


A Comparative Analysis of Models for AAV-Mediated Gene Therapy for Inherited Retinal Diseases

 , , ,
, , ,
Abstract

1. Introduction
2. Mouse Models
2.1. Pathogenesis of Mouse Models for Retinitis Pigmentosa Type 28 (RP28) and X-Linked Retinoschisis (XLRS)
2.2. Advancements in Therapeutic Approaches for Retinal Degenerative Diseases in Mouse Models
2.3. Advancements in AAV-Mediated Gene Therapy for Inherited Retinal Diseases in Mouse Models
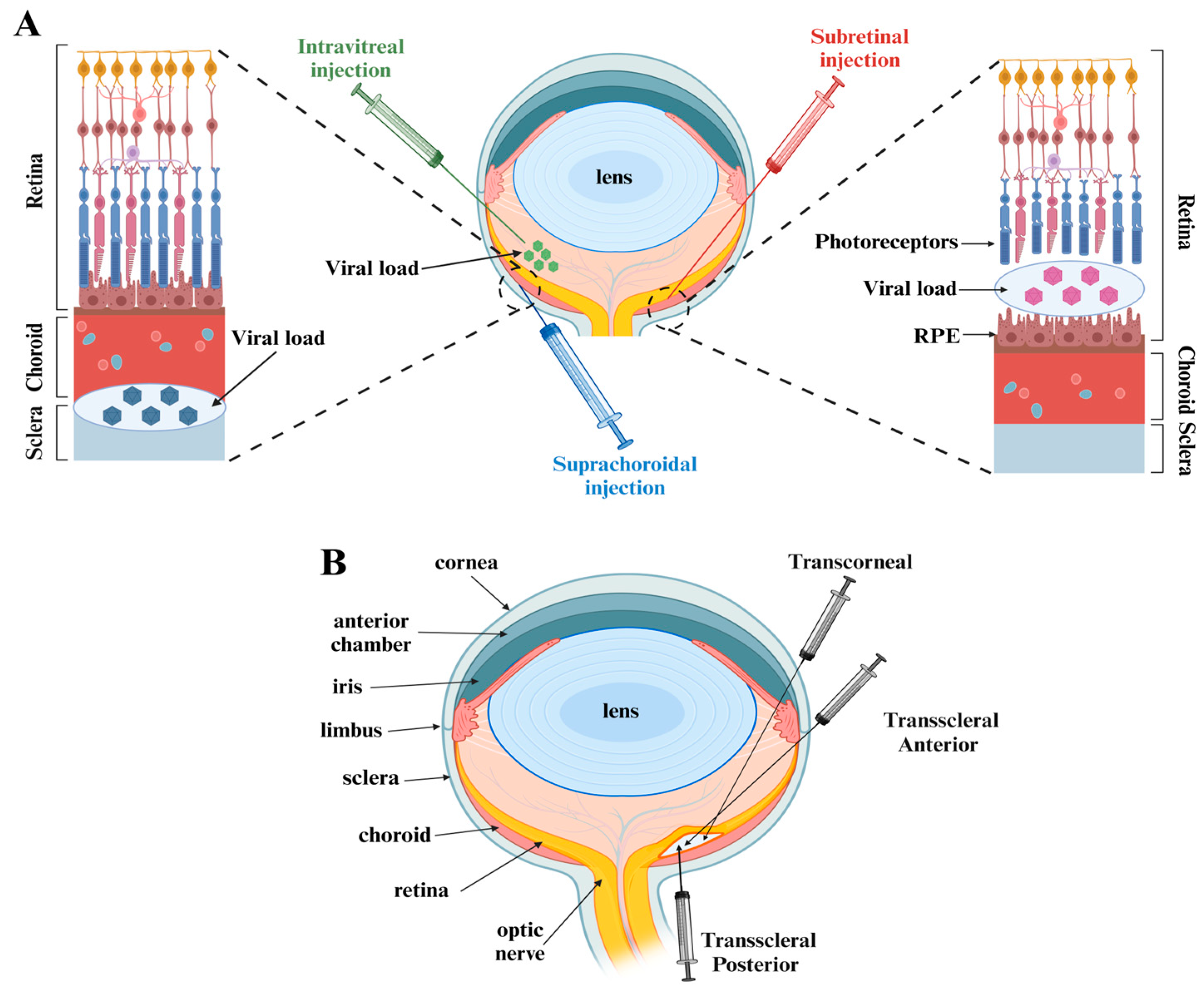
2.4. Analysis of AAV Vector Delivery Methods for Retinal Gene Therapy in Mouse Models
3. Primary Human Retinal Model
3.1. Human Retinal Architecture and Explant Cultures in Retinal Disease Research
3.2. Human Retinal Explants for AAV Vector Evaluation
3.3. Challenges and Limitations of Human Retinal Explants
4. Human Retinal Organoid Models
4.1. Innovative Approaches to the Development of Retinal Organoids
4.2. Exploring Retinal Organoids in the Study and Treatment of Genetic Retinal Diseases
4.3. Application of Retinal Organoids in AAV-Based Gene Therapy for Inherited Retinal Diseases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Benhar, I.; London, A.; Schwartz, M. The privileged immunity of immune privileged organs: The case of the eye. Front. Immunol. 2012, 3, 296. [Google Scholar] [CrossRef] [PubMed]
- Ladha, R.; Caspers, L.E.; Willermain, F.; de Smet, M.D. Subretinal Therapy: Technological Solutions to Surgical and Immunological Challenges. Front. Med. 2022, 9, 846782. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.W. Retinal gene therapy using adeno-associated viral vectors: Multiple applications for a small virus. Hum. Gene Ther. 2014, 25, 671–678. [Google Scholar] [CrossRef] [PubMed]
- FDA approves hereditary blindness gene therapy. Nat. Biotechnol. 2018, 36, 6. [CrossRef]
- Fauser, S.; Luberichs, J.; Schüttauf, F. Genetic animal models for retinal degeneration. Surv. Ophthalmol. 2002, 47, 357–367. [Google Scholar] [CrossRef]
- Barnett, J.M.; Yanni, S.E.; Penn, J.S. The development of the rat model of retinopathy of prematurity. Doc. Ophthalmol. 2010, 120, 3–12. [Google Scholar] [CrossRef]
- LaVail, M.M.; Nishikawa, S.; Steinberg, R.H.; Naash, M.I.; Duncan, J.L.; Trautmann, N.; Matthes, M.T.; Yasumura, D.; Lau-Villacorta, C.; Chen, J.; et al. Phenotypic characterization of P23H and S334ter rhodopsin transgenic rat models of inherited retinal degeneration. Exp. Eye Res. 2018, 167, 56–90. [Google Scholar] [CrossRef]
- Mage, R.G.; Esteves, P.J.; Rader, C. Rabbit models of human diseases for diagnostics and therapeutics development. Dev. Comp. Immunol. 2019, 92, 99–104. [Google Scholar] [CrossRef]
- Miyadera, K.; Acland, G.M.; Aguirre, G.D. Genetic and phenotypic variations of inherited retinal diseases in dogs: The power of within- and across-breed studies. Mamm. Genome 2012, 23, 40–61. [Google Scholar] [CrossRef]
- McCall, M.A. Pig Models in Retinal Research and Retinal Disease. Cold Spring Harb. Perspect. Med. 2024, 14, a041296. [Google Scholar] [CrossRef]
- Alsalloum, A.; Khubetsova, M.; Mityaeva, O.; Volchkov, P. Culture of human retinal explants as an ex vivo model for retinal gene therapy. bioRxiv 2024, preprint. [Google Scholar] [CrossRef]
- Dalkara, D.; Byrne, L.C.; Klimczak, R.R.; Visel, M.; Yin, L.; Merigan, W.H.; Flannery, J.G.; Schaffer, D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013, 5, 189ra76. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Colón, A.; Bighinati, A.; Parween, S.; Debnath, S.; Piano, I.; Adani, E.; Corsi, F.; Gargini, C.; Vergara, N.; Marigo, V.; et al. H105A peptide eye drops promote photoreceptor survival in murine and human models of retinal degeneration. bioRxiv 2024, preprint. [Google Scholar] [CrossRef]
- Peirson, S.N.; Brown, L.A.; Pothecary, C.A.; Benson, L.A.; Fisk, A.S. Light and the laboratory mouse. J. Neurosci. Methods 2018, 300, 26–36. [Google Scholar] [CrossRef]
- Perlman, R.L. Mouse models of human disease: An evolutionary perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef]
- Ren, D.; Fisson, S.; Dalkara, D.; Ail, D. Immune Responses to Gene Editing by Viral and Non-Viral Delivery Vectors Used in Retinal Gene Therapy. Pharmaceutics 2022, 14, 1973. [Google Scholar] [CrossRef]
- Karlstetter, M.; Sorusch, N.; Caramoy, A.; Dannhausen, K.; Aslanidis, A.; Fauser, S.; Boesl, M.R.; Nagel-Wolfrum, K.; Tamm, E.R.; Jägle, H.; et al. Disruption of the retinitis pigmentosa 28 gene Fam161a in mice affects photoreceptor ciliary structure and leads to progressive retinal degeneration. Hum. Mol. Genet. 2014, 23, 5197–5210. [Google Scholar] [CrossRef]
- Zach, F.; Grassmann, F.; Langmann, T.; Sorusch, N.; Wolfrum, U.; Stöhr, H. The retinitis pigmentosa 28 protein FAM161A is a novel ciliary protein involved in intermolecular protein interaction and microtubule association. Hum. Mol. Genet. 2012, 21, 4573–4586. [Google Scholar] [CrossRef]
