


Unraveling Histone Loss in Aging and Senescence

Abstract
:1. Introduction
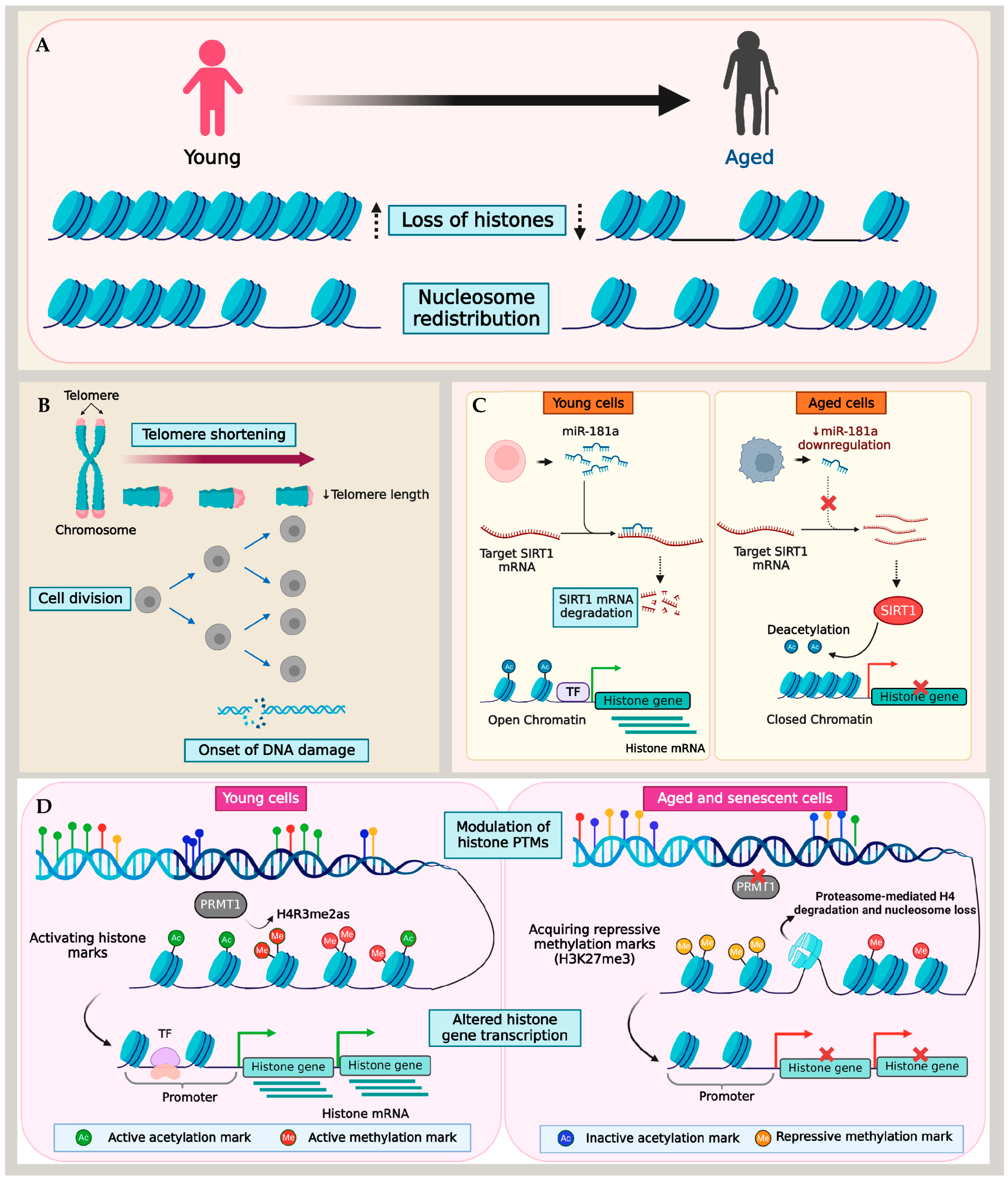
2. Age-Related Histone Loss and Altered Nucleosome Occupancy in Non-Mammalian Models
3. Histone Loss in Mammalian Models of Replicative and Chronological Aging
4. Alterations in Nucleosome Landscape in Aging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Part B | Histone Alteration | Organism or Cell Line | Reason for Histone Alterations | Cellular Alterations | Reference |
|---|---|---|---|---|---|
| 1 | Histone H1 and histone H4 | Human fetal lung fibroblast (TIG-1) | Replicative senescence | Age-related increase in nuclear proteins; decrease in histone H1 biosynthesis in senescent fibroblasts | Mitsui et al., 1980 [65] |
| 2 | Reduction in nucleosome occupancy | Human fibroblast | In vitro and in vivo aging of human diploid fibroblasts | Changes in chromatin structure | Ishimi et al., 1987 [67] |
| 3 | Reduction in nucleosome occupancy | Human donor fibroblast | In vitro and in vivo aging of human diploid fibroblasts | Changes in chromatin structure and cytoskeletal elements | Macieira-Coelho, 1991 [68] |
| 4 | Loss of histone H1 | Human fibroblasts WI-38, MRC-5, IMR-90, and BJ | Cellular senescence induced by retroviral expression of oncogenic Ras (oncogene-induced senescence) | Exhibit unique chromatin condensation, termed senescence-associated heterochromatic foci (SAHF); loss of linker histone H1 accompanied by increased chromatin-bound high mobility group A2 (HMGA2) competitor protein | Funayama, 2006 [88] |
| 5 | Histone H3 and H4 | Human diploid fibroblast IMR90 and WI38, fibroblast from young (9 y.o.) and 92 y.o. individual | Replicative aging in cell culture models and normal aging in primary cells | Downregulation of histone regulatory factors and histone chaperones (SLBP, Asf1a, Asf1b, CAF1-p150, and CAF1-p60); several altered histone modifications; accumulation of DNA damage and DNA damage response; telomere dysfunction | O’Sullivan et al., 2010 [69] |
| 6 | H1, H2A, H2B, H3, H4, and the variant histone H2AX | HeLa Hmgb1−/− | HMGB1 knockdown HeLa cells were generated using shRNA/siRNA | Increased global transcription and altered transcriptome profile in cells undergoing histone depletion | Celona et al., 2011 [60] |
