Abstract
TRPM3 belongs to the melastatin sub-family of transient receptor potential (TRPM) cation channels and has been shown to function as a steroid-activated, heat-sensitive calcium ion (Ca2+) channel. A missense substitution (p.I65M) in the TRPM3 gene of humans (TRPM3) and mice (Trpm3) has been shown to underlie an inherited form of early-onset, progressive cataract. Here, we model the pathogenetic effects of this cataract-causing mutation using ‘knock-in’ mutant mice and human cell lines. Trpm3 and its intron-hosted micro-RNA gene (Mir204) were strongly co-expressed in the lens epithelium and other non-pigmented and pigmented ocular epithelia. Homozygous Trpm3-mutant lenses displayed elevated cytosolic Ca2+ levels and an imbalance of sodium (Na+) and potassium (K+) ions coupled with increased water content. Homozygous TRPM3-mutant human lens epithelial (HLE-B3) cell lines and Trpm3-mutant lenses exhibited increased levels of phosphorylated mitogen-activated protein kinase 1/extracellular signal-regulated kinase 2 (MAPK1/ERK2/p42) and MAPK3/ERK1/p44. Mutant TRPM3-M65 channels displayed an increased sensitivity to external Ca2+ concentration and an altered dose response to pregnenolone sulfate (PS) activation. Trpm3-mutant lenses shared the downregulation of genes involved in insulin/peptide secretion and the upregulation of genes involved in Ca2+ dynamics. By contrast, Trpm3-deficient lenses did not replicate the pathophysiological changes observed in Trpm3-mutant lenses. Collectively, our data suggest that a cataract-causing substitution in the TRPM3 cation channel elicits a deleterious gain-of-function rather than a loss-of-function mechanism in the lens.
1. Introduction
First discovered in a spontaneous, light-sensitive, retinal mutant strain of the fruit fly D. melanogaster, the transient receptor potential (TRP) superfamily of cation channels serve as polymodal sensors in myriad physiological processes, including the perception of light, temperature, pressure, and pain—rendering TRP channels therapeutic targets of interest for human disease [1,2,3,4,5]. Of the 28 mammalian (27 human) TRP channels, which are divided into six sequence-related sub-families, eight belong to the melastatin sub-family (TRPM1-8) that share homology with the charter member TRPM1 (melastatin-1) [6]. In addition to serving as a diagnostic and prognostic marker for primary cutaneous metastatic melanoma [7], mutations in the TRPM1 gene underlie several inherited eye disorders in humans, including night-blindness (nyctalopia), high myopia, and involuntary eye movements (strabismus, nystagmus), along with nyctalopia and leopard complex coat-spotting in Appaloosa horses [8,9,10,11]. TRPM3 (melastatin-2), which shares most homology with TRPM1 (~57% amino acid Identity), has been shown to function as a calcium ion (Ca2+) channel with permeability to other cations influenced by extensive alternative splicing and/or activation of non-canonical channel activity [12,13,14,15,16,17]. Like other TRP-channels and voltage-gated ion channels, TRPM3 channels share a multi-pass (type-2) transmembrane topology, typically assembled from tetramers of identical (homomeric) or similar (heteromeric) subunits around a central pore [18,19,20]. Each TRPM3 subunit possesses a cytoplasmic amino (N) terminus with four characteristic melastatin homology regions (MHR1-4) and several calmodulin binding sites, a transmembrane domain comprising six α-helical segments (S1-6) with the central cation pore lined by α-helices S5 and S6 connected by the hydrophobic pore-forming (P) loop, and a large cytoplasmic carboxy (C) terminal domain with a conserved TRP motif and a coiled-coil region [18,19,21,22]. Functional expression studies have shown that TRPM3 channels can be activated by several physico-chemical stimuli, including hypotonic extracellular challenge, the neuro-steroid pregnenolone sulfate (PS), a potent synthetic agonist (CIM0216), noxious heat, cell membrane phosphoinositol phosphates, and sex hormone steroids [15,23,24,25,26,27,28,29]. PS activation of TRPM3 channels elicits a strong increase in intracellular Ca2+ that activates a downstream signaling cascade involving mitogen-activated protein kinases (MAPKs), calmodulin, several nuclear phosphatases, the stimulus-response transcription factors (e.g., AP1), and epigenetic factors to regulate gene transcription [30,31,32,33,34,35]. Conversely, TRPM3 channels can be inhibited by several clinically approved drugs (e.g., non-steroidal anti-inflammatory drugs), antibiotics (e.g., voriconazole), volatile anesthetics (e.g., isoflurane), plant-derived secondary metabolites (e.g., citrus flavanones), and G-protein-coupled receptor βγ subunits [22,36,37,38,39,40,41]. TRPM3 channels are widely expressed in neurons and other cell types and have been implicated in a variety of cellular processes, including insulin/peptide secretion, vascular constriction and dilation, noxious heat sensing, inflammatory and spontaneous pain sensitivity, and tumorigenesis [24,25,28,42,43,44].
Genetic variations in the human TRPM3 gene (TRPM3) have been associated with a broad spectrum of inherited “channelopathies” and acquired diseases or conditions. Non-coding single-nucleotide variants (SNVs) in TRPM3 have been associated with longevity, elevated low-density lipoproteins and triglycerides, systemic sclerosis, and thyroid nodules in humans [4,5] along with racing speed in dogs [45]. Rare deletions involving coding exons in TRPM3 have been reported in Kabuki syndrome and autism [46,47], and a TRPM3 variant has been implicated in an autosomal dominant form of centrofacial pruritis [48]. Recently, de novo missense mutations in TRPM3 have been found to underlie a spectrum of neurological disorders, referred to as TRPM3-related neurodevelopmental disorder (TRPM3-NDD), that includes congenital hypotonia, developmental delay, intellectual disability, seizures, musculoskeletal, and ophthalmological findings [49,50,51]. Like TRPM1, TRPM3 has also been associated with human eye diseases. A missense mutation near the 5′-end of TRPM3 has been linked with autosomal dominant forms of cataract with or without glaucoma and anterior eye defects (e.g., persistent pupillary membrane) [52,53,54], and SNVs in TRPM3 have been associated with age-related nuclear cataract, incipient senile cataract, age-related macular degeneration (AMD), and glaucoma [55,56,57,58]. Notably, the ophthalmological findings reported in TRPM3-NDD include strabismus, nystagmus, and refractive errors but not cataracts, AMD, or glaucoma [50]. Finally, mutation of the gene for micro-RNA 204 (MIR204), which is embedded within an intron of TRPM3, underlies autosomal dominant retinal dystrophy and iris coloboma with or without congenital cataract (RDICC) [59].
The vertebrate lens is a highly transparent cellular structure that plays a central role in eye development and—along with the cornea—serves to focus light onto the photosensitive retina [60,61]. Surrounded by a basement membrane or capsule, the ellipsoidal lens is composed of an anterior monolayer of lens epithelial cells that terminally differentiate into elongated lens fiber cells arranged in tightly packed, concentric growth shells to generate the refractive mass of the lens [62,63]. Trpm3 has been identified as one of 65 “signature” genes for mouse lens epithelial cells [64] and, along with Mir204, is co-regulated by the paired-box transcription factor, PAX6, during eye development [65]. Mice lacking Trpm3 have been found to display an attenuated pupillary response to dim light, whereas mice lacking Mir204 acquire an AMD-like phenotype associated with pathogenic accumulation of rhodopsin and altered barrier function in the retinal pigment epithelium [66,67]. Previously, we have shown that Trpm3-null mice fail to develop early-onset cataract, whereas knock-in of a human cataract-associated mutation in Trpm3 (p.I65M) elicits early-onset, progressive cataract with lens calcification and pro-fibrotic immune responses [68]. Here, we determine the ocular co-expression profile of Trpm3 and Mir204 and model the pathogenetic effects of the TRPM3 p.I65M substitution expressed in the mouse lens and human cell lines.
2. Materials and Methods
2.1. Mice and Lenses
C57BL6/J mice (B6J, strain #000664), B6(Cg)-Tyrc−2J/J mice (B6 albino, strain #000058), and B6J.Cg-Gt(ROSA)26Sortm96(CAG-GCaMP6s)Hze/MwarJ mice (Ai96(RCL-GCaMP6s) or Ai96, strain #028866) [69] were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Trpm3-mutant mice were generated by CRISPR/Cas-9 gene editing as described by [68]. Trpm3-knockout (KO) mice were generated by homologous recombination as described by [70] and re-derived from frozen embryos obtained from the European Mutant Mouse Archive (EMMA) using standard techniques [68]. Transgenic Cre-recombinase (MLR10) mice were generously provided by Dr. M.L. Robinson (Ohio State University) [71]. Mir204-KO and Mir204_Mir211 double KO (dKO) mice were generously provided by Dr. C. Zhang (National Eye Institute, National Institutes of Health, Bethesda, MD, USA) [67]. All experimental mouse strains were maintained on the B6J genetic background to avoid a deletion mutation in Bfsp1 carried by certain inbred strains [72] and genotyped using the polymerase chain reaction (PCR) and gene-specific primers (Table S1) and as described by [68]. Mice were sacrificed in accordance with the American Veterinary Medical Association (AVMA) guidelines. Intact eyes or dissected lenses immersed in phosphate-buffered saline (PBS, #P4417-100TAB, MilliporeSigma, Burlington, MA, USA) were imaged with a digital camera (Spot Insight, Sterling Heights, MI, USA) fitted to a dissecting microscope (Stemi 2000, Zeiss, Thornwood, NY, USA). All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University School of Medicine (Protocol No. 22-0114).
2.2. Mutant Cell Lines
Human lens epithelial B3 (HLE-B3) cells (CRL-11421) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Eagle’s minimum essential medium (EMEM) supplemented with 20% fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) under standard conditions (5% CO2, 37 °C). Mutant cell lines were produced using CRISPR/Cas9 (Clustered Regularly Interspersed Short Palindromic Repeats and CRISPR-associated protein 9) gene editing in our Genome Engineering and Stem Cell Center (geneediting.wustl.edu). Briefly, guide ribonucleic acids (gRNAs) and a single-stranded donor oligonucleotide (ssODN) were designed in silico, synthesized in vitro, and validated by sequencing to introduce a missense transition (c.195A > G) into exon-4 of TRPM3 with ~30% non-homologous end joining (NHEJ) frequency. Sequences of the gRNA and ssODN were as follows: hTRPM3.sp13: 5′-ataaaatgctctttctatccngg-3′ and hTRPM3.ssODN: 5′-tacctatgggggtctttg gtgctgggtatgatgtggacacattctcttttataaaatgccctttccatccaggatttctgagcctgaaaaagaaaacaaaaaaaaaaaaaaaagaaaaaagaaagaa-3′. Of the five resulting cell lines, one was homozygous mutant, one was heterozygous mutant, and three were hemizygous mutants.
2.3. In Situ Hybridization
Lens RNA transcripts were localized in situ using custom-synthesized oligonucleotide probes to Trpm3 (NM_001362504.1, target region 2400–3563 bp, Mm Trpm3-O5-C1, Cat. No. 1114341-C1) and the RNAscope 2.5 HD Detection Reagent—RED (Cat. No. 322360, Advanced Cell Diagnostics, ACD, Inc, Hayward, CA, USA) and to Mir204 (NR_029591, SR-mmu-miR-204-5p-S1, Cat. no. 885551-S1) and the miRNAscope HD Detection Reagent—RED (Cat. No. 324510, ACD) according to the manufacturer’s instructions and as described by [73].
2.4. Immunofluorescence Confocal Microscopy
Whole eyes were processed using standard cryo-section techniques, and immunofluorescence microscopy was performed as described by [68,73,74,75,76,77]. Briefly, whole eyes were fixed in 4% paraformaldehyde and cryo-protected by serial incubation in 15% and 30% sucrose/PBS, then embedded in Tissue-Tek OCT compound (EMS, 62550-01) and saggital sections cut using a cryostat (Cryotome E, Thermo Fisher Scientific). Serial cryo-sections of eyes from at least three animals (n ≥ 6) were used to obtain reproducible images. TRPM3 was labeled with a polyclonal primary antibody (bs-9046R, Bioss Antibodies, Woburn, MA, USA) followed by Alexa-fluor conjugated secondary antibody (Thermo Fisher Scientific), and the actin cytoskeleton was labeled with phalloidin (ActinGreen 488, Cat no. R37110, Invitrogen, Thermo Fisher Scientific). Cell nuclei were counterstained with 4′,6-diaminido-2-phenylindole (DAPI, Millipore Sigma, Burlington, MA, USA) and sections imaged using a confocal microscope (FV1000, Olympus, Center Valley, PA, USA; Zeiss LSM800 with Airyscan, Carl Zeiss, White Plains, NY, USA).
2.5. Cation and Water Content
Lens cation content was measured using atomic absorption spectrometry (AAS) as described by [78]. Briefly, freshly dissected lenses were blotted dry and weighed, then lyophilized for 16 h (Freeze Dry System 7754000, LabConco Corporation, Kansas City, MO, USA) and re-weighed to determine the water content. Dried lenses were digested with 30% nitric acid (16 h, 20 °C), and dilutions of digest in ultra-pure water (>18.18 MΩ·cm) were quantified for Na+, K+, and Ca2+ using an atomic absorption spectrometer (Analyst 100, Perkin-Elmer, Norwalk, CT, USA) at wavelengths of 589.0 nm, 766.5 nm, or 422.7 nm, respectively. Data were presented as ion concentrations with units of millimoles per Kg of lens water and were not corrected for extracellular space since the lens extracellular space accounts for less than 2% of its volume [79].
2.6. Immunoblot Analysis
Immunoblot analysis of MAP kinase activation was performed with a polyclonal antibody to a synthetic peptide derived from the C-terminus of p44/42 MAPK (Erk1/2) (#9102, CST), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (E10) Mouse mAb (#9106, CST), and IRdye-labelled secondary antibody and size-markers using the Odyssey Infrared (IR) imaging System (Li-Cor, Lincoln, NE, USA) according to the manufacturer’s protocol and as described by [68,76].