- Di Gioia, S.A.; Letteboer, S.J.; Kostic, C.; Bandah-Rozenfeld, D.; Hetterschijt, L.; Sharon, D.; Arsenijevic, Y.; Roepman, R.; Rivolta, C. FAM161A, associated with retinitis pigmentosa, is a component of the cilia-basal body complex and interacts with proteins involved in ciliopathies. Hum. Mol. Genet. 2012, 21, 5174–5184. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Matsevich, C.; Obolensky, A.; Kostic, C.; Arsenijevic, Y.; Wolfrum, U.; Banin, E.; Sharon, D. A new mouse model for retinal degeneration due to Fam161a deficiency. Sci. Rep. 2021, 11, 2030. [Google Scholar] [CrossRef] [PubMed]
- Giménez, E.; Montoliu, L. A simple polymerase chain reaction assay for genotyping the retinal degeneration mutation (Pdeb(rd1)) in FVB/N-derived transgenic mice. Lab. Anim. 2001, 35, 153–156. [Google Scholar] [CrossRef]
- Mustafi, D.; Engel, A.H.; Palczewski, K. Structure of cone photoreceptors. Prog. Retin. Eye Res. 2009, 28, 289–302. [Google Scholar] [CrossRef]
- Gharib, W.H.; Robinson-Rechavi, M. When orthologs diverge between human and mouse. Brief. Bioinform. 2011, 12, 436–441. [Google Scholar] [CrossRef]
- Xiao, S.; Sun, W.; Xiao, X.; Li, S.; Luo, H.; Jia, X.; Ouyang, J.; Li, X.; Wang, Y.; Jiang, Y.; et al. Clinical and genetic features of retinoschisis in 120 families with RS1 mutations. Br. J. Ophthalmol. 2023, 107, 367–372. [Google Scholar] [CrossRef]
- Chiang, H.; Chang, E.; Harper, C.A., 3rd. Retinal detachment repair with perfluoro-N-octane endotamponade in an infant with juvenile X-linked retinoschisis. Am. J. Ophthalmol. Case Rep. 2020, 20, 100975. [Google Scholar] [CrossRef]
- Duan, C.; Ding, C.; Sun, X.; Mao, S.; Liang, Y.; Liu, X.; Ding, X.; Chen, J.; Tang, S. Retinal organoids with X-linked retinoschisis RS1 (E72K) mutation exhibit a photoreceptor developmental delay and are rescued by gene augmentation therapy. Stem Cell Res. Ther. 2024, 15, 152. [Google Scholar] [CrossRef]
- Weber, B.H.; Schrewe, H.; Molday, L.L.; Gehrig, A.; White, K.L.; Seeliger, M.W.; Jaissle, G.B.; Friedburg, C.; Tamm, E.; Molday, R.S. Inactivation of the murine X-linked juvenile retinoschisis gene, Rs1h, suggests a role of retinoschisin in retinal cell layer organization and synaptic structure. Proc. Natl. Acad. Sci. USA 2002, 99, 6222–6227. [Google Scholar] [CrossRef]
- Zeng, Y.; Takada, Y.; Kjellstrom, S.; Hiriyanna, K.; Tanikawa, A.; Wawrousek, E.; Smaoui, N.; Caruso, R.; Bush, R.A.; Sieving, P.A. RS-1 Gene Delivery to an Adult Rs1h Knockout Mouse Model Restores ERG b-Wave with Reversal of the Electronegative Waveform of X-Linked Retinoschisis. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3279–3285. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kinoshita, J.; Ivanova, E.; Sun, D.; Li, H.; Liao, T.; Cao, J.; Bell, B.A.; Wang, J.M.; Tang, Y.; et al. Mouse models of X-linked juvenile retinoschisis have an early onset phenotype, the severity of which varies with genotype. Hum. Mol. Genet. 2019, 28, 3072–3090. [Google Scholar] [CrossRef] [PubMed]
- Vijayasarathy, C.; Sardar Pasha, S.P.B.; Sieving, P.A. Of men and mice: Human X-linked retinoschisis and fidelity in mouse modeling. Prog. Retin. Eye Res. 2022, 87, 100999. [Google Scholar] [CrossRef] [PubMed]
- Mouse Genome Sequencing Consortium; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Coscas, G.; Cunha-Vaz, J.; Loewenstein, A.; Soubrane, G. Macular Edema: A Practical Approach; Karger Publishers: Basel, Switzerland, 2010; p. 59. ISBN 978-3-8055-9434-9. [Google Scholar]
- Siqueira, R.C.; Voltarelli, J.C.; Messias, A.M.; Jorge, R. Possible mechanisms of retinal function recovery with the use of cell therapy with bone marrow-derived stem cells. Arq. Bras. Oftalmol. 2010, 73, 474–479. [Google Scholar] [CrossRef]
- Machalińska, A.; Baumert, B.; Kuprjanowicz, L.; Wiszniewska, B.; Karczewicz, D.; Machaliński, B. Potential application of adult stem cells in retinal repair--challenge for regenerative medicine. Curr. Eye Res. 2009, 34, 748–760. [Google Scholar] [CrossRef]
- Jindal, N.; Banik, A.; Prabhakar, S.; Vaiphie, K.; Anand, A. Alteration of Neurotrophic Factors After Transplantation of Bone Marrow Derived Lin-ve Stem Cell in NMDA-Induced Mouse Model of Retinal Degeneration. J. Cell Biochem. 2017, 118, 1699–1711. [Google Scholar] [CrossRef]
- Dahlmann-Noor, A.; Vijay, S.; Jayaram, H.; Limb, A.; Khaw, P.T. Current approaches and future prospects for stem cell rescue and regeneration of the retina and optic nerve. Can. J. Ophthalmol. 2010, 45, 333–341. [Google Scholar] [CrossRef]
- Halioua-Haubold, C.L.; Peyer, J.G.; Smith, J.A.; Arshad, Z.; Scholz, M.; Brindley, D.A.; MacLaren, R.E. Regulatory Considerations for Gene Therapy Products in the US, EU, and Japan. Yale J. Biol. Med. 2017, 90, 683–693. [Google Scholar] [PubMed] [PubMed Central]
- Han, Z.; Conley, S.M.; Naash, M.I. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv. Exp. Med. Biol. 2014, 801, 719–724. [Google Scholar] [CrossRef]
- Cashman, S.M.; Gracias, J.; Adhi, M.; Kumar-Singh, R. Adenovirus-mediated delivery of Factor H attenuates complement C3 induced pathology in the murine retina: A potential gene therapy for age-related macular degeneration. J. Gene Med. 2015, 17, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Matet, A.; Kostic, C.; Bemelmans, A.P.; Moulin, A.; Rosolen, S.G.; Martin, S.; Mavilio, F.; Amirjanians, V.; Stieger, K.; Lorenz, B.; et al. Evaluation of tolerance to lentiviral LV-RPE65 gene therapy vector after subretinal delivery in non-human primates. Transl. Res. 2017, 188, 40–57.e4. [Google Scholar] [CrossRef] [PubMed]