| 7 | Altered histone expression and changes in nucleosome occupancy | Liver tissue from young (3 months) and old (21 months) C57BL6 mouse | Age-dependent changes | Age-related activation of lipogenesis and inflammatory genes; nucleosome occupancy changes with age selectively repress or derepress genes involved in lipid metabolism; histone isoforms are differentially regulated | Bochkis et al., 2014 [84] |
| 8 | HIST1H3D, HIST1H3E, and HIST4H4 | CD8+ T cells from healthy young (22–40 yr) and old (65+ yr) individuals | Closed chromatin associated with aging | Age-associated changes to chromatin accessibility | Ucar et al., 2017 [89] |
| 9 | Altered histone H3 expression and changes in H3 nucleosome occupancy | Tissues (i.e., heart, liver, cerebellum, and olfactory bulb) and one primary cell type (i.e., primary neural stem cell cultures from the subventricular zone) from 3-, 12-, and 29-month-old mice | Chronological aging | Age-related alterations in nucleosome positioning exhibit localized changes in genomic loci linked to DNA remodeling factors; these changes also extend their influence to the regulation of inflammatory genes | Chen et al., 2020 [87] |
| 10 | H4 depletion and degradation of nucleosomes | Human fibroblasts IMR90, CRL-1474, and HEK-293T | Senescence-mediated H4 loss; proteasome-mediated H4 degradation | H4 and H3 concentrations are reduced at the promoter regions of cell cycle inhibitor genes, SASP-related genes, and anti-apoptotic genes | Lin et al., 2020 [90] |
| 11 | Global loss of core histones | Human naive CD4+ T cell from young (20–35 years) and old (65–85 years) adults | Age-related reduction of miR-181ab1 and consequent increase of its target histone deacetylase, SIRT1, leads to deacetylation of histone gene promoters and downregulation of histone genes | Replication stress, delayed S-phase progression, and activation of proinflammatory pathways | Kim et al., 2021 [70] |
| 12 | Histone reduction | Human fetal lung IMR90 diploid fibroblasts, neonatal human epidermal melanocytes | Senescent cells form cytoplasmic chromatin fragments (CCFs), and proteolysis of CCFs depletes total histones in a lysosome-dependent manner | Genomic DNA damage; deterioration of nuclear integrity; CCFs transition to the cytoplasm and subsequent histone loss from CCFs | Ivanov et al., 2013 [80] |