2.7. Expression Constructs
Wild-type (I65) and mutant (M65) open reading frames for human TRPM3 transcript variant-9 (NM_001007471.2) (Figure S1) were custom synthesized using de novo gene synthesis chemistry and ligated into the pReceiver-M61 (CMV-IRES-eGFP) mammalian expression vector (GeneCopia, Rockville, MD, USA).
2.8. Cell Culture and Transfection
Human embryonic kidney 293T (HEK293T) cells (CRL-1573, ATCC), maintained in 6-well tissue culture dishes in Dulbecco’s modified Eagle’s media (DMEM) supplemented with 10% FBS (Gibco, Thermo Fisher Scientific), were transfected with TRPM3-IRES-eGFP constructs using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. One day post-transfection, HEK-293 cells were seeded on Matrigel-coated coverslips (BD Biosciences, Bedford, MA, USA), and transfected cells were identified by membrane-targeted eGFP fluorescence prior to Ca2+ imaging analysis 24–48 h later.
2.9. Cytoplasmic Ca2+ Imaging
Fluorescent calcium measurement was performed using the Fura-2 ratiometric assay, as described in detail in [80]. Briefly, coverslips were loaded (45 min, 37 °C) with Fura-2 AM (2.5 µM) and 10% Pluronic F-68 (Molecular Probes, Eugene, OR, USA) in HBSS/HEPES buffer then washed in the same buffer and de-esterified in the dark (15 min, 37 °C) and used for Ca2+ imaging within 1 hr of loading. Fura-2-loaded cells were perfused in Tyrode’s solution (containing in mM: 130 NaCl, 2 KCl, 2 CaCl2, 2 MgCl2, 25 Hepes, 30 glucose, pH 7.3 to 7.4 with NaOH, and 310 mosmol/kgH2O) and alternately excited by 340 nm and 380 nm light with emission detected at 510 nm and the fluorescence ratio (R340/380) determined in >150 cells. After first establishing a baseline fluorescence ratio, the cells were exposed to (1 min) pregnenolone sulfate (PS, 10–50 µM, Sigma-Aldrich, St. Louis, MO, USA) or high (10 mM) external Ca2+ concentration. Cells were washed with Tyrode’s solution for 2–3 min between stimuli.
2.10. Mouse RNA-Sequencing Analysis
Mouse lens RNA samples (4 male and 4 female lenses per sample) were prepared in triplicate, and cDNA libraries were synthesized, indexed, pooled, and then sequenced using an Illumina NovaSeq 6000 in our Genome Technology Access Center (GTAC) according to the manufacturer’s protocols (Illumina, San Diego, CA, USA) and as described by [76]. Illumina’s bcl2fastq2 software version 2.20 was used to perform basecalls and demultiplexing. An Illumina DRAGEN Bio-IT on-premise server running version 3.9.3-8 software was used to align and quantify RNA-seq reads to the Ensembl release 101 primary assembly. All gene counts were then imported into the R/Bioconductor package EdgeR [81] for read depth normalization and pairwise comparison of each line to the appropriate control group. Only genes with counts-per-million (CPM) ≥ 5 within at least 3 samples were included in the analysis. Samples were validated for consistency across biological replicates by plotMDS function of the R package Limma [82]. To analyze for differences between conditions, we performed differential expression analysis, and the results were filtered for only those genes with Benjamini–Hochberg false-discovery rate-adjusted p-values less than or equal to (≤) 0.05 and with fold-change greater than or equal to (≥) 2. Normalized gene counts were then clustered using the R ComplexHeatmap package [83] before genes with similar differential expression patterns were analyzed for functional classification with the Gene Ontology knowledgebase [84,85].
2.11. Statistical Analysis
One-way analysis of variance (ANOVA) was used to determine statistical significance (p) ± standard error (SE) or standard deviation (SD).
3. Results
3.1. Ocular Expression Profiles of Trpm3 and Mir204
In order to determine the distribution of transcripts for Trpm3 in the wild-type mouse eye, we performed in situ hybridization (ISH) with an RNAscope probe that spanned the coding region for the transmembrane channel domain of TRPM3. ISH revealed that Trpm3 transcripts were strongly localized to the postnatal day 5 (P5) lens epithelium and nascent lens fiber cells (Figure 1). Trpm3 transcripts were also localized to other ocular tissues at P5, including the non-pigmented and pigmented iris and ciliary epithelia, the corneal epithelium, the retinal pigmented epithelium, the inner neuro-retina, and the optic nerve (Figure S2). We note that Trpm3 transcripts were also localized to the mature (P21) Trpm3-KO lens epithelium (Figure S3), suggesting that the ISH probe cross-hybridizes with Trpm3 transcripts interrupted by the IRES-LacZ-neo cassette (Figure S1), which enables expression of the β-galactosidase reporter gene [68]. Since Trpm3-cassette “fusion” transcripts were present in Trpm3-KO lenses, we sought to compare the localization of TRPM3 channels in wild-type versus Trpm3-KO lenses using a polyclonal antibody that recognizes a peptide derived from the C-terminal region of human TRPM3. Immunofluorescence confocal microscopy confirmed strong localization of TRPM3 to the wild-type lens epithelium that was absent from the Trpm3-KO lens epithelium (Figure 2).
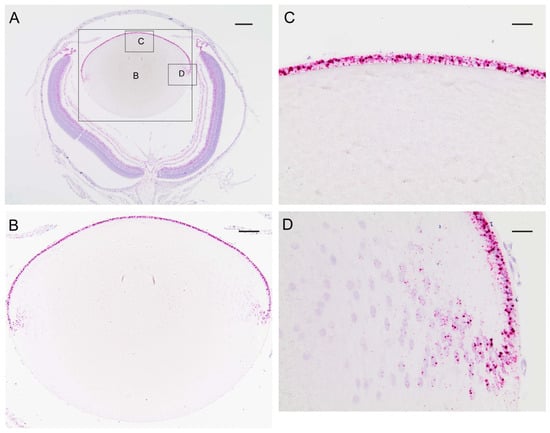
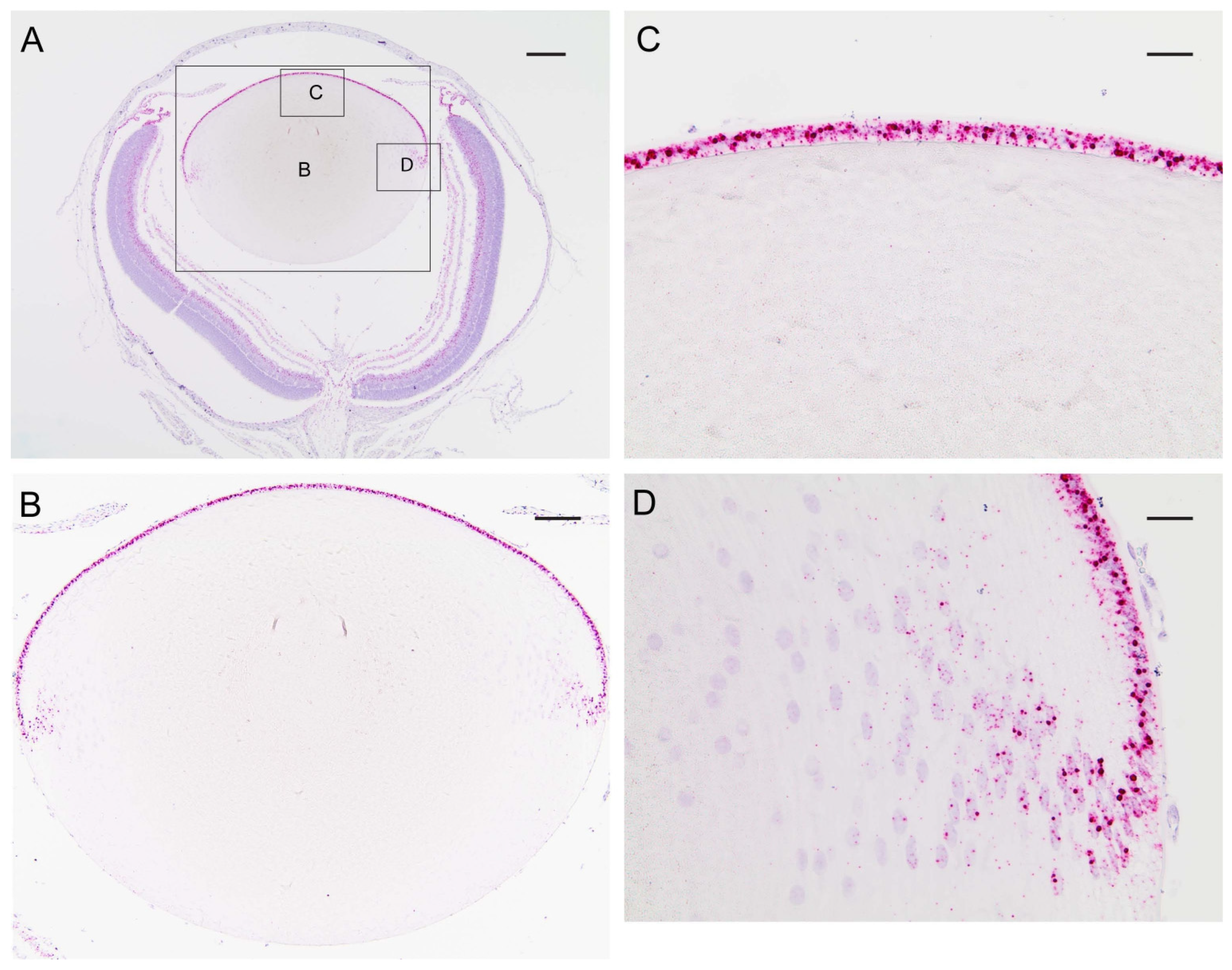
Figure 1.
In situ hybridization of Trpm3 transcripts in the neonatal mouse lens. Representative chromogenic labeling of Trpm3 transcripts (red dots) in the albino mouse eye (A) and lens (B–D) at P5 showing strong localization to the anterior epithelium (B,C) and the equatorial epithelium and nascent lens fiber cells (D). Cell nuclei were counterstained with hematoxylin (blue). Scale bar: 200 µm (A), 100 µm (B), 50 µm (C,D).
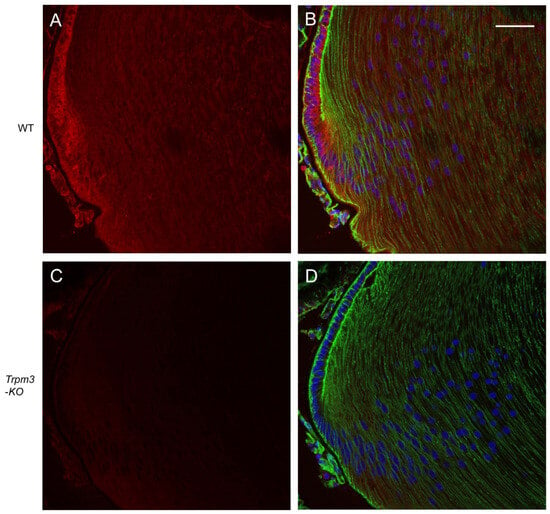
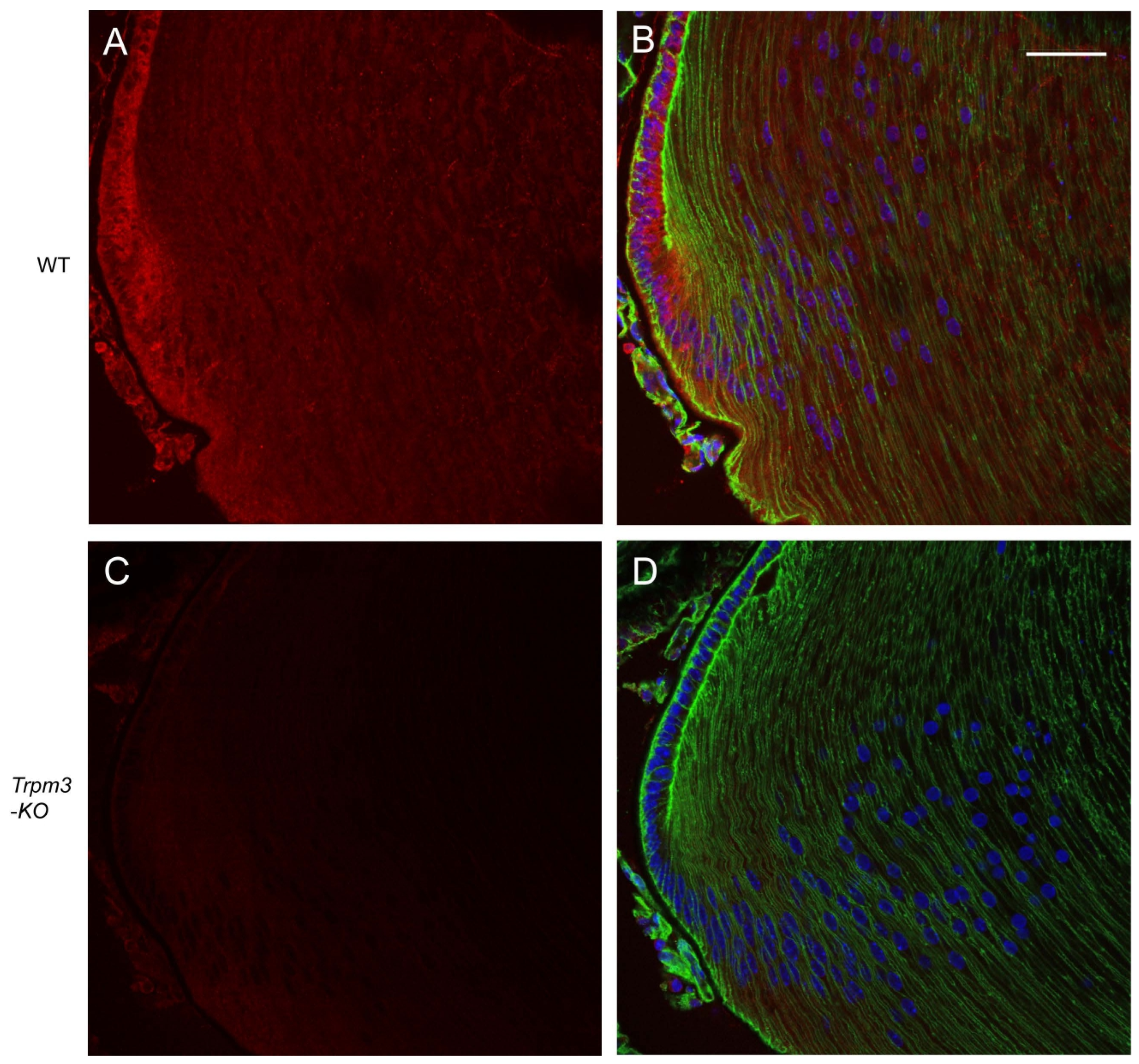
Figure 2.