- Jomary, C.; Vincent, K.; Grist, J.; Neal, M.; Jones, S. Rescue of photoreceptor function by AAV-mediated gene transfer in a mouse model of inherited retinal degeneration. Gene Ther. 1997, 4, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Fenner, B.J.; Tan, T.E.; Barathi, A.V.; Tun, S.B.B.; Yeo, S.W.; Tsai, A.S.H.; Lee, S.Y.; Cheung, C.M.G.; Chan, C.M.; Mehta, J.S.; et al. Gene-Based Therapeutics for Inherited Retinal Diseases. Front. Genet. 2022, 12, 794805. [Google Scholar] [CrossRef]
- Martin, K.R.; Quigley, H.A.; Zack, D.J.; Levkovitch-Verbin, H.; Kielczewski, J.; Valenta, D.; Baumrind, L.; Pease, M.E.; Klein, R.L.; Hauswirth, W.W. Gene therapy with brain-derived neurotrophic factor as a protection: Retinal ganglion cells in a rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4357–4365. [Google Scholar] [CrossRef]
- Nickells, R.W.; Schmitt, H.M.; Maes, M.E.; Schlamp, C.L. AAV2-Mediated Transduction of the Mouse Retina After Optic Nerve Injury. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6091–6104. [Google Scholar] [CrossRef]
- Han, I.C.; Cheng, J.L.; Burnight, E.R.; Ralston, C.L.; Fick, J.L.; Thomsen, G.J.; Tovar, E.F.; Russell, S.R.; Sohn, E.H.; Mullins, R.F.; et al. Retinal Tropism and Transduction of Adeno-Associated Virus Varies by Serotype and Route of Delivery (Intravitreal, Subretinal, or Suprachoroidal) in Rats. Hum. Gene Ther. 2020, 31, 1288–1299. [Google Scholar] [CrossRef]
- Erles, K.; Sebökovà, P.; Schlehofer, J.R. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV). J. Med. Virol. 1999, 59, 406–411. [Google Scholar] [CrossRef]
- Fuller-Carter, P.I.; Basiri, H.; Harvey, A.R.; Carvalho, L.S. Focused Update on AAV-Based Gene Therapy Clinical Trials for Inherited Retinal Degeneration. BioDrugs 2020, 34, 763–781. [Google Scholar] [CrossRef]
- Ferla, R.; Dell’Aquila, F.; Doria, M.; Ferraiuolo, M.; Noto, A.; Grazioli, F.; Ammendola, V.; Testa, F.; Melillo, P.; Iodice, C.; et al. Efficacy, pharmacokinetics, and safety in the mouse and primate retina of dual AAV vectors for Usher syndrome type 1B. Mol. Ther. Methods Clin. Dev. 2023, 28, 396–411. [Google Scholar] [CrossRef] [PubMed]
- Vendomèle, J.; Chauveau, G.A.; Dalkara, D.; Galy, A.; Fisson, S. Peripheral Cellular Immune Responses Induced by Subretinal Adeno-Associated Virus Gene Transfer Can Be Restrained by the Subretinal-Associated Immune Inhibition Mechanism. Hum. Gene Ther. 2024, 35, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Long, B.R.; Sandza, K.; Holcomb, J.; Crockett, L.; Hayes, G.M.; Arens, J.; Fonck, C.; Tsuruda, L.S.; Schweighardt, B.; O’Neill, C.A.; et al. The Impact of Pre-existing Immunity on the Non-clinical Pharmacodynamics of AAV5-Based Gene Therapy. Mol. Ther. Methods Clin. Dev. 2019, 13, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Datta, S.; Brabbit, E.; Love, Z.; Woytowicz, V.; Flattery, K.; Capri, J.; Yao, K.; Wu, S.; Imboden, M.; et al. Nr2e3 is a genetic modifier that rescues retinal degeneration and promotes homeostasis in multiple models of retinitis pigmentosa. Gene Ther. 2021, 28, 223–241. [Google Scholar] [CrossRef] [PubMed]
- McNamee, S.M.; Chan, N.P.; Akula, M.; Avola, M.O.; Whalen, M.; Nystuen, K.; Singh, P.; Upadhyay, A.K.; DeAngelis, M.M.; Haider, N.B. Preclinical dose response study shows NR2E3 can attenuate retinal degeneration in the retinitis pigmentosa mouse model RhoP23H+/−. Gene Ther. 2024, 31, 255–262. [Google Scholar] [CrossRef]
- Olivares, A.M.; Jelcick, A.S.; Reinecke, J.; Leehy, B.; Haider, A.; Morrison, M.A.; Cheng, L.; Chen, D.F.; DeAngelis, M.M.; Haider, N.B. Multimodal Regulation Orchestrates Normal and Complex Disease States in the Retina. Sci. Rep. 2017, 7, 690. [Google Scholar] [CrossRef]
- Westhaus, A.; Eamegdool, S.S.; Fernando, M.; Fuller-Carter, P.; Brunet, A.A.; Miller, A.L.; Rashwan, R.; Knight, M.; Daniszewski, M.; Lidgerwood, G.E.; et al. AAV capsid bioengineering in primary human retina models. Sci. Rep. 2023, 13, 21946. [Google Scholar] [CrossRef]
- Pavlou, M.; Schön, C.; Occelli, L.M.; Rossi, A.; Meumann, N.; Boyd, R.F.; Bartoe, J.T.; Siedlecki, J.; Gerhardt, M.J.; Babutzka, S.; et al. Novel AAV capsids for intravitreal gene therapy of photoreceptor disorders. EMBO Mol. Med. 2021, 13, e13392. [Google Scholar] [CrossRef]
- Drag, S.; Dotiwala, F.; Upadhyay, A.K. Gene Therapy for Retinal Degenerative Diseases: Progress, Challenges, and Future Directions. Investig. Ophthalmol. Vis. Sci. 2023, 64, 39. [Google Scholar] [CrossRef]
- Dhurandhar, D.; Sahoo, N.K.; Mariappan, I.; Narayanan, R. Gene therapy in retinal diseases: A review. Indian. J. Ophthalmol. 2021, 69, 2257–2265. [Google Scholar] [CrossRef]