| 13 | H1, H2A, H2B, H3, and H4 | Primary human umbilical vein endothelial cells (HUVECs) | Replicative senescence and inflammatory cytokine TNF-α-mediated senescence | Cell cycle arrest due to decline in cell cycle and mitosis regulatory factors; deterioration of DNA repair mechanisms; loss of chromatin architecture | Kandhaya-Pillai et al., 2023 [76] |
5. Multiple Mechanisms of Histone Loss during Aging
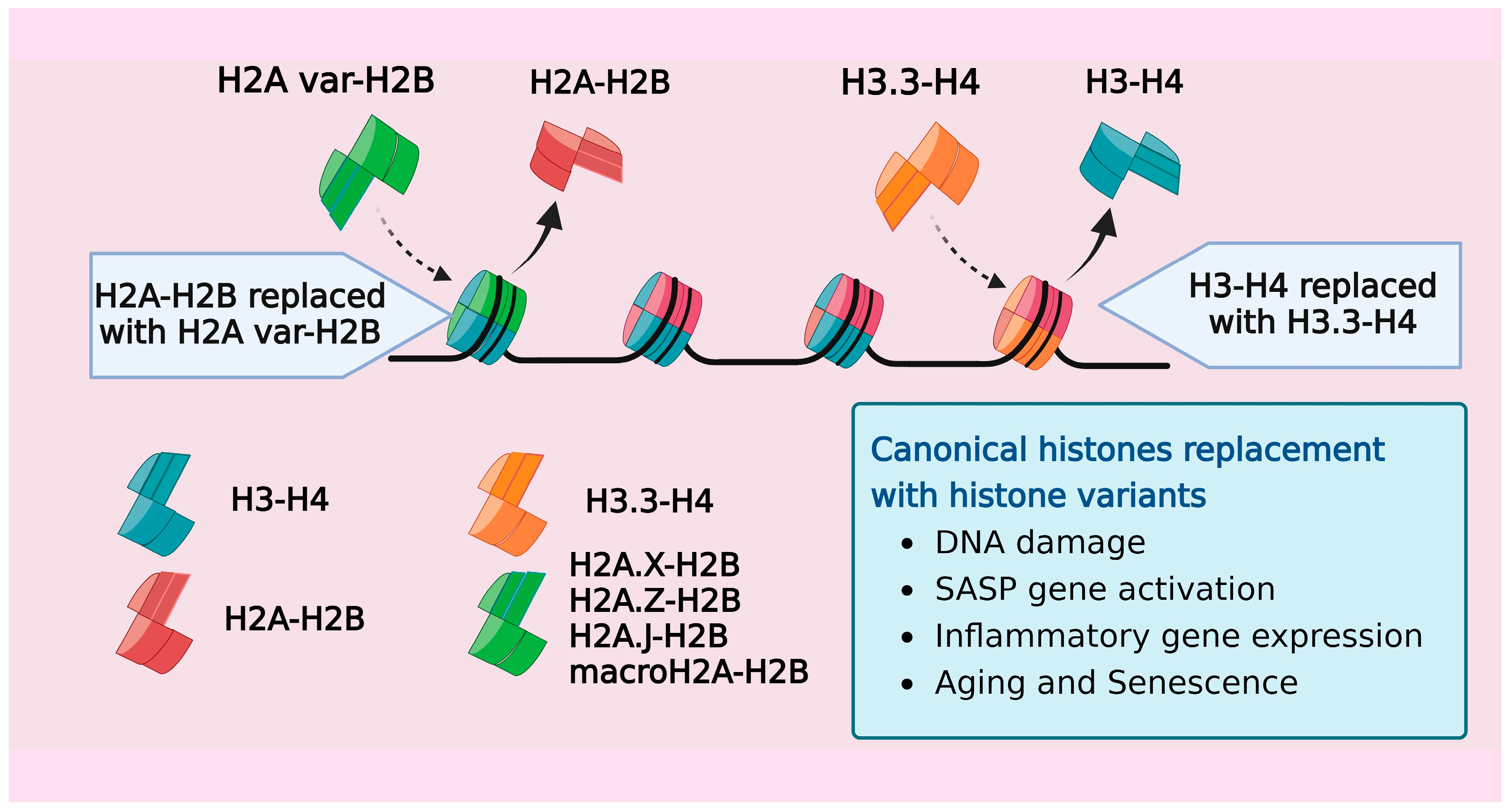
6. Age-Associated Replacement of Canonical Isoforms by Histone Variants
| Human H1 Histones | ||||
|---|---|---|---|---|
| Variant Symbol | Previous Symbol | HGNC Gene Symbol | HGNC ID | HGNC Gene Name |
| H1.0 | H1F0 | H1-0 | HGNC:4714 | H1.0 linker histone |
| H1.1 | HIST1H1A | H1-1 | HGNC:4715 | H1.1 linker histone, cluster member |
| H1.2 | HIST1H1C | H1-2 | HGNC:4716 | H1.2 linker histone, cluster member |
| H1.3 | HIST1H1D | H1-3 | HGNC:4717 | H1.3 linker histone, cluster member |
| H1.4 | HIST1H1E | H1-4 | HGNC:4718 | H1.4 linker histone, cluster member |
| H1.5 | HIST1H1B | H1-5 | HGNC:4719 | H1.5 linker histone, cluster member |
| H1.6 | HIST1H1T | H1-6 | HGNC:4720 | H1.6 linker histone, cluster member |
| H1.7 | H1FNT | H1-7 | HGNC:24893 | H1.7 linker histone |
| H1.8 | H1FOO | H1-8 | HGNC:18463 | H1.8 linker histone |
| NA | HILS1 | H1-9P | HGNC:30616 | H1.9 linker histone, pseudogene |
| H1.10 | H1FX | H1-10 | HGNC:4722 | H1.10 linker histone |
| NA | HIST1H1PS1 | H1-12P | HGNC:19163 | H1.12 linker histone, cluster member pseudogene |
| Human H2A Histones | ||||
| Variant Symbol | Previous Symbol | HGNC Gene Symbol | HGNC ID | HGNC Gene Name |
| H2A | HIST1H2AA | H2AC1 | HGNC:18729 | H2A clustered histone 1 |
| NA | HIST1H2APS1 | H2AC2P | HGNC:18720 | H2A clustered histone 2, pseudogene |
| NA | HIST1H2APS2 | H2AC3P | HGNC:18804 | H2A clustered histone 3, pseudogene |
| H2A | HIST1H2AB | H2AC4 | HGNC:4734 | H2A clustered histone 4 |
| NA | HIST1H2APS5 | H2AC5P | HGNC:4728 | H2A clustered histone 5, pseudogene |
| H2A | HIST1H2AC | H2AC6 | HGNC:4733 | H2A clustered histone 6 |
| H2A | HIST1H2AD | H2AC7 | HGNC:4729 | H2A clustered histone 7 |
| H2A | HIST1H2AE | H2AC8 | HGNC:4724 | H2A clustered histone 8 |
