Cytokine 2023, 164, 156144. [Google Scholar] [CrossRef]
- Sharifi, S.; Fathi, N.; Memar, M.Y.; Hosseiniyan Khatibi, S.M.; Khalilov, R.; Negahdari, R.; Zununi Vahed, S.; Maleki Dizaj, S. Anti-microbial activity of curcumin nanoformulations: New trends and future perspectives. Phytother. Res. 2020, 34, 1926–1946. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Bagheri, H.; Ghasemi, F.; Khoi, J.M.; Pourhanifeh, M.H.; Heyden, Y.V.; Mortezapour, E.; Nikdasti, A.; Jeandet, P.; Khan, H.; et al. Anti-Cancer Activity of Curcumin on Multiple Myeloma. Anticancer Agents Med. Chem. 2021, 21, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V. Curcumin as a clinically-promising anti-cancer agent: Pharmacokinetics and drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 953–972. [Google Scholar] [CrossRef] [PubMed]
- Ammon, H.P.; Wahl, M.A. Pharmacology of Curcuma longa. Planta Med. 1991, 57, 1–7. [Google Scholar] [CrossRef]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 2001, 7, 1894–1900. [Google Scholar]
- Wahlstrom, B.; Blennow, G. A study on the fate of curcumin in the rat. Acta Pharmacol. Toxicol. 1978, 43, 86–92. [Google Scholar] [CrossRef]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef]
- Sazdova, I.; Keremidarska-Markova, M.; Dimitrova, D.; Mitrokhin, V.; Kamkin, A.; Hadzi-Petrushev, N.; Bogdanov, J.; Schubert, R.; Gagov, H.; Avtanski, D.; et al. Anticarcinogenic Potency of EF24: An Overview of Its Pharmacokinetics, Efficacy, Mechanism of Action, and Nanoformulation for Drug Delivery. Cancers 2023, 15, 5478. [Google Scholar] [CrossRef]
- Vareed, S.K.; Kakarala, M.; Ruffin, M.T.; Crowell, J.A.; Normolle, D.P.; Djuric, Z.; Brenner, D.E. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1411–1417. [Google Scholar] [CrossRef]
- Cunningham, R.P.; Moore, M.P.; Moore, A.N.; Healy, J.C.; Roberts, M.D.; Rector, R.S.; Martin, J.S. Curcumin supplementation mitigates NASH development and progression in female Wistar rats. Physiol. Rep. 2018, 6, e13789. [Google Scholar] [CrossRef]
- Surh, Y.J.; Chun, K.S.; Cha, H.H.; Han, S.S.; Keum, Y.S.; Park, K.K.; Lee, S.S. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: Down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat. Res. 2001, 480–481, 243–268. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.T.; Lysz, T.; Ferraro, T.; Abidi, T.F.; Laskin, J.D.; Conney, A.H. Inhibitory effects of curcumin on in vitro lipoxygenase and cyclooxygenase activities in mouse epidermis. Cancer Res. 1991, 51, 813–819. [Google Scholar]
- Dickinson, D.A.; Iles, K.E.; Zhang, H.; Blank, V.; Forman, H.J. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003, 17, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Eckert, R.L. Curcumin suppresses AP1 transcription factor-dependent differentiation and activates apoptosis in human epidermal keratinocytes. J. Biol. Chem. 2007, 282, 6707–6715. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Mechta-Grigoriou, F.; Gerald, D.; Yaniv, M. The mammalian Jun proteins: Redundancy and specificity. Oncogene 2001, 20, 2378–2389. [Google Scholar] [CrossRef]
- Shepard, A.; Hoxha, S.; Troutman, S.; Harbaugh, D.; Kareta, M.S.; Kissil, J.L. Transcriptional regulation of miR-30a by YAP impacts PTPN13 and KLF9 levels and Schwann cell proliferation. J. Biol. Chem. 2021, 297, 100962. [Google Scholar] [CrossRef]
- Ray, S.L.; Coulson, D.J.; Yeoh, M.L.Y.; Tamara, A.; Latief, J.S.; Bakhashab, S.; Weaver, J.U. The Role of miR-342 in Vascular Health. Study in Subclinical Cardiovascular Disease in Mononuclear Cells, Plasma, Inflammatory Cytokines and PANX2. Int. J. Mol. Sci. 2020, 21, 7217. [Google Scholar] [CrossRef]
- Dashek, R.J.; Cunningham, R.P.; Taylor, C.L.; Alessi, I.; Diaz, C.; Meers, G.M.; Wheeler, A.A.; Ibdah, J.A.; Parks, E.J.; Yoshida, T.; et al. Hepatocellular RECK as a Critical Regulator of Metabolic Dysfunction-associated Steatohepatitis Development. Cell. Mol. Gastroenterol. Hepatol. 2024, 18, 101365. [Google Scholar] [CrossRef]
- Huang, C.; Han, X.; Yang, L.; Song, W.; Zhang, H.; Zhu, X.; Huang, G.; Xu, J. Exosomal miR-129 and miR-342 derived from intermittent hypoxia-stimulated vascular smooth muscle cells inhibit the eIF2alpha/ATF4 axis from preventing calcified aortic valvular disease. J. Cell Commun. Signal. 2023, 17, 1449–1467. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higashi, Y.; Dashek, R.; Delafontaine, P.; Rector, R.S.; Chandrasekar, B. EF24, a Curcumin Analog, Reverses Interleukin-18-Induced miR-30a or miR-342-Dependent TRAF3IP2 Expression, RECK Suppression, and the Proinflammatory Phenotype of Human Aortic Smooth Muscle Cells. Cells 2024, 13, 1673. https://doi.org/10.3390/cells13201673
Higashi Y, Dashek R, Delafontaine P, Rector RS, Chandrasekar B. EF24, a Curcumin Analog, Reverses Interleukin-18-Induced miR-30a or miR-342-Dependent TRAF3IP2 Expression, RECK Suppression, and the Proinflammatory Phenotype of Human Aortic Smooth Muscle Cells. Cells. 2024; 13(20):1673. https://doi.org/10.3390/cells13201673
Chicago/Turabian StyleHigashi, Yusuke, Ryan Dashek, Patrice Delafontaine, Randy Scott Rector, and Bysani Chandrasekar. 2024. "EF24, a Curcumin Analog, Reverses Interleukin-18-Induced miR-30a or miR-342-Dependent TRAF3IP2 Expression, RECK Suppression, and the Proinflammatory Phenotype of Human Aortic Smooth Muscle Cells" Cells 13, no. 20: 1673. https://doi.org/10.3390/cells13201673