- Irigoyen, C.; Amenabar Alonso, A.; Sanchez-Molina, J.; Rodríguez-Hidalgo, M.; Lara-López, A.; Ruiz-Ederra, J. Subretinal Injection Techniques for Retinal Disease: A Review. J. Clin. Med. 2022, 11, 4717. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, I.; Heredero Berzal, A.; Koster, C.; Ten Asbroek, A.L.M.A.; Bergen, A.A.; Boon, C.J.F. The Road towards Gene Therapy for X-Linked Juvenile Retinoschisis: A Systematic Review of Preclinical Gene Therapy in Cell-Based and Rodent Models of XLRS. Int. J. Mol. Sci. 2024, 25, 1267. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Shen, J.; Hafiz, Z.; Hackett, S.F.; Silva, R.L.E.; Khan, M.; Lorenc, V.E.; Chen, D.; Chadha, R.; Zhang, M.; et al. AAV8-vectored suprachoroidal gene transfer produces widespread ocular transgene expression. J. Clin. Investig. 2019, 129, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- Madrakhimov, S.B.; Yang, J.Y.; Ahn, D.H.; Han, J.W.; Ha, T.H.; Park, T.K. Peripapillary Intravitreal Injection Improves AAV-Mediated Retinal Transduction. Mol. Ther. Methods Clin. Dev. 2020, 17, 647–656. [Google Scholar] [CrossRef]
- Igarashi, T.; Miyake, K.; Asakawa, N.; Miyake, N.; Shimada, T.; Takahashi, H. Direct comparison of administration routes for AAV8-mediated ocular gene therapy. Curr. Eye Res. 2013, 38, 569–577. [Google Scholar] [CrossRef]
- Takahashi, K.; Igarashi, T.; Miyake, K.; Kobayashi, M.; Yaguchi, C.; Iijima, O.; Yamazaki, Y.; Katakai, Y.; Miyake, N.; Kameya, S.; et al. Improved Intravitreal AAV-Mediated Inner Retinal Gene Transduction after Surgical Internal Limiting Membrane Peeling in Cynomolgus Monkeys. Mol. Ther. 2017, 25, 296–302. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Milosavljevic, N.; Bedford, R.A.; Lucas, R.J.; Bishop, P.N. Efficacy and Safety of Glycosidic Enzymes for Improved Gene Delivery to the Retina following Intravitreal Injection in Mice. Mol. Ther. Methods Clin. Dev. 2017, 9, 192–202. [Google Scholar] [CrossRef]
- Carvalho, C.; Lemos, L.; Antas, P.; Seabra, M.C. Gene therapy for inherited retinal diseases: Exploiting new tools in genome editing and nanotechnology. Front. Ophthalmol. 2023, 3, 1270561. [Google Scholar] [CrossRef]
- Peng, Y.; Tang, L.; Zhou, Y. Subretinal Injection: A Review on the Novel Route of Therapeutic Delivery for Vitreoretinal Diseases. Ophthalmic Res. 2017, 58, 217–226. [Google Scholar] [CrossRef]
- Qi, Y.; Dai, X.; Zhang, H.; He, Y.; Zhang, Y.; Han, J.; Zhu, P.; Zhang, Y.; Zheng, Q.; Li, X.; et al. Trans-Corneal Subretinal Injection in Mice and Its Effect on the Function and Morphology of the Retina. PLoS ONE 2015, 10, e0136523. [Google Scholar] [CrossRef]
- Butler, M.C.; Sullivan, J.M. Ultrahigh Resolution Mouse Optical Coherence Tomography to Aid Intraocular Injection in Retinal Gene Therapy Research. J. Vis. Exp. 2018, 141, e55894. [Google Scholar] [CrossRef]
- Kansara, V.; Muya, L.; Wan, C.R.; Ciulla, T.A. Suprachoroidal Delivery of Viral and Nonviral Gene Therapy for Retinal Diseases. J. Ocul. Pharmacol. Ther. 2020, 36, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Ciulla, T.; Yeh, S. Microinjection via the suprachoroidal space: A review of a novel mode of administration. Am. J. Manag. Care 2022, 28, S243–S252. [Google Scholar] [CrossRef] [PubMed]
- Hancock, S.E.; Wan, C.R.; Fisher, N.E.; Andino, R.V.; Ciulla, T.A. Biomechanics of suprachoroidal drug delivery: From benchtop to clinical investigation in ocular therapies. Expert Opin. Drug Deliv. 2021, 18, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.Y.; Fujioka, J.K.; Gholamian, T.; Zaharia, M.; Tran, S.D. Suprachoroidal Injection: A Novel Approach for Targeted Drug Delivery. Pharmaceuticals 2023, 16, 1241. [Google Scholar] [CrossRef]
- Seung, H.S.; Sümbül, U. Neuronal cell types and connectivity: Lessons from the retina. Neuron 2014, 83, 1262–1272. [Google Scholar] [CrossRef]
- Anderson, D.E.; Holstein, S.A.; Kedar, S. Visual Pathway Degeneration in Chemotherapy-Related Neurotoxicity: A Review and Directions for Future Research. Neuroophthalmology 2020, 44, 139–147. [Google Scholar] [CrossRef]
- Masri, R.A.; Weltzien, F.; Purushothuman, S.; Lee, S.C.S.; Martin, P.R.; Grünert, U. Composition of the Inner Nuclear Layer in Human Retina. Investig. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef]
- Lujan, B.J.; Roorda, A.; Croskrey, J.A.; Dubis, A.M.; Cooper, R.F.; Bayabo, J.K.; Duncan, J.L.; Antony, B.J.; Carroll, J. Directional optical coherence tomography provides accurate outer nuclear layer and Henle fiber layer measurements. Retina 2015, 35, 1511–1520. [Google Scholar] [CrossRef]
- Engelsberg, K.; Ehinger, B.; Ghosh, F. Early development of retinal subtypes in long-term cultures of human embryonic retina. Curr. Eye Res. 2008, 33, 185–191. [Google Scholar] [CrossRef]