Immunofluorescence confocal microscopy of TRPM3 channels in the mature mouse lens. Representative immuno-fluorescent labeling of TRPM3 (red) in the wild-type lens (A,B) and Trpm3-KO lens (C,D) at P21 showing localization to the lens equatorial epithelium in (A,B) but not (C,D). Actin cytoskeleton and cell nuclei were labeled with phalloidin (green) and DAPI (blue), respectively. Scale bar: 50 µm.
To compare the ocular distribution of Trpm3 with that of its intron-hosted micro-RNA gene, Mir204 (on mouse chromosome 19), we performed ISH with an RNAscope probe spanning the 5p arm of Mir204. Since Mir204 shares strong homology with Mir211, which is hosted in an intron of Trpm1 (on mouse chromosome 7), the ISH probe may not discriminate between these miRNAs. Like Trpm3 transcripts, Mir204/211 transcripts were strongly localized to the neonatal (P5) lens epithelium, non-pigmented and pigmented epithelia of the iris and ciliary body, and the retinal pigmented epithelium (Figures S4 and S5). Overall, our data suggest that co-incident expression of Trpm3 and Mir204/211 in the eye was strongest in epithelia from the lens, iris, ciliary body, and retina.
3.2. Lens Phenotype of Mir204- and Mir211-Deficient Mice
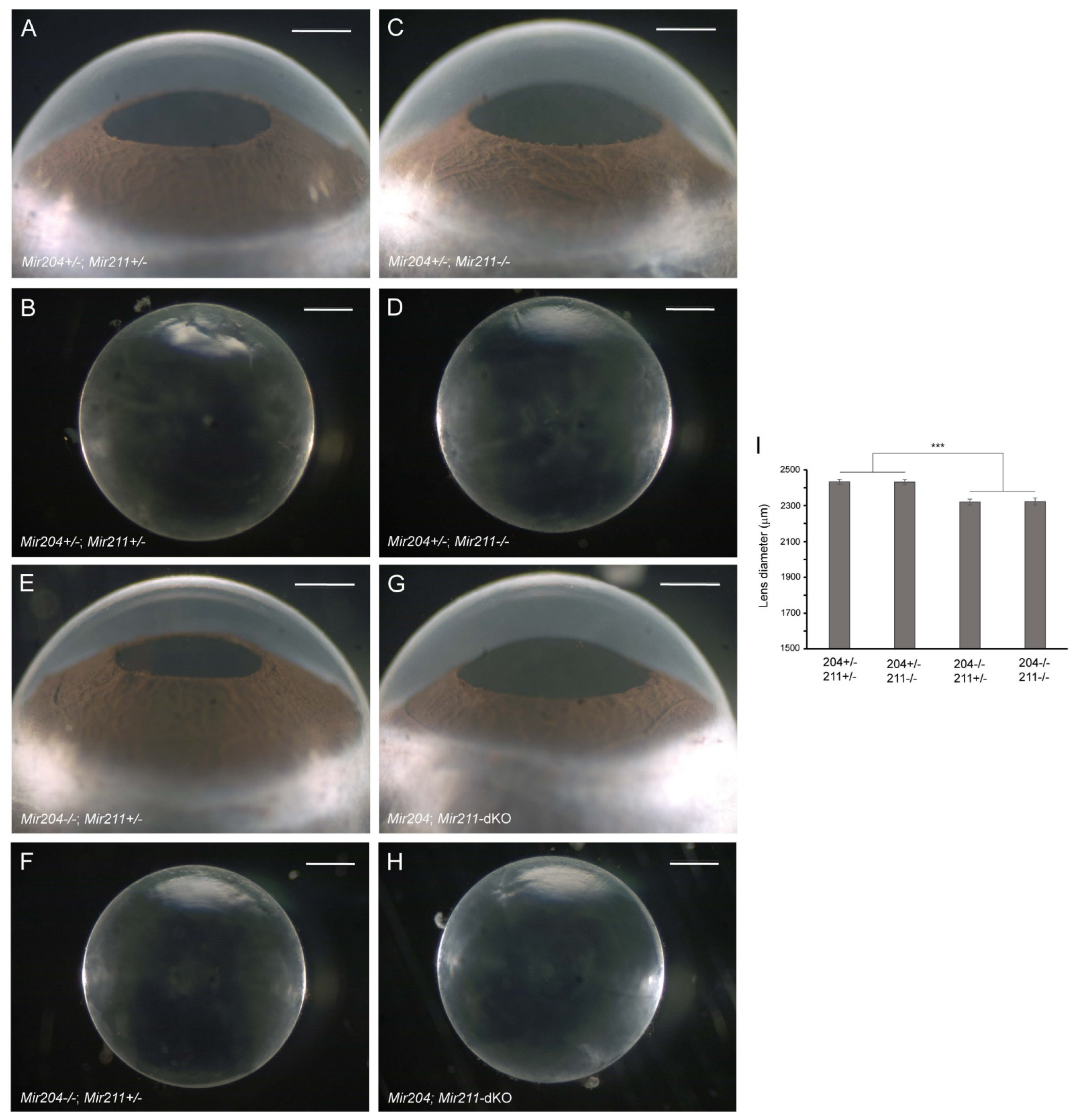
Since Mir204 is hosted within an intron of Trpm3 and a mutation in MIR204 has been associated with RDICC in humans [59], we next sought to determine the lens phenotype of Mir204-KO mice. At 4–6 months of age, Mir204-KO mice displayed a grossly normal eye phenotype with clear lenses similar to wild-type mice (data not shown). Thus, Mir204-KO mice—like Trpm3-KO mice—maintained clear lenses well beyond early-onset cataract in Trpm3-mutant (I/M and M/M) mice [68]. Since Mir204 shares homology with Mir211, we sought to address the possibility of functional redundancy between the two micro-RNAs by examining the lens phenotype of Mir204; Mir211-dKO mice (Mir204−/−; Mir211−/−). Littermates hemizygous for Mir204 and either hemizygous or null for Mir211 (Mir204+/−; Mir211+/− or Mir204+/−; Mir211−/−) developed a grossly normal eye phenotype with clear lenses at 4 months of age (Figure 3A–D). Similarly, Mir204; Mir211-dKO and Mir204−/−; Mir211+/− littermates displayed a grossly normal eye phenotype with clear lenses at 4 months of age (Figure 3E–H). However, Mir204; Mir211-dKO and Mir204−/−; Mir211+/− lenses were smaller than those from Mir204+/−; Mir211+/− and Mir204+/−; Mir211−/− littermates (Figure 3I). Thus, the loss of both Mir204 copies and at least one Mir211 copy resulted in a lens growth deficit in the absence of early-onset cataract. We note that hyper-mature cataracts have been reported in Mir204; Mir211-dKO mice at 10–15 months of age [86]. Regardless, such a late onset contrasts with the early cataract development in Trpm3-mutant (I/M, M/M) mice. Overall, our data suggest that, like loss of Trpm3, loss of Mir204 and/or Mir211 did not cause early-onset cataract similar to that found in Trpm3-mutant mice [68].
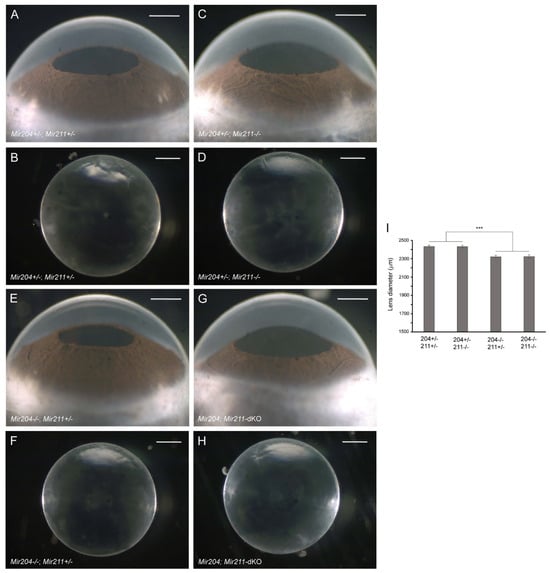
Figure 3.
Lens phenotype of Mir204- and Mir211-deficient mice. Representative dark-field images of eyes (A,C,E,G) and corresponding lenses (B,D,F,H) from Mir204+/−; Mir211+/− mice (A,B), Mir204+/−; Mir211−/− mice (C,D), Mir204−/−; Mir211+/− mice (E,F), and Mir204; Mir211-dKO mice (G,H) at 4 months of age. Scale bar: 500 µm (A–H). (I) Lens diameter for each genotype ± SD. *** p < 0.001.