| NA | HIST1H2APS3 | H2AC9P | HGNC:18805 | H2A clustered histone 9, pseudogene |
| NA | HIST1H2APS4 | H2AC10P | HGNC:4732 | H2A clustered histone 10, pseudogene |
| H2A | HIST1H2AG | H2AC11 | HGNC:4737 | H2A clustered histone 11 |
| H2A | HIST1H2AH | H2AC12 | HGNC:13671 | H2A clustered histone 12 |
| H2A | HIST1H2AI | H2AC13 | HGNC:4725 | H2A clustered histone 13 |
| H2A | HIST1H2AJ | H2AC14 | HGNC:4727 | H2A clustered histone 14 |
| H2A | HIST1H2AK | H2AC15 | HGNC:4726 | H2A clustered histone 15 |
| H2A | HIST1H2AL | H2AC16 | HGNC:4730 | H2A clustered histone 16 |
| H2A | HIST1H2AM | H2AC17 | HGNC:4735 | H2A clustered histone 17 |
| H2A | HIST2H2AA3 | H2AC18 | HGNC:4736 | H2A clustered histone 18 |
| H2A | HIST2H2AA4 | H2AC19 | HGNC:29668 | H2A clustered histone 19 |
| H2A | HIST2H2AC | H2AC20 | HGNC:4738 | H2A clustered histone 20 |
| H2A | HIST2H2AB | H2AC21 | HGNC:20508 | H2A clustered histone 21 |
| H2A | HIST3H2A | H2AC25 | HGNC:20507 | H2A clustered histone 25 |
| H2A.Z.1 | H2AFZ | H2AZ1 | HGNC:4741 | H2A.Z variant histone 1 |
| H2A.Z.2 | H2AFV | H2AZ2 | HGNC:20664 | H2A.Z variant histone 2 |
| macroH2A.1 | H2AFY | MACROH2A1 | HGNC:4740 | macroH2A.1 histone |
| macroH2A.2 | H2AFY2 | MACROH2A2 | HGNC:14453 | macroH2A.2 histone |
| H2A.X | H2AFX | H2AX | HGNC:4739 | H2A.X variant histone |
| H2A.J | H2AFJ | H2AJ | HGNC:14456 | H2A.J histone |
| H2A.B | H2AFB1 | H2AB1 | HGNC:22516 | H2A.B variant histone 1 |
| H2A.B | H2AFB2 | H2AB2 | HGNC:18298 | H2A.B variant histone 2 |
| H2A.B | H2AFB3 | H2AB3 | HGNC:14455 | H2A.B variant histone 3 |
| H2A.P | HYPM | H2AP | HGNC:18417 | H2A.P histone |
| NA | NA | H2AQ1P | HGNC:53962 | H2A.Q variant histone 1, pseudogene |
| H2A.L | NA | H2AL1Q | HGNC:53959 | H2A.L variant histone 1Q |
| NA | NA | H2AL1MP | HGNC:53961 | H2A.L variant histone 1 M, pseudogene |
| H2A.L | NA | H2AL3 | HGNC:53960 | H2A.L variant histone 3 |
| Human H2B Histones | ||||
| Variant Symbol | Previous Symbol | HGNC Gene Symbol | HGNC ID | HGNC Gene Name |
| H2B | HIST1H2BA | H2BC1 | HGNC:18730 | H2B clustered histone 1 |
| NA | HIST1H2BPS1 | H2BC2P | HGNC:18719 | H2B clustered histone 2, pseudogene |
| H2B | HIST1H2BB | H2BC3 | HGNC:4751 | H2B clustered histone 3 |
| H2B | HIST1H2BC | H2BC4 | HGNC:4757 | H2B clustered histone 4 |