- Osborne, A.; Sanderson, J.; Martin, K.R. Neuroprotective Effects of Human Mesenchymal Stem Cells and Platelet-Derived Growth Factor on Human Retinal Ganglion Cells. Stem Cells 2018, 36, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Duebel, J.; Balya, D.; Fradot, M.; Viney, T.J.; Siegert, S.; Groner, A.C.; Cabuy, E.; Forster, V.; Seeliger, M.; et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010, 329, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Murali, A.; Ramlogan-Steel, C.A.; Andrzejewski, S.; Steel, J.C.; Layton, C.J. Retinal explant culture: A platform to investigate human neuro-retina. Clin. Exp. Ophthalmol. 2019, 47, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Orlans, H.O.; Edwards, T.L.; De Silva, S.R.; Patrício, M.I.; MacLaren, R.E. Human Retinal Explant Culture for Ex Vivo Validation of AAV Gene Therapy. Methods Mol. Biol. 2018, 1715, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, K.; Valtink, M. RPE cell cultivation. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 65–67. [Google Scholar] [CrossRef]
- Remington, L.A.; Goodwin, D. Clinical Anatomy and Physiology of the Visual System, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 61–92. ISBN 9780323711685. [Google Scholar]
- Fronk, A.H.; Vargis, E. Methods for culturing retinal pigment epithelial cells: A review of current protocols and future recommendations. J. Tissue Eng. 2016, 7, 2041731416650838. [Google Scholar] [CrossRef]
- Lynn, S.A.; Ward, G.; Keeling, E.; Scott, J.A.; Cree, A.J.; Johnston, D.A.; Page, A.; Cuan-Urquizo, E.; Bhaskar, A.; Grossel, M.C.; et al. Ex-vivo models of the Retinal Pigment Epithelium (RPE) in long-term culture faithfully recapitulate key structural and physiological features of native RPE. Tissue Cell 2017, 49, 447–460. [Google Scholar] [CrossRef]
- Becker, J.; Fakhiri, J.; Grimm, D. Fantastic AAV Gene Therapy Vectors and How to Find Them-Random Diversification, Rational Design and Machine Learning. Pathogens 2022, 11, 756. [Google Scholar] [CrossRef]
- Hickey, D.G.; Edwards, T.L.; Barnard, A.R.; Singh, M.S.; de Silva, S.R.; McClements, M.E.; Flannery, J.G.; Hankins, M.W.; MacLaren, R.E. Tropism of engineered and evolved recombinant AAV serotypes in the rd1 mouse and ex vivo primate retina. Gene Ther. 2017, 24, 787–800. [Google Scholar] [CrossRef]
- Dalkara, D.; Kolstad, K.D.; Caporale, N.; Visel, M.; Klimczak, R.R.; Schaffer, D.V.; Flannery, J.G. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol. Ther. 2009, 17, 2096–2102. [Google Scholar] [CrossRef]
- Xi, Z.; Öztürk, B.E.; Johnson, M.E.; Turunç, S.; Stauffer, W.R.; Byrne, L.C. Quantitative single-cell transcriptome-based ranking of engineered AAVs in human retinal explants. Mol. Ther. Methods Clin. Dev. 2022, 25, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, B.E.; Johnson, M.E.; Kleyman, M.; Turunç, S.; He, J.; Jabalameli, S.; Xi, Z.; Visel, M.; Dufour, V.L.; Iwabe, S.; et al. scAAVengr, a transcriptome-based pipeline for quantitative ranking of engineered AAVs with single-cell resolution. Elife 2021, 10, e64175. [Google Scholar] [CrossRef]
- Westhaus, A.; Cabanes-Creus, M.; Jonker, T.; Sallard, E.; Navarro, R.G.; Zhu, E.; Torres, G.B.; Lee, S.; Wilmott, P.; Gonzalez-Cordero, A.; et al. AAV-p40 Bioengineering Platform for Variant Selection Based on Transgene Expression. Hum. Gene Ther. 2022, 33, 664–682. [Google Scholar] [CrossRef]
- Khabou, H.; Garita-Hernandez, M.; Chaffiol, A.; Reichman, S.; Jaillard, C.; Brazhnikova, E.; Bertin, S.; Forster, V.; Desrosiers, M.; Winckler, C.; et al. Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight 2018, 3, e96029. [Google Scholar] [CrossRef] [PubMed]
- Hulliger, E.C.; Hostettler, S.M.; Kleinlogel, S. Empowering Retinal Gene Therapy with a Specific Promoter for Human Rod and Cone ON-Bipolar Cells. Mol. Ther. Methods Clin. Dev. 2020, 17, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, B.; Laperrousaz, E.; Tribble, J.R.; Verhaagen, J.; Fawcett, J.W.; Martin, K.R.; Williams, P.A.; Osborne, A. Improving adeno-associated viral (AAV) vector-mediated transgene expression in retinal ganglion cells: Comparison of five promoters. Gene Ther. 2023, 30, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.S.; Lee, V.; Wei, Z.; Song, J.Y.; Casal, G.; Cronin, T.; Willett, K.; Huckfeldt, R.; Morgan, J.I.; Aleman, T.S.; et al. Evaluation of Dose and Safety of AAV7m8 and AAV8BP2 in the Non-Human Primate Retina. Hum. Gene Ther. 2017, 28, 154–167. [Google Scholar] [CrossRef]
- Armitage, W.J.; Jones, M.N.; Zambrano, I.; Carley, F.; Tole, D.M. The suitability of corneas stored by organ culture for penetrating keratoplasty and influence of donor and recipient factors on 5-year graft survival. Investig. Ophthalmol. Vis. Sci. 2014, 55, 784–791. [Google Scholar] [CrossRef]
- Wiley, L.A.; Burnight, E.R.; Kaalberg, E.E.; Jiao, C.; Riker, M.J.; Halder, J.A.; Luse, M.A.; Han, I.C.; Russell, S.R.; Sohn, E.H.; et al. Assessment of Adeno-Associated Virus Serotype Tropism in Human Retinal Explants. Hum. Gene Ther. 2018, 29, 424–436. [Google Scholar] [CrossRef]
- Ghareeb, A.E.; Lako, M.; Steel, D.H. Coculture techniques for modeling retinal development and disease, and enabling regenerative medicine. Stem Cells Transl. Med. 2020, 9, 1531–1548. [Google Scholar] [CrossRef]