3.3. Cation and Water Content of Trpm3-Mutant Lenses
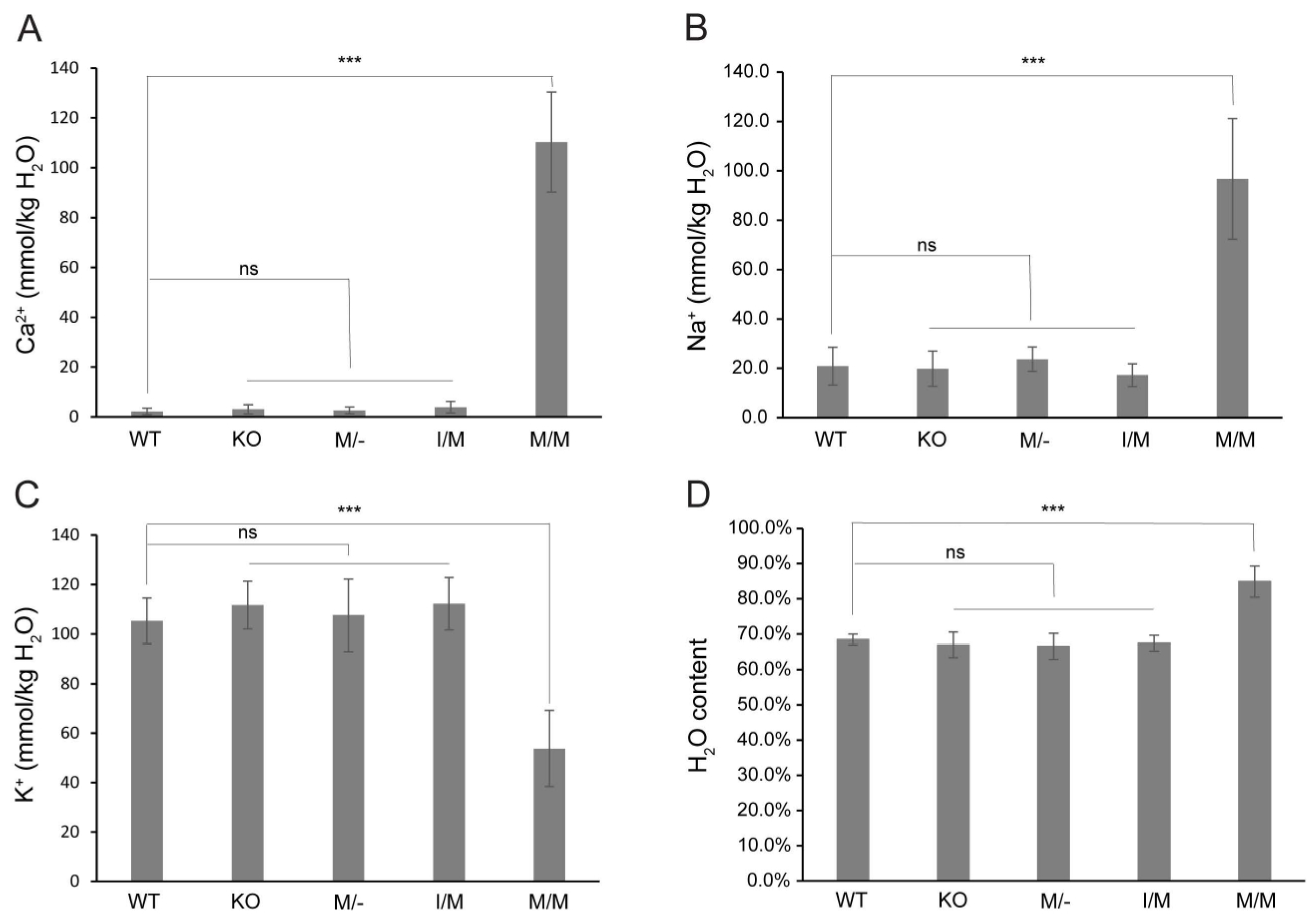
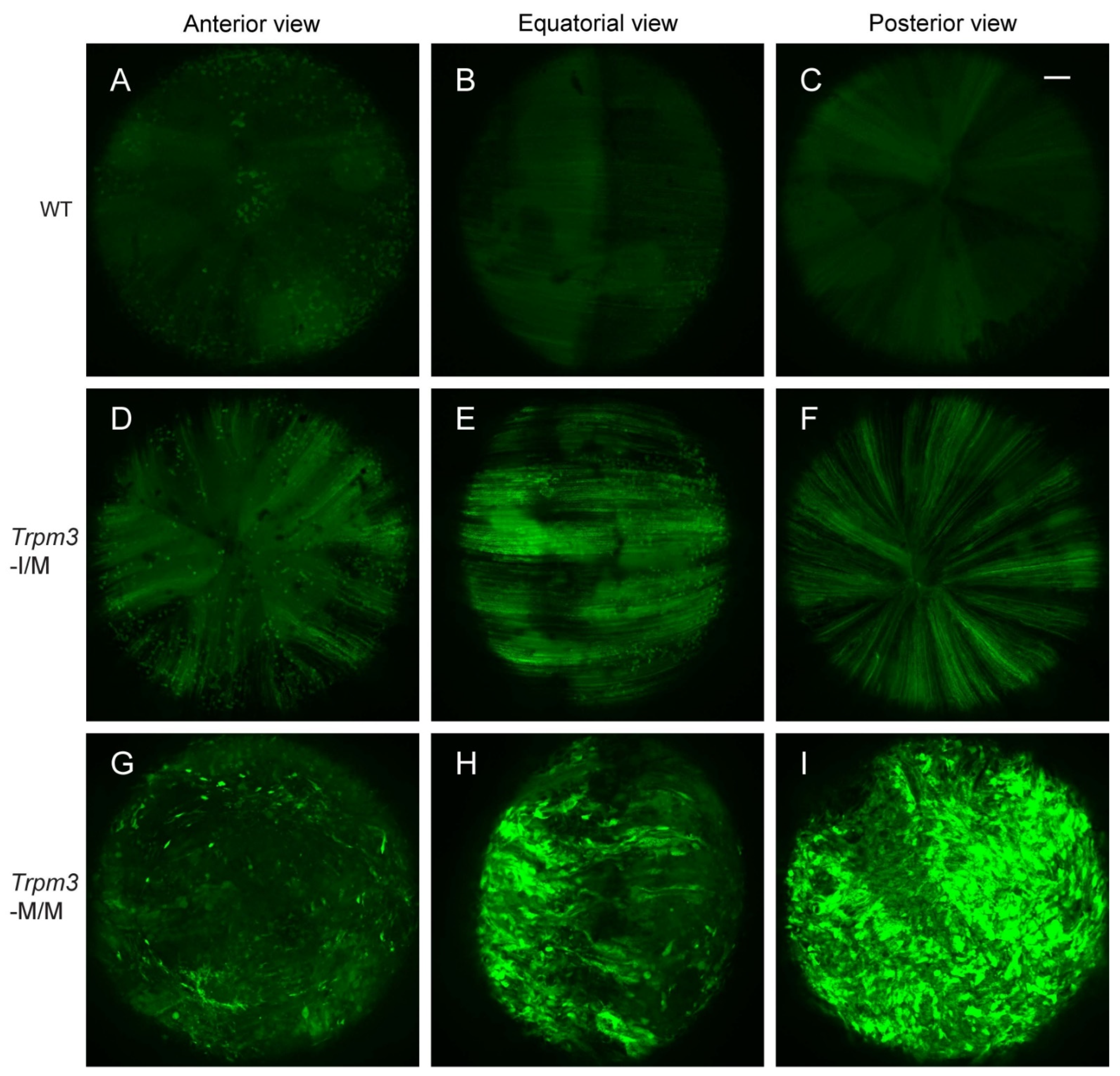
Previously, we provided histochemical evidence for Ca2+ accumulation in Trpm3-mutant lenses [68]. In order to further quantify the cation content of Trpm3-mutant and Trpm3-KO lenses, we undertook atomic absorption spectrometry (AAS). AAS revealed that Trpm3-KO, hemizygous Trpm3-mutant (M/−), and heterozygous Trpm3-mutant (I/M) lenses had intracellular cation (Ca2+, Na+, K+) levels and water content similar to those of wild-type lenses (Figure 4). By contrast, homozygous Trpm3-mutant (M/M) lenses had dramatically elevated intracellular Ca2+ concentrations ([Ca2+]i) coupled with increased ([Na+]i), reduced ([K+]i), and an increased water content (85–90%) compared to wild-type lenses (66–70%) (Figure 4)—consistent with a failure of Trpm3-M/M lenses to osmoregulate. To further visualize Ca2+ levels in intact Trpm3-mutant lenses ex vivo, we crossed Trpm3-mutant mice with mice transgenic for the genetically encoded, Cre-dependent fluorescent Ca2+ indicator variant (GCaMP6s) that is characterized by high sensitivity with slow decay and response kinetics [87]. Subsequent breeding of GCaMP6s;Trpm3-mutant mice with lens-specific Cre mice [71] enabled deletion of a floxed-STOP cassette preventing GCaMP6s expression and resulted in excitation of enhanced green fluorescent protein (EGFP) proportional to the binding of cytosolic Ca2+ to the calmodulin and calmodulin-interacting M13 peptide (CaMP) complex. Figure 5 revealed mildly increased fluorescence in Trpm3-I/M lenses and intense fluorescence in the Trpm3-M/M lens compared to the wild-type lens—supporting Ca2+ influx. Overall, these cation data were consistent with Ca2+ accumulation and failed osmoregulation, contributing to cataract development in Trpm3-mutant lenses.
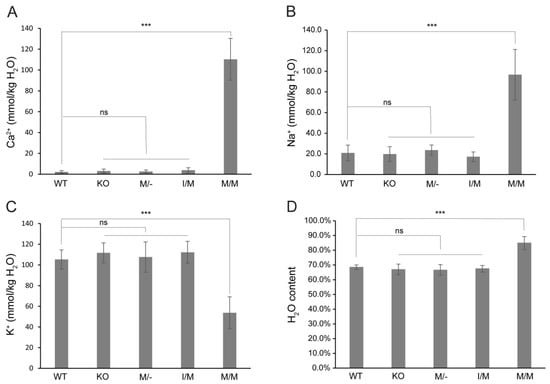
Figure 4.
Cation and water content of Trpm3-mutant and Trpm3-KO lenses. Atomic absorption spectroscopy measurements of Ca2+ (A), Na+ (B), and K+ (C) concentrations and water content (D) in lenses (P21) from wild-type (WT), Trpm3-KO, hemizygous (M/−), heterozygous (I/M), and homozygous (M/M) Trpm3-mutant mice. *** p < 0.001; ns—not significant; error bars ± SD.
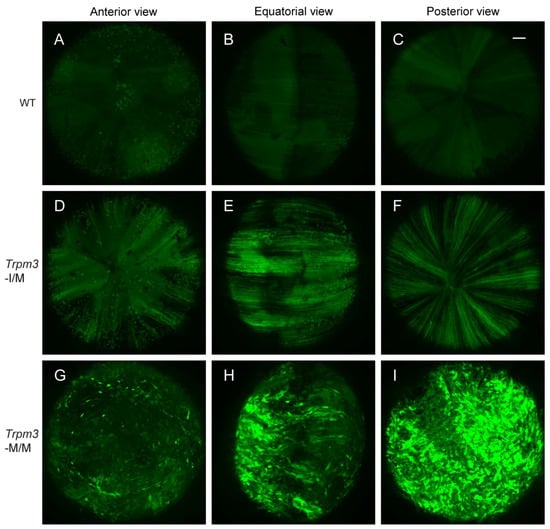
Figure 5.
Elevated calcium in Trpm3-mutant lenses determined using a genetically encoded calcium sensor. Representative confocal fluorescence microscopy images of Cre-dependent GCaMP6s activation in wild-type (A–C), heterozygous Trpm3-I/M (D–F), and homozygous Trpm3-M/M (G–I) lenses at P21 showing anterior (A,D,G), equatorial (B,E,H), and posterior (C,F,I) views. Scale bar: 100 µm.
3.4. Functional Expression of Mutant TRPM3 Channels
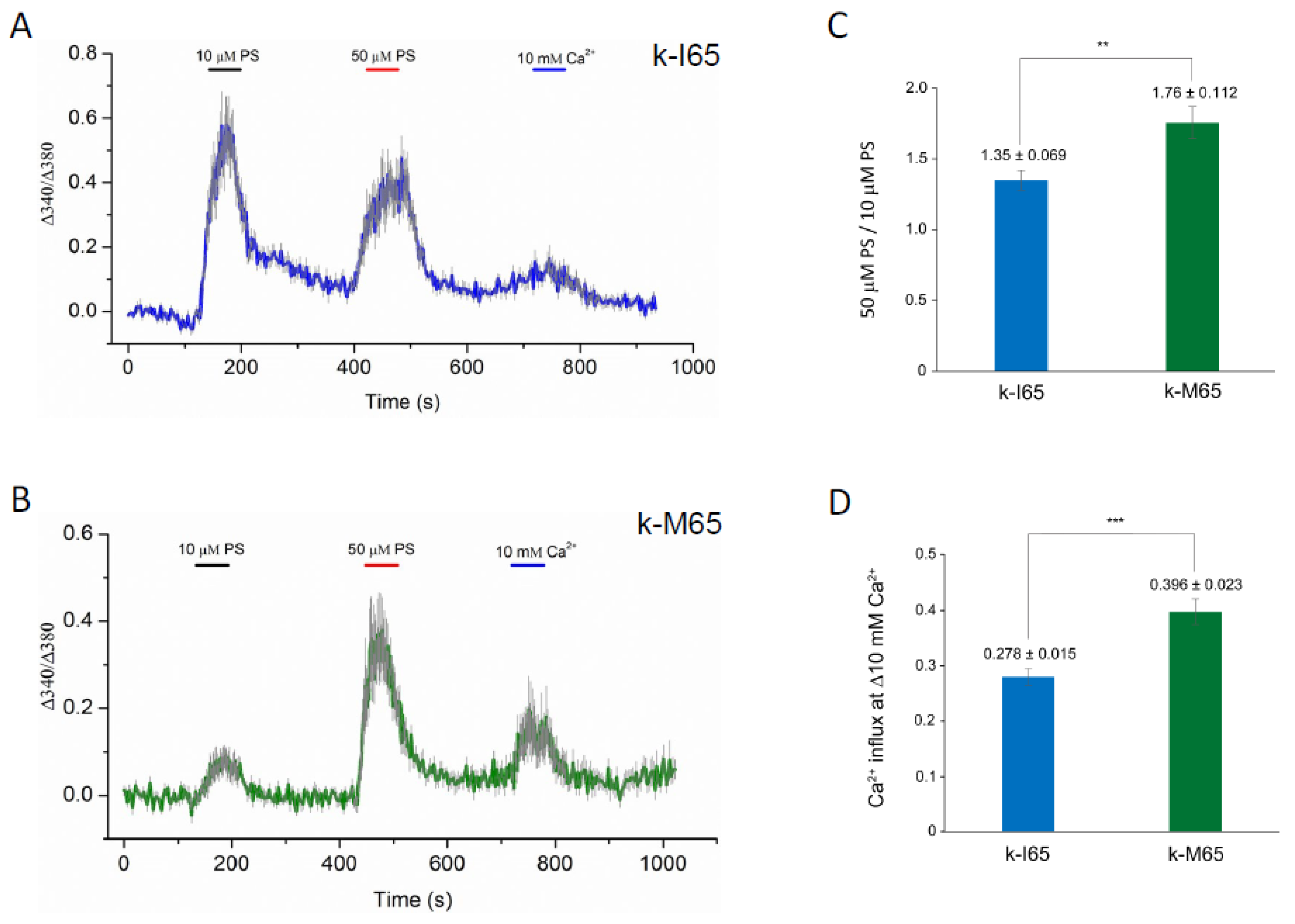
TRPM3 was originally shown to function as a spontaneous Ca2+ channel that can be activated by PS in vitro [23,24,88]. In order to determine the pathogenic effects of the cataract-causing p.I65M substitution on TRPM3 channel properties, we undertook transient (over-)expression of wild-type (I65) and mutant (M65) channels in HEK-293T cells followed by fluorescent Ca2+ imaging with Fura-2. According to the current human reference sequence (GRCh39p.14), TRPM3 comprises 30 exons that generate at least 23 transcript variants (1–23) and channel isoforms (a–w) via alternative exon splicing [54] (https://www.ncbi.nlm.nih.gov/gene/80036; accessed on 5 December 2023). Of the 23 predicted channel isoforms (a–w), 19 are full-length with a transmembrane channel pore domain (a, b, d–g, i–k, m–t, v, w), and 4 are truncated lacking a channel pore (h, c, l, u). Since most of the full-length channel isoforms start translation in exon-2, skip exon-3 (non-coding), and include exon-4 containing the p.I65M substitution (codons 60–86), we selected variant-9 (NM_001007471) for custom synthesis of wild-type and mutant expression plasmids (Figure S1). Variant-9 is expressed in the human lens [52] and encodes channel isoform-k (NP_001007472), comprising 1707 amino acid residues. Following transient expression of wild-type (k-I65) and mutant (k-M65) channels, the Fura-2 ratio-metric assay revealed robust and long-lasting Ca2+ influx in response to 50 µM PS activation (Figure 6A,B). However, compared to wild-type k-I65 channels, the mutant k-M65 channels exhibited a higher ratio of peak Ca2+ influx in response to 50 μM PS versus 10 µM PS, suggesting a steeper dose–response relationship at this concentration range (Figure 6A,C). In addition, the mutant k-M65 channels elicited an increased Ca2+ influx response to elevated extracellular Ca2+ (from 2 mM to 10 mM) compared to wild-type k-I65 channels (Figure 6B,D). Overall, while the significance of the altered sensitivity of mutant k-M65 channels to PS activation in the lens is unclear, the increased sensitivity of the mutant k-M65 channels to extracellular Ca2+ concentration may contribute to cataract formation.