| H2B | HIST1H2BD | H2BC5 | HGNC:4747 | H2B clustered histone 5 |
| H2B | HIST1H2BE | H2BC6 | HGNC:4753 | H2B clustered histone 6 |
| H2B | HIST1H2BF | H2BC7 | HGNC:4752 | H2B clustered histone 7 |
| H2B | HIST1H2BG | H2BC8 | HGNC:4746 | H2B clustered histone 8 |
| H2B | HIST1H2BH | H2BC9 | HGNC:4755 | H2B clustered histone 9 |
| H2B | HIST1H2BI | H2BC10 | HGNC:4756 | H2B clustered histone 10 |
| H2B | HIST1H2BJ | H2BC11 | HGNC:4761 | H2B clustered histone 11 |
| H2B | HIST1H2BK | H2BC12 | HGNC:13954 | H2B clustered histone 12 |
| H2B | HIST1H2BL | H2BC13 | HGNC:4748 | H2B clustered histone 13 |
| H2B | HIST1H2BM | H2BC14 | HGNC:4750 | H2B clustered histone 14 |
| H2B | HIST1H2BN | H2BC15 | HGNC:4749 | H2B clustered histone 15 |
| NA | HIST1H2BPS2 | H2BC16P | HGNC:4754 | H2B clustered histone 16, pseudogene |
| H2B | HIST1H2BO | H2BC17 | HGNC:4758 | H2B clustered histone 17 |
| H2B | HIST2H2BF | H2BC18 | HGNC:24700 | H2B clustered histone 18 |
| NA | HIST2H2BD | H2BC19P | HGNC:20517 | H2B clustered histone 19, pseudogene |
| NA | HIST2H2BC | H2BC20P | HGNC:20516 | H2B clustered histone 20, pseudogene |
| H2B | HIST2H2BE | H2BC21 | HGNC:4760 | H2B clustered histone 21 |
| H2B | HIST3H2BB | H2BC26 | HGNC:20514 | H2B clustered histone 26 |
| NA | HIST3H2BA | H2BC27P | HGNC:20515 | H2B clustered histone 27, pseudogene |
| H2B.K | H2BE1 | H2BK1 | HGNC:53833 | H2B.K variant histone 1 |
| NA | H2BP4 | H2BL1P | HGNC:54442 | H2B.L histone variant 1, pseudogene |
| H2B.W | H2BFWT | H2BW1 | HGNC:27252 | H2B.W histone 1 |
| H2B.W | H2BFM | H2BW2 | HGNC:27867 | H2B.W histone 2 |
| NA | NA | H2BW3P | HGNC:44390 | H2B.W histone 3, pseudogene |
| NA | H2BFXP | H2BW4P | HGNC:25757 | H2B.W histone 4, pseudogene |
| H2B.N | NA | H2BN1 | HGNC:56200 | H2B.N variant histone 1 |
| H2B | H2BFS | H2BC12L | HGNC:4762 | H2B clustered histone 12 like |
| Human H3 Histones | ||||
| Variant Symbol | Previous Symbol | HGNC Gene Symbol | HGNC ID | HGNC Gene Name |
| H3.1 | HIST1H3A | H3C1 | HGNC:4766 | H3 clustered histone 1 |
| H3.1 | HIST1H3B | H3C2 | HGNC:4776 | H3 clustered histone 2 |
| H3.1 | HIST1H3C | H3C3 | HGNC:4768 | H3 clustered histone 3 |
| H3.1 | HIST1H3D | H3C4 | HGNC:4767 | H3 clustered histone 4 |
| NA | NA | H3C5P | HGNC:54427 | H3 clustered histone 5, pseudogene |