- Schnichels, S.; Paquet-Durand, F.; Löscher, M.; Tsai, T.; Hurst, J.; Joachim, S.C.; Klettner, A. Retina in a dish: Cell cultures, retinal explants and animal models for common diseases of the retina. Prog. Retin. Eye Res. 2021, 81, 100880. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Nasonkin, I.O. Limitations and Promise of Retinal Tissue From Human Pluripotent Stem Cells for Developing Therapies of Blindness. Front. Cell Neurosci. 2020, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Diacou, R.; Nandigrami, P.; Fiser, A.; Liu, W.; Ashery-Padan, R.; Cvekl, A. Cell fate decisions, transcription factors and signaling during early retinal development. Prog. Retin. Eye Res. 2022, 91, 101093. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, R.; Kageyama, R. Regulation of retinal cell fate specification by multiple transcription factors. Brain Res. 2008, 1192, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef]
- Cowan, C.S.; Renner, M.; De Gennaro, M.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikram, T.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e34. [Google Scholar] [CrossRef]
- Sridhar, A.; Hoshino, A.; Finkbeiner, C.R.; Chitsazan, A.; Dai, L.; Haugan, A.K.; Eschenbacher, K.M.; Jackson, D.L.; Trapnell, C.; Bermingham-McDonogh, O.; et al. Single-Cell Transcriptomic Comparison of Human Fetal Retina, hPSC-Derived Retinal Organoids, and Long-Term Retinal Cultures. Cell Rep. 2020, 30, 1644–1659.e4. [Google Scholar] [CrossRef]
- Wahle, P.; Brancati, G.; Harmel, C.; He, Z.; Gut, G.; Del Castillo, J.S.; Xavier da Silveira Dos Santos, A.; Yu, Q.; Noser, P.; Fleck, J.S.; et al. Multimodal spatiotemporal phenotyping of human retinal organoid development. Nat. Biotechnol. 2023, 41, 1765–1775. [Google Scholar] [CrossRef]
- Heredero Berzal, A.; Wagstaff, E.L.; Ten Asbroek, A.L.M.A.; Ten Brink, J.B.; Bergen, A.A.; Boon, C.J.F. The Analysis of Embryoid Body Formation and Its Role in Retinal Organoid Development. Int. J. Mol. Sci. 2024, 25, 1444. [Google Scholar] [CrossRef]
- Wagstaff, E.L.; Heredero Berzal, A.; Boon, C.J.F.; Quinn, P.M.J.; Ten Asbroek, A.L.M.A.; Bergen, A.A. The Role of Small Molecules and Their Effect on the Molecular Mechanisms of Early Retinal Organoid Development. Int. J. Mol. Sci. 2021, 22, 7081. [Google Scholar] [CrossRef]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A look into retinal organoids: Methods, analytical techniques, and applications. Cell Mol. Life Sci. 2021, 78, 6505–6532. [Google Scholar] [CrossRef]
- Kuwahara, A.; Ozone, C.; Nakano, T.; Saito, K.; Eiraku, M.; Sasai, Y. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 2015, 6, 6286. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, P.; Ma, J.H.; Cui, Z.; Yu, Q.; Liu, S.; Xue, Y.; Zhu, D.; Cao, J.; Li, Z.; et al. Modeling Retinitis Pigmentosa: Retinal Organoids Generated From the iPSCs of a Patient With the USH2A Mutation Show Early Developmental Abnormalities. Front. Cell Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Breuer, D.K.; Yashar, B.M.; Filippova, E.; Hiriyanna, S.; Lyons, R.H.; Mears, A.J.; Asaye, B.; Acar, C.; Vervoort, R.; Wright, A.F.; et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 2002, 70, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Kondkar, A.A.; Abu-Amero, K.K. Leber congenital amaurosis: Current genetic basis, scope for genetic testing and personalized medicine. Exp. Eye Res. 2019, 189, 107834. [Google Scholar] [CrossRef]
- Lukovic, D.; Artero Castro, A.; Kaya, K.D.; Munezero, D.; Gieser, L.; Davó-Martínez, C.; Corton, M.; Cuenca, N.; Swaroop, A.; Ramamurthy, V.; et al. Retinal Organoids derived from hiPSCs of an AIPL1-LCA Patient Maintain Cytoarchitecture despite Reduced levels of Mutant AIPL1. Sci. Rep. 2020, 10, 5426. [Google Scholar] [CrossRef]
- Buck, T.M.; Wijnholds, J. Recombinant Adeno-Associated Viral Vectors (rAAV)-Vector Elements in Ocular Gene Therapy Clinical Trials and Transgene Expression and Bioactivity Assays. Int. J. Mol. Sci. 2020, 21, 4197. [Google Scholar] [CrossRef]
- McClements, M.E.; Steward, H.; Atkin, W.; Goode, E.A.; Gándara, C.; Chichagova, V.; MacLaren, R.E. Tropism of AAV Vectors in Photoreceptor-Like Cells of Human iPSC-Derived Retinal Organoids. Transl. Vis. Sci. Technol. 2022, 11, 3. [Google Scholar] [CrossRef]
- Garita-Hernandez, M.; Routet, F.; Guibbal, L.; Khabou, H.; Toualbi, L.; Riancho, L.; Reichman, S.; Duebel, J.; Sahel, J.-A.; Goureau, O.; et al. AAV-Mediated Gene Delivery to 3D Retinal Organoids Derived from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 994. [Google Scholar] [CrossRef]
- Boon, N.; Lu, X.; Andriessen, C.A.; Moustakas, I.; Buck, T.M.; Freund, C.; Arendzen, C.H.; Böhringer, S.; Mei, H.; Wijnholds, J. AAV-mediated gene augmentation therapy of CRB1 patient-derived retinal organoids restores the histological and transcriptional retinal phenotype. Stem Cell Rep. 2023, 18, 1123–1137. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed]
- West, E.L.; Majumder, P.; Naeem, A.; Fernando, M.; O’Hara-Wright, M.; Lanning, E.; Kloc, M.; Ribeiro, J.; Ovando-Roche, P.; Shum, I.O.; et al. Antioxidant and lipid supplementation improve the development of photoreceptor outer segments in pluripotent stem cell-derived retinal organoids. Stem Cell Rep. 2022, 17, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Perdigão, P.R.L.; Ollington, B.; Sai, H.; Leung, A.; Sacristan-Reviriego, A.; van der Spuy, J. Retinal Organoids from an AIPL1 CRISPR/Cas9 Knockout Cell Line Successfully Recapitulate the Molecular Features of LCA4 Disease. Int. J. Mol. Sci. 2023, 24, 5912. [Google Scholar] [CrossRef] [PubMed]