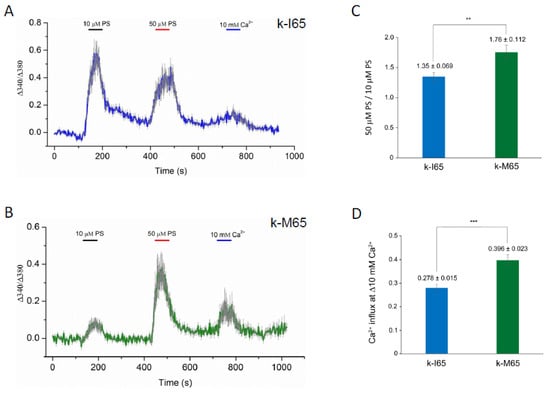
Figure 6.
Cytoplasmic calcium responses in Fura-2-loaded HEK-293T cells that express either wild-type (I65) or mutant (M65) TRPM3 (k-isoform) channels. (A,B) Representative traces of Ca2+ transients evoked by 10 μM and 50 μM pregnenolone sulfate (PS) or by changes of extracellular Ca2+ (2 mM to 10 mM). Traces are averaged from cells in one region of interest expressing wild-type k-I65 (A) and mutant k-M65 (B) TRPM3 channels, respectively. (C) Ratio of peak Ca2+ influx in response to 50 μM PS versus 10 μM PS. (D) Peak Ca2+ influx in response to increase in extracellular Ca2+ concentration. ** p < 0.01; *** p < 0.001; error bars ± SD.
3.5. MAP Kinase Activation in TRPM3-Mutant Cell Lines and Lenses
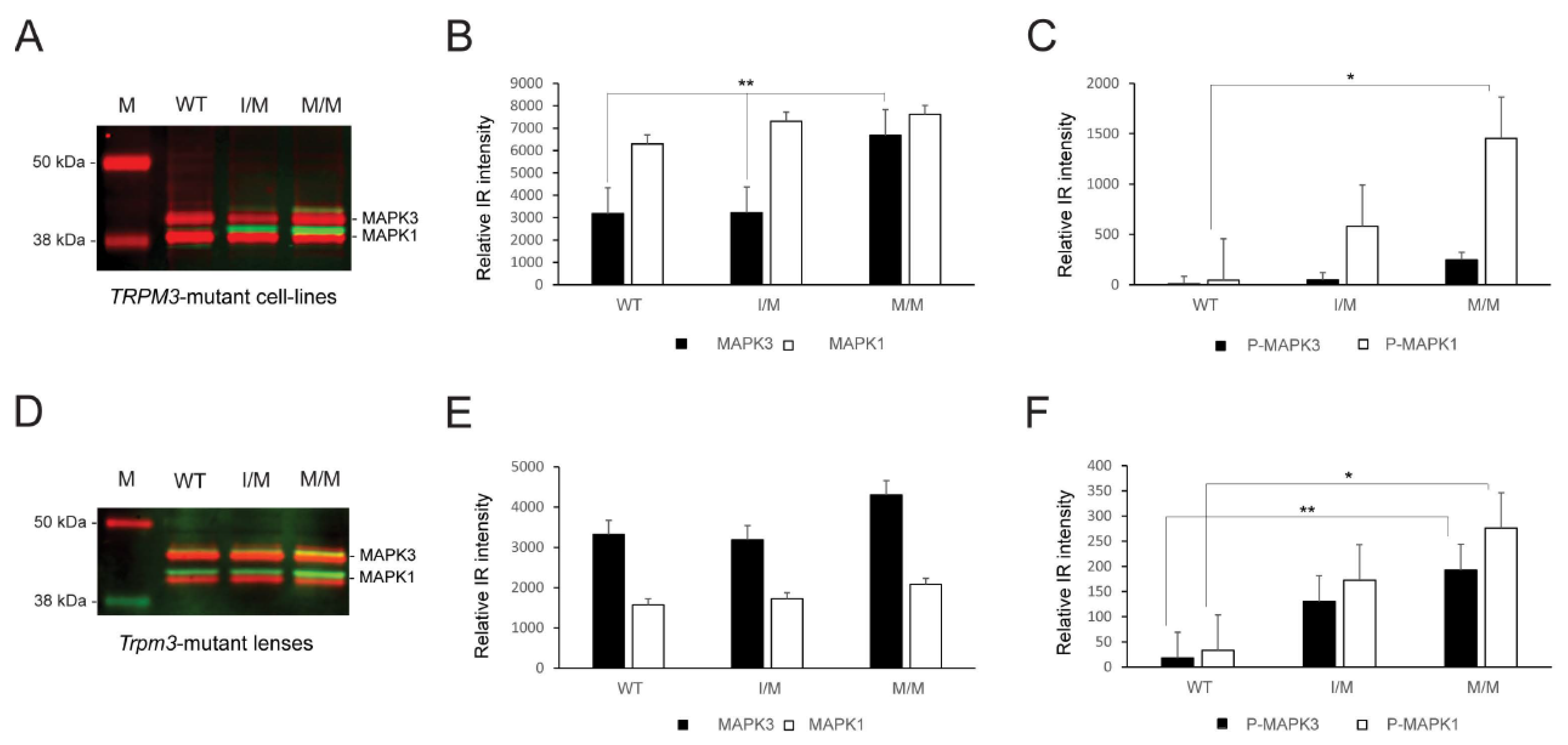
Since TRPM3-facilitated Ca2+ influx has been associated with the activation of mitogen-activated protein kinase-1 MAPK1/ERK2/p42 and MAPK3/ERK1/p44 in vitro [32,33], we performed immunoblot analysis to compare MAP kinase status in knock-in TRPM3-mutant HLE-B3 cell lines and Trpm3-mutant lenses versus wild-type lenses. Immunoblot analysis revealed that constitutive MAPK1 levels were significantly greater than MAPK3 levels in wild-type and heterozygous TRPM3-mutant (I/M) HLE-B3 cells, whereas constitutive MAPK3 levels were significantly elevated and similar to MAPK1 levels in homozygous TRPM3-mutant (M/M) HLE-B3 cells (Figure 7A,B). Moreover, phospho-activation of MAPK1 was significantly greater in TRPM3-M/M HLE-B3 cells compared to wild-type cells (Figure 7A,C). By contrast, constitutive MAPK3 levels were greater than MAPK1 levels in wild-type, Trpm3-I/M, and Trpm3-M/M lenses (Figure 7D,E), and phosphorylated MAPK1 and MAPK3 levels were both significantly elevated in Trpm3-M/M lenses compared to wild-type lenses (Figure 6D,F). Overall, while the relative basal levels of MAPK1 and MAPK3 differed between HLE-B3 cells and mouse lenses, phospho-activation of MAPK3 and/or MAPK1 was increased in the presence of mutant TRPM3 channels.
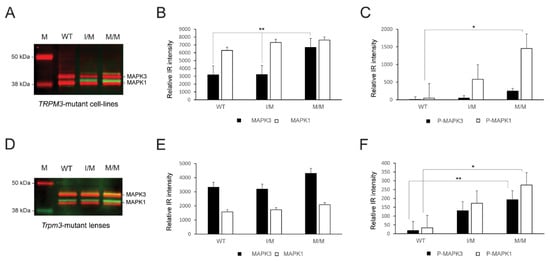
Figure 7.
MAP kinase activation in the TRPM3-mutant human lens epithelial B3 (HLE-B3) cells and Trpm3-mutant mouse lenses. Quantitative immunoblot analysis of MAPK3/p44 and MAPK1/p42 in heterozygous TRPM3-I/M and homozygous TRPM3-M/M mutant HLE-B3 cell-lines (A–C) and in Trpm3-mutant mouse lenses at P21 (D–F). Constitutive MAPK3 and MAPK1 levels are shown in (A,D) (red bands) and in (B,E) (black and white bars). Phosphorylated (P) MAPK3 and MAPK1 levels are shown in (A,D) (green/yellow bands) and in (C,F) (black and white bars). WT, wild-type. M, molecular weight markers. * p < 0.05; ** p < 0.01; error bars ± SE.
3.6. Gene Expression Profiles of Trpm3-Mutant and Trpm3-KO Lenses
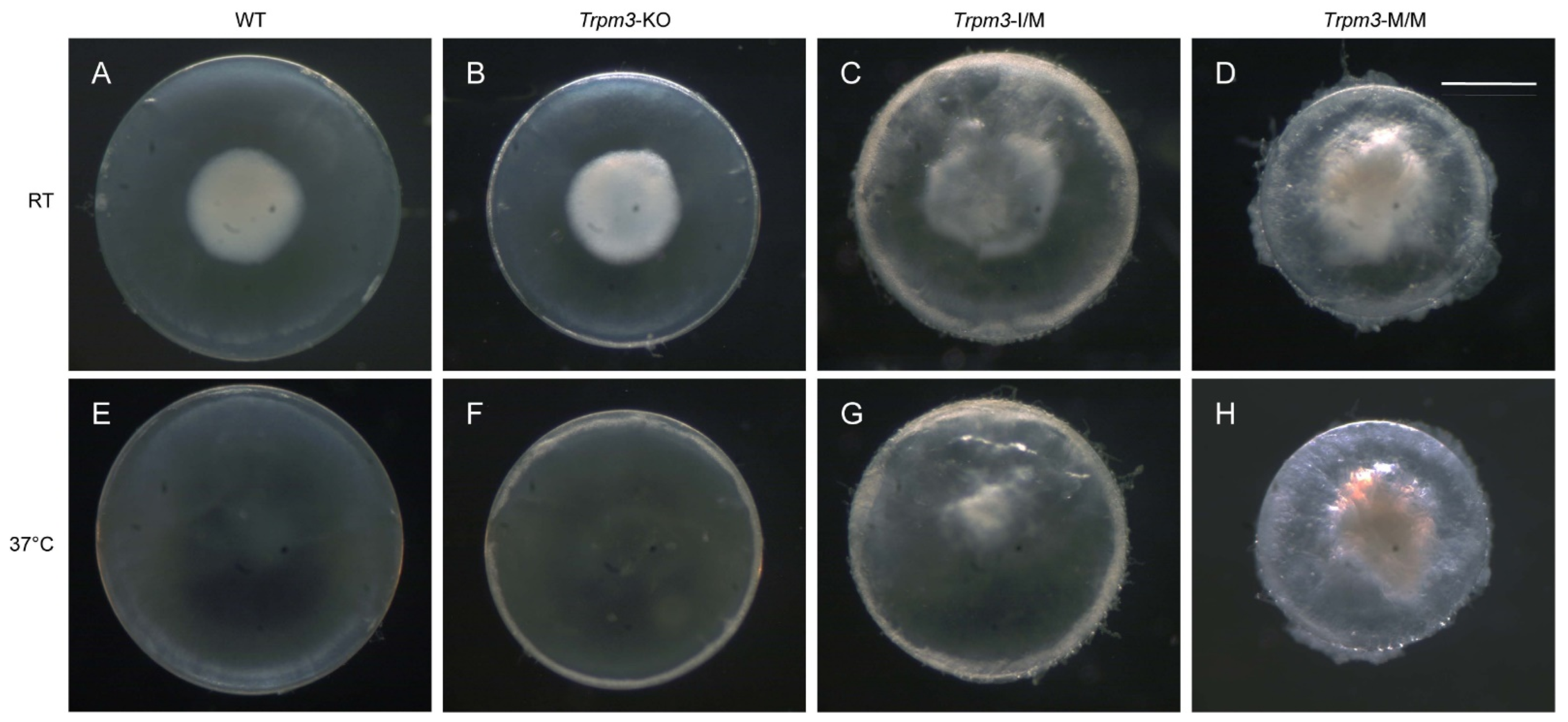
To determine the effects of the Trpm3 mutation versus Trpm3 deficiency on lens gene expression, we performed RNA-seq analysis to compare global transcriptional changes in Trpm3-mutant (I/M, M/M) lenses during early cataract development versus non-cataractous Trpm3-KO and wild-type lenses at P7. We selected lenses at P7 since resorption of the embryonic tunica vasculosa lentis (TVL) was largely complete, and at ages beyond P7, Trpm3-M/M lenses became severely opacified, fragile, and prone to rupture since they could not be cleanly dissected away from adherent contamination of other ocular tissues including the iris anteriorly and the retina posteriorly. During the dissection of P7 eyes at room temperature (20–22 °C), we observed so-called “cold cataract” formation in the core regions of wild-type, Trpm3-KO, and Trpm3-mutant lenses (Figure 8A–D). A cold cataract is a natural phenomenon of neonatal rodent lenses believed to result from changes in the supramolecular organization of specific lens proteins, including the beaded filament-specific proteins BFSP1 and BFSP2 [89], which can be effectively reversed by warming to 37 °C (Figure 8E–H). Despite the role of TRPM3 in heat detection by sensory neurons [28,70], neither Trpm3 deficiency nor Trpm3 mutation appeared to impact the reversal of cold cataracts in the lens.
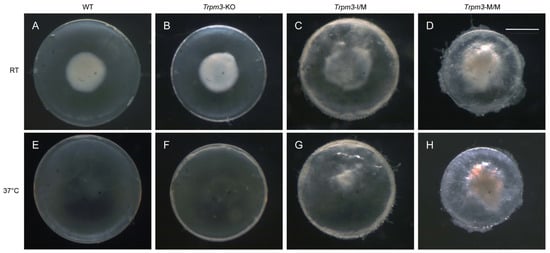
Figure 8.