- Sladen, P.E.; Naeem, A.; Adefila-Ideozu, T.; Vermeule, T.; Busson, S.L.; Michaelides, M.; Naylor, S.; Forbes, A.; Lane, A.; Georgiadis, A. AAV-RPGR Gene Therapy Rescues Opsin Mislocalisation in a Human Retinal Organoid Model of RPGR-Associated X-Linked Retinitis Pigmentosa. Int. J. Mol. Sci. 2024, 25, 1839. [Google Scholar] [CrossRef]
- Leung, A.; Sacristan-Reviriego, A.; Perdigão, P.R.L.; Sai, H.; Georgiou, M.; Kalitzeos, A.; Carr, A.F.; Coffey, P.J.; Michaelides, M.; Bainbridge, J.; et al. Investigation of PTC124-mediated translational readthrough in a retinal organoid model of AIPL1-associated Leber congenital amaurosis. Stem Cell Rep. 2022, 17, 2187–2202. [Google Scholar] [CrossRef]
- Sai, H.; Ollington, B.; Rezek, F.O.; Chai, N.; Lane, A.; Georgiadis, T.; Bainbridge, J.; Michaelides, M.; Sacristan-Reviriego, A.; Perdigão, P.R.L.; et al. Effective AAV-mediated gene replacement therapy in retinal organoids modeling AIPL1-associated LCA4. Mol. Ther. Nucleic Acids 2024, 35, 102148. [Google Scholar] [CrossRef]
- Kaya, K.D.; Chen, H.Y.; Brooks, M.J.; Kelley, R.A.; Shimada, H.; Nagashima, K.; de Val, N.; Drinnan, C.T.; Gieser, L.; Kruczek, K.; et al. Transcriptome-based molecular staging of human stem cell-derived retinal organoids uncovers accelerated photoreceptor differentiation by 9-cis retinal. Mol. Vis. 2019, 25, 663–678. [Google Scholar] [PubMed] [PubMed Central]
- Kruczek, K.; Qu, Z.; Welby, E.; Shimada, H.; Hiriyanna, S.; English, M.A.; Zein, W.M.; Brooks, B.P.; Swaroop, A. In vitro modeling and rescue of ciliopathy associated with IQCB1/NPHP5 mutations using patient-derived cells. Stem Cell Rep. 2022, 17, 2172–2186. [Google Scholar] [CrossRef]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef]
- Slembrouck-Brec, A.; Rodrigues, A.; Rabesandratana, O.; Gagliardi, G.; Nanteau, C.; Fouquet, S.; Thuret, G.; Reichman, S.; Orieux, G.; Goureau, O. Reprogramming of Adult Retinal Müller Glial Cells into Human-Induced Pluripotent Stem Cells as an Efficient Source of Retinal Cells. Stem Cells Int. 2019, 2019, 7858796. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.; Slembrouck-Brec, A.; Nanteau, C.; Terray, A.; Tymoshenko, Y.; Zagar, Y.; Reichman, S.; Xi, Z.; Sahel, J.A.; Fouquet, S.; et al. Modeling PRPF31 retinitis pigmentosa using retinal pigment epithelium and organoids combined with gene augmentation rescue. NPJ Regen. Med. 2022, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Kruczek, K.; Qu, Z.; Gentry, J.; Fadl, B.R.; Gieser, L.; Hiriyanna, S.; Batz, Z.; Samant, M.; Samanta, A.; Chu, C.J.; et al. Gene Therapy of Dominant CRX-Leber Congenital Amaurosis using Patient Stem Cell-Derived Retinal Organoids. Stem Cell Rep. 2021, 16, 252–263. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Inheritance Type | Retinal Organoid Differentiation Procedure | Neural Induction Media (NIM) | Retinal Differentiation Media | AAV Type | Transduction-Readout Time Point | Main Results | Reference |
|---|---|---|---|---|---|---|---|---|---|
| RP or LCA | CRB1 | autosomal recessive disease | EB formation then transferred to adherent culture, NRV isolation and cultivation in suspension. Retinal organoid differentiation was carried out as reported previously with some modifications [125] | D1: mTesR/NIM 3:1 (DMEM/F12 1:1, N2, NEAA, 2 μg/mL heparin) D2: mTesR1/NIM 1:1 D3–D15: 100% NIM | D16–D34: DMEM/F12 3:1, B27, NEAA D35–D6\3: +FBS, 100 µM Taurine D64–D84: +1 µM RA D84–D119: +0.5 µM RA Dd120–end—without RA | AAV5.CMVmin.hCRB1 or AAV5.CMVmin.hCRB2 | D120–D180, D210 | Increased number of photoreceptor nuclei. Increased CRB1 localization at the OLM after AAV.hCRB1 treatment. Overexpression of CRB2 in photoreceptor cells after AAV.hCRB2 treatment. Increased thickness of the ONL, but not the retina or the INL. Restoration of gene expression related to the endosomal system to isogenic control levels. | [124] |
| X-linked RP | RP2 | X-linked recessive disease | EB formation then transferred to adherent culture, NRV isolation and cultivation in suspension. Retinal organoid differentiation was carried out as reported previously with some modifications [125]. | D1: E8/NIM (DMEM/F12 1:1, N2, NEAA, 2 μg/mL heparin) 3:1 D2: E8/NIM 1:1 D3–D15: 100% NIM | D16–D41: DMEM/F12 3:1, B27, NEAA D42–D63: +FBS, 100 μM Taurine D63–D91: + 1 μM RA D92–D139: +0.5 μM RA D140–end: without RA | AAV5.CAGp.RP2 | D140–D180 | ONL thickening close to endogenous control. The percentage of rhodopsin-positive cells was above average in the non transduced RP2 KO. The percentage of cone arrestin-positive cells was reduced after AAV transduction. The rescue of the ONL thinning phenotype in RP2 KO ROs suggests a protective effect of RP2 overexpression in photoreceptor cells. | [117] |
| X-linked RP | RPGR | X-linked recessive disease | Adherent culture, NRV isolation and cultivation in suspension. Retinal organoid differentiation was carried out as previously reported [126,127]. | D3–D28-42: DMEM/F12 (1:1), NEAA, N2 before NRV formation | D28–42–D69: DMEM/F12 3:81, FBS, B27, 100 μM Taurine D70–D83: +1 μM retinoic acid D84–D99: +N2, 0.5 μM RA D100–end of differentiation: without B27 and RA | AAV2.7m8.hRKp.PGRORF15 | D135–D160 | CRX-positive photoreceptor cells within the ONL. The extension of recoverin positive OS into the peripheral space. Significantly upregulated expression of the RPGRORF15 transcript. | [128] |
| LCA4 | AIPL1 | autosomal recessive disease | Adherent culture, NRV isolation and cultivation in suspension. Retinal organoid differentiation was carried out as previously reported [127,129]. | D3–D28-42: DMEM/F12 (1:1), NEAA, N2 before NRV formation | D28–42–D69: DMEM/F12 3:1, FBS, B27, 100 μM Taurine D70–D83: +1 μM RA D84–D99: +N2, 0.5 μM RA D100–end: without B27 and RA | AAV2.7m8.hRKp.AIPL1 | D161–D175, D219, D231 | Rod OS structures increased significantly in length, and the abnormal accumulation of rhodopsin in the somas of patient rods was abolished. L/M-opsin cone OSs also recovered significantly and CEP290 protein became detectable. Localization of other OS proteins, including visual arrestin, peripherin2 phosphodiesterase 6B, and rod α-transducin, was restored to varying degrees. Reduced levels of S-opsin were mislocalized to axons and synaptic pedicles. OS biogenesis was at least partially rescued. | [130] |
| LCA and renal-retinal Senior-Løken syndrome | IQCB1/NPHP5 | autosomal recessive disease | EB formation then transferred to adherent culture, NRV isolation and cultivation in suspension. Retinal organoid differentiation was carried out as previously reported, with some modifications [131]. | D1: E8/NIM 3:1 (DMEM/F12 1:1, N2, NEAA, 2 μg/mL heparin) D2: E8/NIM 1:1 D3–D15: 100% NIM | D16–D28: DMEM/F12 3:1, B27, NEAA/NIM 3:1 D28–D41: +20 ng/mL IGF-1 D42—D62: +FBS, 100 µM Taurine D63—D90: +1 µM 9-cis-retinaldehyde D91–D119: +0.5 µM 9-cis-retinaldehyde D120–end: replacing FBS with KSR | AAV2.CMV.NPHP5 | D120–D150, D200 | Upregulation of genes primarily associated with innate immunity or interferon-induced viral responses. Remarkable recovery of PDE6α and PDE6β was observed in both models, indicating that AIPL1 function in the ONL was restored. | [132] |
| RP | PRPF31 | autosomal dominant disease | Adherent culture, NRV isolation and cultivation in suspension. Retinal organoid differentiation was carried out as previously reported [133,134]. | D0–D2: TeSR-E6 D3–D27: +N2 | D28–D34: DMEM/F-12, 1:1, NEAA, B27, 10 ng/mL FGF2 D35–D83: +FBS, without FGF2 D84–D200: DMEM/F-12, B27, NEAA | AAV2.7m8.CAG.PRPF31 | D85–D175 | Displayed ~40% NRL-positive rods and 20% hCAR-positive cones. Increasing PRPF31 expression levels directly, preventing photoreceptor degeneration in mature retinal organoids transduced before the first signs of degeneration. | [135] |
| LCA | CRX | autosomal dominant disease | EB formation then transferred to adherent culture, NRV isolation and cultivation in suspension. Retinal organoid differentiation was carried out as previously reported with modification of culturing dissected retinal organoids individually in a 96-well plate format [131]. | D1: E8/NIM 3:1 (DMEM/F12 1:1, N2, NEAA, 2 μg/mL heparin) D2: E8/NIM 1:1 D3–D15: 100% NIM | D16–D28: DMEM/F12 3:1, B27, NEAA/NIM 3:1 D28–D41: +20 ng/mL IGF-1 D42–D62: +FBS, 100 µM Taurine D63–D90: +1 µM 9-cis-retinaldehyde D91–end: +0.5 µM 9-cis-retinaldehyde | AAV2.CRX.CRX | D120–D150, D180 | Increased CRX mRNA and protein in treated organoids. Rescued rhodopsin expression. Partially restored L/M opsin expression. Long-term expression of CRX did not result in activation of the apoptotic marker Caspase3. | [136] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsalloum, A.; Gornostal, E.; Mingaleva, N.; Pavlov, R.; Kuznetsova, E.; Antonova, E.; Nadzhafova, A.; Kolotova, D.; Kadyshev, V.; Mityaeva, O.; et al. A Comparative Analysis of Models for AAV-Mediated Gene Therapy for Inherited Retinal Diseases. Cells 2024, 13, 1706. https://doi.org/10.3390/cells13201706
Alsalloum A, Gornostal E, Mingaleva N, Pavlov R, Kuznetsova E, Antonova E, Nadzhafova A, Kolotova D, Kadyshev V, Mityaeva O, et al. A Comparative Analysis of Models for AAV-Mediated Gene Therapy for Inherited Retinal Diseases. Cells. 2024; 13(20):1706. https://doi.org/10.3390/cells13201706
Chicago/Turabian StyleAlsalloum, Almaqdad, Ekaterina Gornostal, Natalia Mingaleva, Roman Pavlov, Ekaterina Kuznetsova, Ekaterina Antonova, Aygun Nadzhafova, Daria Kolotova, Vitaly Kadyshev, Olga Mityaeva, and et al. 2024. "A Comparative Analysis of Models for AAV-Mediated Gene Therapy for Inherited Retinal Diseases" Cells 13, no. 20: 1706. https://doi.org/10.3390/cells13201706
APA StyleAlsalloum, A., Gornostal, E., Mingaleva, N., Pavlov, R., Kuznetsova, E., Antonova, E., Nadzhafova, A., Kolotova, D., Kadyshev, V., Mityaeva, O., & Volchkov, P. (2024). A Comparative Analysis of Models for AAV-Mediated Gene Therapy for Inherited Retinal Diseases. Cells, 13(20), 1706. https://doi.org/10.3390/cells13201706