Lens phenotype of neonatal Trpm3-mutant and Trpm3-KO mice. Representative dark-field images of wild-type (A,E), Trpm3-KO (B,F), Trpm3-I/M (C,G), and Trpm3-M/M (D,H) lenses at P7 showing cold-cataract formation (A–D) during dissection at room temperature (RT) that was reversible at 37 °C (E–H). Note the Trpm3-mutant cataract was not temperature sensitive (C,D,G,H). Scale bar: 500 µm.
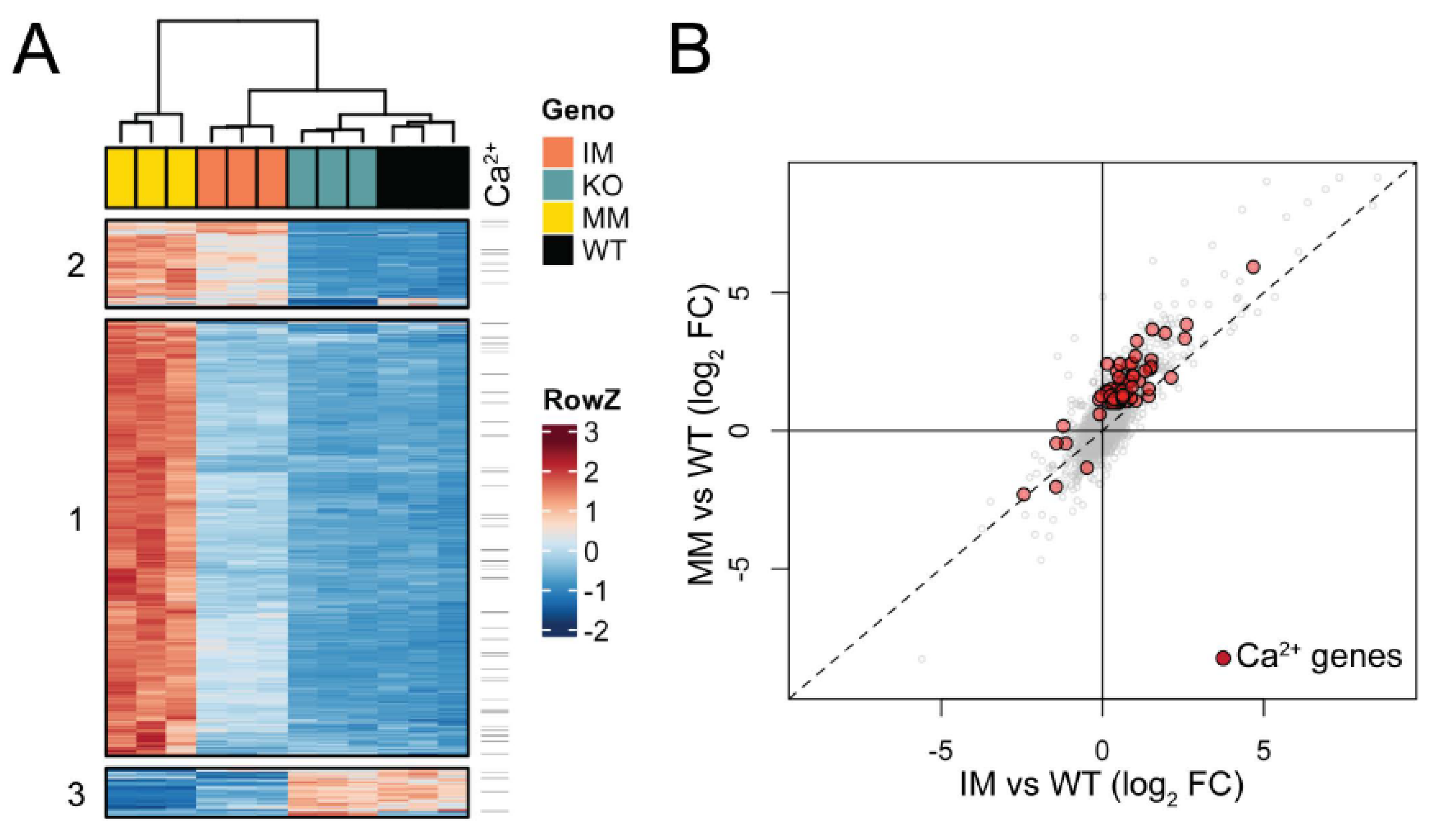
Triplicate lens RNA samples were sequenced to a depth of >60 M reads and aligned to the mouse mm10 genome build with a >98% alignment rate. Multi-dimensional scaling (MDS) analysis of RNA-seq data showed high consistency between biological triplicates at P7, with data from each genotype clustering independent from other samples (Figure S6A). Pairwise differential expression analysis comparing each Trpm3 genotype to wild-type identified 1249 genes with significant expression changes (fold-change, FC ≥ 2, false discovery rate, FDR ≤ 0.05). Across Trpm3 genotypes, 1131 genes were upregulated, with the majority identified in Trpm3-M/M versus wild-type comparison, while the comparison of Trpm3-I/M and Trpm3-KO to wild type identified few uniquely affected genes (Figure S6B). The 129 downregulated genes were less consistent between genotypes (Figure S6C). To better understand the relative effects of these gene sets in the three mouse models compared to the wild type, we clustered the 1249 mis-expressed genes by their relative expression across all samples (Figure 9A). Generally, the data supported the intermediate phenotype of Trpm3-I/M lenses compared to Trpm3-M/M lenses, as well as the lack of a significant transcriptional phenotype in Trpm3-KO lenses. Hierarchical clustering identified three primary gene sets (Figure 9A). The largest, Cluster 1, comprised 951 genes that were mostly upregulated in Trpm3-M/M lenses, with only moderate effects noted in Trpm3-I/M (Table S2). Clusters 2 (191 genes) and 3 (107 genes) were significantly smaller and comprised up- and downregulated gene sets, respectively, where Trpm3-I/M and Trpm3-M/M lenses were more similar to each other than to WT and Trpm3-KO lenses (Table S2). To identify genes that could be mediating the more severe phenotype in Trpm3-M/M versus Trpm3-I/M lenses, we first focused on the genes within Cluster 1. Gene ontogeny (GO) analysis of genes in Cluster 1 indicated enrichment of at least 16 GO categories comprising 680 genes related to Ca2+ homeostasis and transport (Table S3). To better comprehend the calcium-related transcriptional response in Trpm3-mutant lenses, we identified all genes significantly affected in our dataset that are within those 16 Ca2+-associated GO categories. Of the 80 genes identified, the majority lie within Cluster 1 and show a more severe upregulation in Trpm3-M/M than in Trpm3-I/M lenses (Figure 9, Table S4). Four of the most upregulated genes (>5-fold) in Trpm3-M/M lenses included those coding for the plasma-membrane Ca2+ transporting ATPases Atp2b1 and Atp2b4, the Na+/K+ transporting ATPase beta-1 subunit, Atp1b1, and the Ca2+ binding protein, Cabp5. In addition, the genes for two other TRP channels, Trpv4 and Trpm1, were upregulated (~ 2.4-fold) in Trpm3-M/M lenses but not in Trpm3-KO lenses, suggesting these channels do not compensate for the loss of Trpm3 function in the lens (Table S4). Further, since the relative expression level of Trpm3 in mutant lenses is over 25-fold greater than those of Trpm1 and Trpv4 (Figure S7), it is unlikely that mild upregulation of the latter channels contributes greatly to the lens phenotype of Trpm3-mutant mice.
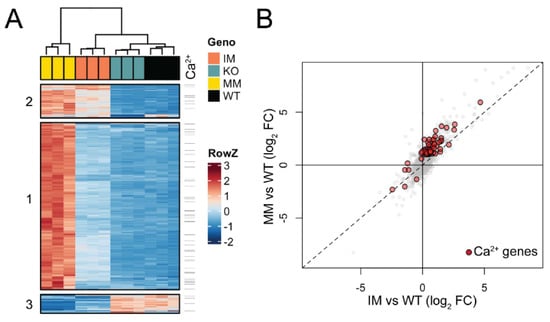
Figure 9.
Gene expression changes in Trpm3-mutant and Trpm3-KO lenses using RNA-seq analysis. (A) Heatmap showing unique expression changes (≥2-fold) in Trpm3-mutant (I/M, M/M) and Trpm3-KO lenses compared to wild-type lenses at P7. (B) Scatterplot highlighting genes involved in Ca2+-associated processes (red) that are differentially expressed in Trpm3-I/M and Trpm3-M/M lenses.
GO analysis of genes within Cluster 2 suggested that genes related to membrane potential and development are upregulated in both Trpm3-I/M and Trpm3-M/M lenses at P7 (Table S3). GO analysis of the downregulated genes in Cluster 3 showed enrichment for genes involved in the regulation of insulin and peptide secretion (Table S3)—a known function of TRPM3 [24,25,44]. Overall, the relative upregulation of gene expression in Trpm3-mutant lenses revealed a progressive transcriptional phenotype in which Trpm3-I/M lenses were moderately affected and Trpm3-M/M lenses were severely affected with significant activation of genes underlying Ca2+ homeostasis/dynamics (Figure 9B).
4. Discussion
Trpm3 has been identified as a signature gene for the lens epithelium and, along with its intron-hosted miRNA gene, Mir204, has been implicated in eye development and disease [52,53,54,55,56,57,58,59,64,65,68]. In this study, we have shown that Trpm3 and Mir204/211 were strongly co-expressed in the lens epithelium and other non-pigmented and pigmented ocular epithelia from the iris, ciliary body, and retina. Further, we found that a cataract-causing substitution (p.I65M) in Trpm3 resulted in (1) altered sensitivity of mutant TRPM3-M65 channels to extracellular [Ca2+] and PS activation in vitro and (2) disturbed cation (Ca2+, Na+, K+) and water homeostasis, increased MAPK1/3 phospho-activation, and upregulation of genes involved in Ca2+-dependent processes in the lens. By contrast, Trpm3-KO lenses did not replicate the pathophysiological changes found in Trpm3-mutant (I/M, M/M) lenses and, like Trpm3-KO lenses, neither Mir204-KO nor Mir204; Mir211-dKO lenses developed early-onset cataract. While the combined loss-of-function for Trpm3 and Mir204/211 on lens phenotype remains to be determined, our data suggest that early-onset cataract in Trpm3-mutant lenses resulted from a deleterious gain-of-function mechanism rather than a loss-of-function mechanism in the lens. Further, since the signature function of TRPM3 channels in the lens remains unclear, future studies will be needed to examine whether and how other mechanisms might compensate for the absence of the channel.
Elevated cytoplasmic Ca2+ concentrations, believed to result from an increase in non-selective cation conductance, have long been implicated in the pathophysiology of lens aging and cataract formation in humans and experimental animals [90]. Atomic absorption spectroscopy and fluorescent calcium imaging (GCaMP6s) studies of ex vivo Trpm3-mutant mouse lenses revealed that while cytosolic Ca2+ levels in heterozygous (I/M) and hemizygous (M/−) Trpm3-mutant lenses were mildly elevated, those in homozygous (M/M) Trpm3-mutant lenses were dramatically higher (~50-fold) than wild-type lenses (Figure 4A and Figure 5)—consistent with previously reported histochemical calcification of Trpm3-M/M lenses [68]. The high ([Ca2+]i) in Trpm3-M/M lenses was coupled with elevated ([Na+]i), decreased ([K+]i), and increased water content compared to wild-type lenses, further suggesting that osmoregulation had failed in Trpm3-M/M lenses (Figure 4B–D). In addition, functional over-expression studies in transfected HLE-B3 cells revealed that recombinant mutant TRPM3-M65 channels displayed a steeper dose–response to PS activation (between 10 and 50 μM) and an increased sensitivity to extracellular [Ca2+] (10 mM) when compared with wild-type TRPM3-I65 channels in vitro. Further, constitutive expression of knock-in mutant TRPM3-M65 channels in HLE-B3 cells and Trpm3-M/M lenses significantly increased phospho-activation of MAPK1/3 (Figure 7)—a known downstream target of TRPM3-mediated Ca2+ influx [32,33]. While PS is an effective experimental agonist for TRPM3 channels, the role of this neuro-steroid in lens biology is unclear, as is the significance of an altered sensitivity to PS activation. The true physiological agonist(s) of TRPM3 in the eye/lens remains elusive. Regardless, our data suggest that mutant TRPM3-M65 channels ‘leak’ inwardly when exposed to elevated extracellular [Ca2+], leading to cytoplasmic Ca2+ overload coupled with MAPK1/3 activation. Since the extracellular Ca2+ levels in the eye’s aqueous humor (1–2 mM), which bathes the anterior lens epithelium, are ~10,000-fold higher than intracellular Ca2+ levels in the lens (100 nM range) [90], it is likely that this steep inward concentration gradient causes increased Ca2+ entry via mutant TRPM3-M65 channels resulting in chronic Ca2+ accumulation in the developing lens triggering early-onset, progressive cataract. Beyond a role for TRPM3 in rare forms of inherited pediatric cataract, it is conceivable that aging human TRPM3 channels may also contribute to the historically enigmatic, non-selective cation leak implicated in the much more common forms of age-related cataract.
Recently, functional expression studies of de novo missense mutations underlying TRPM3-NDD, which include ophthalmological findings other than cataract, have been shown to increase basal channel activity leading to cellular calcium overload and enhanced response to PS activation—consistent with a deleterious gain-of-function effect(s) [51]. These TRPM3-NDD studies focused on transcript variant-14 (NM_001366145.2) coding for channel isoform-m (NP_ 1353074.2, 1719 amino-acids) that starts in exon-2 but contains one more coding exon (exon-16, 12 amino acids) than the variant-9/isoform-k (1707 amino-acids) used here (Figure S1). In addition to the different variants/isoforms studied, the TRPM3-NDD missense mutations lie in or near the transmembrane (TM) pore domain toward the C-terminus of channel isoform-m, whereas the p.I65M mutation underlying TRPM3 cataract lies distant to the pore near the N-terminus of channel isoform-k within a predicted calmodulin binding motif [21]. Further, since we have shown that the p.I65M missense mutation may also act as an alternative translation start site in vitro [52], we cannot exclude the possibility that it may generate N-terminal mutant isoforms of TRPM3 (starting at codon 65 of isoform-k) with novel channel properties in vivo. These TRPM3 mutants may include N-terminal truncation mutants of isoforms starting translation upstream in exon-2 (p.M1del-64 mutants) and N-terminal extension mutants of isoforms starting translation downstream at the end of exon-5 (p.M1ext-89 mutants) (Figure S1). Such differences in the location of mutations within functionally distinct regions of TRPM3 coupled with alternative splice and/or start sites between TRPM3 variants and isoforms may contribute to pleiotropic effects that generate variable phenotypes in the brain (TRPM3-NDD) versus the lens (TRPM3 cataract).
Trpm3-mutant lenses undergoing cataract development at P7 revealed >1240 differentially regulated genes, whereas transparent Trpm3-KO lenses at P7 yielded ~50 differentially regulated genes with minimal sharing between mutant and null genotypes (Figure S6B). These data highlight the dramatically distinct effects of Trpm3 mutation versus Trpm3 deficiency on global gene expression in the lens. Trpm3-mutant lenses revealed modest downregulation of genes involved in insulin and peptide secretion (Figure 9A, Tables S2 and S3). Activation of TRPM3 channels with PS or the synthetic agonist CIM0216 has been shown to control insulin release from pancreatic β cells and calcitonin gene-related peptide (CGRP) release from sensory nerve terminals [24,25,44]. However, the precise role of TRPM3-mediated insulin and/or neuro-peptide release in the lens is unclear. Conversely, Trpm3-mutant lenses revealed strong upregulation of genes involved in cation homeostasis—including those for the P-type ion-transport ATPases Atp1b1, Atp2b1, and Atp2b4 (Table S4). Atp1b1 encodes the glycosylated Na+/K+-transporting ATPase subunit β-1 (ATP1B1) that complexes with the large catalytic α-subunit to regulate the formation of functional Na+/K+ pumps, which actively extrude Na+ and uptake K+ in order to establish and maintain electrochemical gradients across the plasma membrane [91]. Atp2b1 and Atp2b4 encode the calmodulin-dependent plasma membrane Ca2+-ATPases ATP2B1 and ATP2B4, respectively, that function in Ca2+ export from eukaryotic cells against steep concentration gradients and play a critical role in intracellular Ca2+ homeostasis [92]. Such upregulation of ion-transport ATPase genes is consistent with attempts to counteract increased Ca2+ influx caused by functionally mutant TRPM3 channels expressed in the anterior epithelium of the lens.
5. Conclusions
Overall, our data support a deleterious gain-of-function mechanism that activates TRPM3 (p.I65M) mutant channels to drive chronic Ca2+-influx in the lens, resulting in an inherited form of early-onset, progressive cataract. Further, they suggest that TRPM3 channels may participate in the non-selective cation leak long associated with lens aging and cataract formation. The fact that TRPM3 channels can be inhibited by certain drugs (e.g., NSAIDs) and nutraceuticals—including citrus flavanones that have been associated with the attenuation of cataract [93,94]—raises the possibility of therapeutic interventions for TRPM3 cataract.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cells13030257/s1, Figure S1: Schematic of exon usage for TRPM3 transcript variant-9 used herein for functional expression studies of channel isoform-k in Figure 6; Figure S2: In situ hybridization (ISH) of Trpm3-transcripts in the neonatal mouse eye; Figure S3: In situ hybridization of Trpm3-transcripts in the mature mouse lens; Figure S4: In situ hybridization of Mir204 transcripts in the neonatal mouse lens; Figure S5: In situ hybridization of Mir204-transcripts in the neonatal mouse eye; Figure S6: Multi-dimensional scaling (MDS) plot (A) and Venn diagrams of upregulated genes (B) and downregulated genes (C) in heterozygous Trpm3-I/M, homozygous Trpm3-M/M, and Trpm3-KO lenses versus wild-type lenses at P7; Figure S7: Scatterplot of differentially regulated genes in Trpm3-KO (A), Trpm3-I/M (B), and Trpm3-M/M (C) lenses highlighting the relative expression levels of Trpm3 versus Trpm1 and Trpv4; Table S1: Primer sequences used for PCR-genotyping of Mir204-KO and Mir204; Mir211-dKO mice; Table S2: Differentially expressed genes (>2-fold) in Trpm3 -mutant lenses summarized in Figure 9A.; Table S3: Enriched GO categories for genes within each cluster in Figure 9A; Table S4: Genes associated with Ca2+ dynamics within Cluster 1 in Figure 9A that were most upregulated in Trpm3-M/M lenses.
Author Contributions
All authors made substantial contributions to the work, and each has read and approved the submitted version of the manuscript. Conceptualization, Y.Z. and A.S.; Methodology, Y.Z., T.M.B., P.A.R., Z.G., Y.-Q.C., M.S., N.A.D. and A.S.; Validation, Y.Z., P.A.R., Z.G., Y.-Q.C., M.S., N.A.D. and A.S.; Formal Analysis, Y.Z., P.A.R., Z.G., Y.-Q.C., N.A.D. and A.S.; Investigation, Y.Z., T.M.B., Z.G. and M.S.; Resources, P.A.R.; Data Curation, Y.Z., T.M.B., P.A.R., Z.G. and M.S.; Writing—Original Draft Preparation, A.S.; Writing—Review and Editing, Y.Z., T.M.B., P.A.R., Z.G., Y.-Q.C., M.S., N.A.D. and A.S.; Visualization, Y.Z., T.M.B., P.A.R., Z.G. and A.S.; Supervision, Y.-Q.C., N.A.D. and A.S.; Project Administration, A.S.; Funding Acquisition, A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NIH/NEI grants EY028899 (to AS) and EY02687 (Core Grant for Vision Research), NS103350 (to Y-QC), and EY009532 and EY029171 (to NAD) and an unrestricted grant to the Department of Ophthalmology and Visual Sciences as well as a Career Development Award (to PAR) from Research to Prevent Blindness (RPB). GTAC is supported in part by NIH Grants P30 CA91842 and UL1TR002345.
Institutional Review Board Statement
The Institutional Animal Care and Use Committee (IACUC) at Washington University approved all mouse procedures (Protocol No. 22-0114) in compliance with the Institute for Laboratory Animal Research (ILAR) guidelines.
Informed Consent Statement
Not applicable.
Data Availability Statement
RNA-seq data files have been deposited in the Gene Expression Omnibus (GEO) database under accession no. GSE250518.
Acknowledgments
We thank C. Zhang (National Eye Institute) for sharing Mir204-KO and Mir204; Mir211-dKO mice, M.L. Robinson (Ohio State) for sharing MLR10-Cre mice, G. Ling for eye histology support, and the Genome Technology Access Center (GTAC) staff at Washington University for RNA sequencing support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Minke, B. The history of the Drosophila TRP channel: The birth of a new channel superfamily. J. Neurogenet. 2010, 24, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Owsianik, G.; Nilius, B.; Voets, T. TRP channels. Compr. Physiol. 2012, 2, 563–608. [Google Scholar] [PubMed]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef]
- Yue, L.; Xu, H. TRP channels in health and disease at a glance. J. Cell Sci. 2021, 134, jcs258372. [Google Scholar] [CrossRef] [PubMed]
- Irie, S.; Furukawa, T. Trpm1. Handb. Exp. Pharmacol. 2014, 222, 387–402. [Google Scholar]
- Guo, H.; Carlson, J.A.; Slominski, A. Role of TRPM in melanocytes and melanoma. Exp. Dermatol. 2012, 21, 650–654. [Google Scholar] [CrossRef]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef]
- Miraldi Utz, V.; Pfeifer, W.; Longmuir, S.Q.; Olson, R.J.; Wang, K.; Drack, A.V. Presentation of TRPM1-Associated Congenital Stationary Night Blindness in Children. JAMA Ophthalmol. 2018, 136, 389–398. [Google Scholar] [CrossRef]
- Iosifidis, C.; Liu, J.; Gale, T.; Ellingford, J.M.; Campbell, C.; Ingram, S.; Chandler, K.; Parry, N.R.A.; Black, G.C.; Sergouniotis, P.I. Clinical and genetic findings in TRPM1-related congenital stationary night blindness. Acta Ophthalmol. 2022, 100, e1332–e1339. [Google Scholar] [CrossRef]
- Bellone, R.R.; Holl, H.; Setaluri, V.; Devi, S.; Maddodi, N.; Archer, S.; Sandmeyer, L.; Ludwig, A.; Foerster, D.; Pruvost, M.; et al. Evidence for a retroviral insertion in TRPM1 as the cause of congenital stationary night blindness and leopard complex spotting in the horse. PLoS ONE 2013, 8, e78280. [Google Scholar] [CrossRef]
- Oberwinkler, J.; Lis, A.; Giehl, K.M.; Flockerzi, V.; Philipp, S.E. Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J. Biol. Chem. 2005, 280, 22540–22548. [Google Scholar] [CrossRef] [PubMed]
- Fruhwald, J.; Camacho Londono, J.; Dembla, S.; Mannebach, S.; Lis, A.; Drews, A.; Wissenbach, U.; Oberwinkler, J.; Philipp, S.E. Alternative splicing of a protein domain indispensable for function of transient receptor potential melastatin 3 (TRPM3) ion channels. J. Biol. Chem. 2012, 287, 36663–36672. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.F.; Drews, A.; Loch, S.; Mohr, F.; Philipp, S.E.; Lambert, S.; Oberwinkler, J. TRPM3 channels provide a regulated influx pathway for zinc in pancreatic beta cells. Pflugers Arch. 2010, 460, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Held, K.; Janssens, A.; Toth, B.I.; Kerselaers, S.; Nilius, B.; Vennekens, R.; Voets, T. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat. Chem. Biol. 2014, 10, 188–195. [Google Scholar] [CrossRef]
- Held, K.; Voets, T.; Vriens, J. TRPM3 in temperature sensing and beyond. Temperature 2015, 2, 201–213. [Google Scholar] [CrossRef]
- Held, K.; Gruss, F.; Aloi, V.D.; Janssens, A.; Ulens, C.; Voets, T.; Vriens, J. Mutations in the voltage-sensing domain affect the alternative ion permeation pathway in the TRPM3 channel. J. Physiol. 2018, 596, 2413–2432. [Google Scholar] [CrossRef] [PubMed]
- Oberwinkler, J.; Phillipp, S.E. Trpm3. Handb. Exp. Pharmacol. 2007, 179, 253–267. [Google Scholar]
- Oberwinkler, J.; Philipp, S.E. Trpm3. Handb. Exp. Pharmacol. 2014, 222, 427–459. [Google Scholar]
- Yu, F.H.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef]
- Przibilla, J.; Dembla, S.; Rizun, O.; Lis, A.; Jung, M.; Oberwinkler, J.; Beck, A.; Philipp, S.E. Ca(2+)-dependent regulation and binding of calmodulin to multiple sites of Transient Receptor Potential Melastatin 3 (TRPM3) ion channels. Cell Calcium 2018, 73, 40–52. [Google Scholar] [CrossRef]
- Zhao, C.; MacKinnon, R. Structural and functional analyses of a GPCR-inhibited ion channel TRPM3. Neuron 2023, 111, 81–91.e7. [Google Scholar] [CrossRef]
- Grimm, C.; Kraft, R.; Sauerbruch, S.; Schultz, G.; Harteneck, C. Molecular and functional characterization of the melastatin-related cation channel TRPM3. J. Biol. Chem. 2003, 278, 21493–21501. [Google Scholar] [CrossRef]
- Wagner, T.F.; Loch, S.; Lambert, S.; Straub, I.; Mannebach, S.; Mathar, I.; Dufer, M.; Lis, A.; Flockerzi, V.; Philipp, S.E.; et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat. Cell Biol. 2008, 10, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Kichko, T.; De Clercq, K.; Klaassen, H.; Van Bree, R.; Vanherck, J.C.; Marchand, A.; Reeh, P.W.; Chaltin, P.; Voets, T.; et al. Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proc. Natl. Acad. Sci. USA 2015, 112, E1363–E1372. [Google Scholar] [CrossRef]
- Toth, B.I.; Konrad, M.; Ghosh, D.; Mohr, F.; Halaszovich, C.R.; Leitner, M.G.; Vriens, J.; Oberwinkler, J.; Voets, T. Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J. Gen. Physiol. 2015, 146, 51–63. [Google Scholar] [CrossRef]
- Badheka, D.; Borbiro, I.; Rohacs, T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J. Gen. Physiol. 2015, 146, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Voets, T. Heat sensing involves a TRiPlet of ion channels. Br. J. Pharmacol. 2019, 176, 3893–3898. [Google Scholar] [CrossRef] [PubMed]
- Persoons, E.; Kerselaers, S.; Voets, T.; Vriens, J.; Held, K. Partial Agonistic Actions of Sex Hormone Steroids on TRPM3 Function. Int. J. Mol. Sci. 2021, 22, 13652. [Google Scholar] [CrossRef] [PubMed]
- Lesch, A.; Rubil, S.; Thiel, G. Activation and inhibition of transient receptor potential TRPM3-induced gene transcription. Br. J. Pharmacol. 2014, 171, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Lesch, A.; Hui, X.; Lipp, P.; Thiel, G. Transient receptor potential melastatin-3 (TRPM3)-induced activation of AP-1 requires Ca2+ ions and the transcription factors c-Jun, ATF2, and ternary complex factor. Mol. Pharmacol. 2015, 87, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Lesch, A.; Rossler, O.G.; Thiel, G. Extracellular Signal-Regulated Protein Kinase, c-Jun N-terminal Protein Kinase, and Calcineurin Regulate Transient Receptor Potential M3 (TRPM3) Induced Activation of AP-1. J. Cell. Biochem. 2017, 118, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rossler, O.G. Transient receptor potential TRPM3 channels: Pharmacology, signaling, and biological functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rossler, O.G. TRPM3-Induced Gene Transcription Is under Epigenetic Control. Pharmaceuticals 2022, 15, 846. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rossler, O.G. Calmodulin Regulates Transient Receptor Potential TRPM3 and TRPM8-Induced Gene Transcription. Int. J. Mol. Sci. 2023, 24, 7902. [Google Scholar] [CrossRef] [PubMed]
- Straub, I.; Krugel, U.; Mohr, F.; Teichert, J.; Rizun, O.; Konrad, M.; Oberwinkler, J.; Schaefer, M. Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol. Pharmacol. 2013, 84, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Krugel, U.; Straub, I.; Beckmann, H.; Schaefer, M. Primidone inhibits TRPM3 and attenuates thermal nociception in vivo. Pain 2017, 158, 856–867. [Google Scholar] [CrossRef]
- Badheka, D.; Yudin, Y.; Borbiro, I.; Hartle, C.M.; Yazici, A.; Mirshahi, T.; Rohacs, T. Inhibition of Transient Receptor Potential Melastatin 3 ion channels by G-protein betagamma subunits. eLife 2017, 6, e26147. [Google Scholar] [CrossRef]
- Dembla, S.; Behrendt, M.; Mohr, F.; Goecke, C.; Sondermann, J.; Schneider, F.M.; Schmidt, M.; Stab, J.; Enzeroth, R.; Leitner, M.G.; et al. Anti-nociceptive action of peripheral mu-opioid receptors by G-beta-gamma protein-mediated inhibition of TRPM3 channels. eLife 2017, 6, e26280. [Google Scholar] [CrossRef]
- Quallo, T.; Alkhatib, O.; Gentry, C.; Andersson, D.A.; Bevan, S. G protein betagamma subunits inhibit TRPM3 ion channels in sensory neurons. eLife 2017, 6, e26138. [Google Scholar] [CrossRef]
- Kelemen, B.; Lisztes, E.; Vladar, A.; Hanyicska, M.; Almassy, J.; Olah, A.; Szollosi, A.G.; Penzes, Z.; Posta, J.; Voets, T.; et al. Volatile anaesthetics inhibit the thermosensitive nociceptor ion channel transient receptor potential melastatin 3 (TRPM3). Biochem. Pharmacol. 2020, 174, 113826. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.P.; Cost, N.G.; Hegde, S.; Kellner, E.; Mikhaylova, O.; Stratton, Y.; Ehmer, B.; Abplanalp, W.A.; Pandey, R.; Biesiada, J.; et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell 2014, 26, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, M. Transient receptor potential channels in the context of nociception and pain-recent insights into TRPM3 properties and function. Biol. Chem. 2019, 400, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Mannebach, S.; Mathar, I.; Weissgerber, P.; Freichel, M.; Loodin, A.P.; Fecher-Trost, C.; Belkacemi, A.; Beck, A.; Philipp, S.E. Control of Insulin Release by Transient Receptor Potential Melastatin 3 (TRPM3) Ion Channels. Cell Physiol. Biochem. 2020, 54, 1115–1131. [Google Scholar]
- Kim, J.; Williams, F.J.; Dreger, D.L.; Plassais, J.; Davis, B.W.; Parker, H.G.; Ostrander, E.A. Genetic selection of athletic success in sport-hunting dogs. Proc. Natl. Acad. Sci. USA 2018, 115, E7212–E7221. [Google Scholar] [CrossRef]
- Kuniba, H.; Yoshiura, K.; Kondoh, T.; Ohashi, H.; Kurosawa, K.; Tonoki, H.; Nagai, T.; Okamoto, N.; Kato, M.; Fukushima, Y.; et al. Molecular karyotyping in 17 patients and mutation screening in 41 patients with Kabuki syndrome. J. Hum. Genet. 2009, 54, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Pagnamenta, A.T.; Holt, R.; Yusuf, M.; Pinto, D.; Wing, K.; Betancur, C.; Scherer, S.W.; Volpi, E.V.; Monaco, A.P. A family with autism and rare copy number variants disrupting the Duchenne/Becker muscular dystrophy gene DMD and TRPM3. J. Neurodev. Disord. 2011, 3, 124–131. [Google Scholar] [CrossRef]
- Thompson, B.A.; Dear, K.; Donaldson, E.; Nixon, R.; Winship, I.M. A novel candidate gene in autosomal dominant facial pruritus. Clin. Exp. Dermatol. 2022, 47, 184–186. [Google Scholar] [CrossRef]
- Lines, M.A.; Goldenberg, P.; Wong, A.; Srivastava, S.; Bayat, A.; Hove, H.; Karstensen, H.G.; Anyane-Yeboa, K.; Liao, J.; Jiang, N.; et al. Phenotypic spectrum of the recurrent TRPM3 p.(Val837Met) substitution in seven individuals with global developmental delay and hypotonia. Am. J. Med. Genet. A 2022, 188, 1667–1675. [Google Scholar] [CrossRef]
- Dyment, D.; Lines, M.; Innes, A.M. TRPM3-Related Neurodevelopmental Disorder. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; NCBI: Seattle, DC, USA, 2023. [Google Scholar]
- Burglen, L.; Van Hoeymissen, E.; Qebibo, L.; Barth, M.; Belnap, N.; Boschann, F.; Depienne, C.; De Clercq, K.; Douglas, A.G.L.; Fitzgerald, M.P.; et al. Gain-of-function variants in the ion channel gene TRPM3 underlie a spectrum of neurodevelopmental disorders. eLife 2023, 12, e81032. [Google Scholar] [CrossRef]
- Bennett, T.M.; Mackay, D.S.; Siegfried, C.J.; Shiels, A. Mutation of the melastatin-related cation channel, TRPM3, underlies inherited cataract and glaucoma. PLoS ONE 2014, 9, e104000. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Leng, Y.; Han, S.; Yan, L.; Lu, C.; Luo, Y.; Zhang, X.; Cao, L. Clinical and genetic characteristics of Chinese patients with familial or sporadic pediatric cataract. Orphanet J. Rare Dis. 2018, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A. TRPM3_miR-204: A complex locus for eye development and disease. Hum. Genom. 2020, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Yonova-Doing, E.; Zhao, W.; Igo, R.P., Jr.; Wang, C.; Sundaresan, P.; Lee, K.E.; Jun, G.R.; Alves, A.C.; Chai, X.; Chan, A.S.Y.; et al. Common variants in SOX-2 and congenital cataract genes contribute to age-related nuclear cataract. Commun. Biol. 2020, 3, 755. [Google Scholar] [CrossRef]
- Wang, Q.; Dhindsa, R.S.; Carss, K.; Harper, A.R.; Nag, A.; Tachmazidou, I.; Vitsios, D.; Deevi, S.V.V.; Mackay, A.; Muthas, D.; et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 2021, 597, 527–532. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes. Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.W.; Kinzy, T.G.; Cooke Bailey, J.N.; Xu, Z.; Hysi, P.; Wiggs, J.L.; Consortium, N. Mechanosensitive ion channel gene survey suggests potential roles in primary open angle glaucoma. Sci. Rep. 2023, 13, 15871. [Google Scholar] [CrossRef] [PubMed]
- Conte, I.; Hadfield, K.D.; Barbato, S.; Carrella, S.; Pizzo, M.; Bhat, R.S.; Carissimo, A.; Karali, M.; Porter, L.F.; Urquhart, J.; et al. MiR-204 is responsible for inherited retinal dystrophy associated with ocular coloboma. Proc. Natl. Acad. Sci. USA 2015, 112, E3236–E3245. [Google Scholar] [CrossRef]
- Beebe, D.C.; Coats, J.M. The lens organizes the anterior segment: Specification of neural crest cell differentiation in the avian eye. Dev. Biol. 2000, 220, 424–431. [Google Scholar] [CrossRef]
- Iribarren, R. Crystalline lens and refractive development. Prog. Retin. Eye Res. 2015, 47, 86–106. [Google Scholar] [CrossRef]
- Bassnett, S.; Shi, Y.; Vrensen, G.F. Biological glass: Structural determinants of eye lens transparency. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 1250–1264. [Google Scholar] [CrossRef]
- Donaldson, P.J.; Grey, A.C.; Maceo Heilman, B.; Lim, J.C.; Vaghefi, E. The physiological optics of the lens. Prog. Retin. Eye Res. 2017, 56, e1–e24. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, D.; Cvekl, A. A comprehensive spatial-temporal transcriptomic analysis of differentiating nascent mouse lens epithelial and fiber cells. Exp. Eye Res. 2018, 175, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Shaham, O.; Gueta, K.; Mor, E.; Oren-Giladi, P.; Grinberg, D.; Xie, Q.; Cvekl, A.; Shomron, N.; Davis, N.; Keydar-Prizant, M.; et al. Pax6 regulates gene expression in the vertebrate lens through miR-204. PLoS Genet. 2013, 9, e1003357. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Pothecary, C.A.; Jagannath, A.; Foster, R.G.; Hankins, M.W.; Peirson, S.N. Profound defects in pupillary responses to light in TRPM-channel null mice: A role for TRPM channels in non-image-forming photoreception. Eur. J. Neurosci. 2012, 35, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Miyagishima, K.J.; Dong, L.; Rising, A.; Nimmagadda, M.; Liang, G.; Sharma, R.; Dejene, R.; Wang, Y.; Abu-Asab, M.; et al. Regulation of phagolysosomal activity by miR-204 critically influences structure and function of retinal pigment epithelium/retina. Hum. Mol. Genet. 2019, 28, 3355–3368. [Google Scholar] [CrossRef]
- Zhou, Y.; Bennett, T.M.; Shiels, A. Mutation of the TRPM3 cation channel underlies progressive cataract development and lens calcification associated with pro-fibrotic and immune cell responses. FASEB J. 2021, 35, e21288. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Garner, A.R.; Shimaoka, D.; Chuong, A.S.; Klapoetke, N.C.; Li, L.; van der Bourg, A.; Niino, Y.; Egolf, L.; Monetti, C.; et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 2015, 85, 942–958. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, Y.; Rizo, C.M.; Overbeek, P.A.; Robinson, M.L. Insertion of a Pax6 consensus binding site into the alphaA-crystallin promoter acts as a lens epithelial cell enhancer in transgenic mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1930–1939. [Google Scholar] [CrossRef]
- Simirskii, V.N.; Lee, R.S.; Wawrousek, E.F.; Duncan, M.K. Inbred FVB/N mice are mutant at the cp49/Bfsp2 locus and lack beaded filament proteins in the lens. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4931–4934. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shiels, A. Epha2 and Efna5 participate in lens cell pattern-formation. Differentiation 2018, 102, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; Shiels, A. Lens ER-stress response during cataract development in Mip-mutant mice. Biochim. Biophys. Acta 2016, 1862, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; Shiels, A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation 2019, 109, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; Ruzycki, P.A.; Shiels, A. Mutation of the EPHA2 Tyrosine-Kinase Domain Dysregulates Cell Pattern Formation and Cytoskeletal Gene Expression in the Lens. Cells 2021, 10, 2606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; White, T.W.; Shiels, A. Charged multivesicular body protein 4b forms complexes with gap junction proteins during lens fiber cell differentiation. FASEB J. 2023, 37, e22801. [Google Scholar] [CrossRef] [PubMed]
- Delamere, N.A.; Dean, W.L.; Stidam, J.M.; Moseley, A.E. Influence of amphotericin B on the sodium pump of porcine lens epithelium. Am. J. Physiol. 1996, 270 Pt 1, C465–C473. [Google Scholar] [CrossRef]
- Yorio, T.; Bentley, P.J. The effects of hyperosmotic agents on the electrical properties of the amphibian lens in vitro. Exp. Eye Res. 1976, 22, 195–208. [Google Scholar] [CrossRef]
- Guo, Z.; Czerpaniak, K.; Zhang, J.; Cao, Y.Q. Increase in trigeminal ganglion neurons that respond to both calcitonin gene-related peptide and pituitary adenylate cyclase-activating polypeptide in mouse models of chronic migraine and posttraumatic headache. Pain 2021, 162, 1483–1499. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; Hill, D.P.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar]
- Huang, J.; Zhao, L.; Fan, Y.; Liao, L.; Ma, P.X.; Xiao, G.; Chen, D. The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat. Commun. 2019, 10, 2876. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef]
- Lee, N.; Chen, J.; Sun, L.; Wu, S.; Gray, K.R.; Rich, A.; Huang, M.; Lin, J.H.; Feder, J.N.; Janovitz, E.B.; et al. Expression and characterization of human transient receptor potential melastatin 3 (hTRPM3). J. Biol. Chem. 2003, 278, 20890–20897. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Xia, C.H.; FitzGerald, P.G.; Li, R.; Wang, J.; Gong, X. CP49 and filensin intermediate filaments are essential for formation of cold cataract. Mol. Vis. 2020, 26, 603–612. [Google Scholar]
- Rhodes, J.D.; Sanderson, J. The mechanisms of calcium homeostasis and signalling in the lens. Exp. Eye Res. 2009, 88, 226–234. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Doki, Y.; Sugiyama, Y.; Kobayashi, R.; Nagai, N.; Morisita, N.; Endo, S.; Funakoshi-Tago, M.; Tamura, H. Effect of Alpha-Glucosyl-Hesperidin Consumption on Lens Sclerosis and Presbyopia. Cells 2021, 10, 382. [Google Scholar] [CrossRef]
- Doki, Y.; Nakazawa, Y.; Morishita, N.; Endo, S.; Nagai, N.; Yamamoto, N.; Tamura, H.; Funakoshi-Tago, M. Hesperetin treatment attenuates glycation of lens proteins and advanced-glycation end products generation. Mol. Med. Rep. 2023, 27, 103. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).