| H3.1 | HIST1H3E | H3C6 | HGNC:4769 | H3 clustered histone 6 |
| H3.1 | HIST1H3F | H3C7 | HGNC:4773 | H3 clustered histone 7 |
| H3.1 | HIST1H3G | H3C8 | HGNC:4772 | H3 clustered histone 8 |
| NA | HIST1H3PS1 | H3C9P | HGNC:18982 | H3 clustered histone 9, pseudogene |
| H3.1 | HIST1H3H | H3C10 | HGNC:4775 | H3 clustered histone 10 |
| H3.1 | HIST1H3I | H3C11 | HGNC:4771 | H3 clustered histone 11 |
| H3.1 | HIST1H3J | H3C12 | HGNC:4774 | H3 clustered histone 12 |
| H3.2 | HIST2H3D | H3C13 | HGNC:25311 | H3 clustered histone 13 |
| H3.2 | HIST2H3C | H3C14 | HGNC:20503 | H3 clustered histone 14 |
| H3.2 | HIST2H3A | H3C15 | HGNC:20505 | H3 clustered histone 15 |
| H3.3 | H3F3, H3F3A | H3-3A | HGNC:4764 | H3.3 histone A |
| H3.3 | H3F3B | H3-3B | HGNC:4765 | H3.3 histone B |
| H3.4 | HIST3H3 | H3-4 | HGNC:4778 | H3.4 histone, cluster member |
| H3.5 | H3F3C | H3-5 | HGNC:33164 | H3.5 histone |
| NA (H3.6) | H3F3AP6 | H3P16 | HGNC:42982 | H3 histone pseudogene 16 |
| H3.7 | HIST2H3PS2 | H3-7 | HGNC:32060 | H3.7 histone (putative) |
| NA (H3.8) | H3F3AP5 | H3P44 | HGNC:42981 | H3 histone pseudogene 44 |
| H3.Y.1 | NA | H3Y1 | HGNC:43735 | H3.Y histone 1 |
| H3.Y.2 | NA | H3Y2 | HGNC:43734 | H3.Y histone 2 |
| cenH3 | NA | CENPA | HGNC:1851 | Centromere protein A |
| Human H4 Histones | ||||
| Variant Symbol | Previous Symbol | HGNC Gene Symbol | HGNC ID | HGNC Gene Name |
| H4 | HIST1H4A | H4C1 | HGNC:4781 | H4 clustered histone 1 |
| H4 | HIST1H4B | H4C2 | HGNC:4789 | H4 clustered histone 2 |
| H4 | HIST1H4C | H4C3 | HGNC:4787 | H4 clustered histone 3 |
| H4 | HIST1H4D | H4C4 | HGNC:4782 | H4 clustered histone 4 |
| H4 | HIST1H4E | H4C5 | HGNC:4790 | H4 clustered histone 5 |
| H4 | HIST1H4F | H4C6 | HGNC:4783 | H4 clustered histone 6 |
| H4 | HIST1H4G | H4C7 | HGNC:4792 | H4 clustered histone 7 |
| H4 | HIST1H4H | H4C8 | HGNC:4788 | H4 clustered histone 8 |
| H4 | HIST1H4I | H4C9 | HGNC:4793 | H4 clustered histone 9 |
| NA | HIST1H4PS1 | H4C10P | HGNC:4786 | H4 clustered histone 10, pseudogene |
| H4 | HIST1H4J | H4C11 | HGNC:4785 | H4 clustered histone 11 |
| H4 | HIST1H4K | H4C12 | HGNC:4784 | H4 clustered histone 12 |
| H4 | HIST1H4L | H4C13 | HGNC:4791 | H4 clustered histone 13 |
| H4 | HIST2H4A | H4C14 | HGNC:4794 | H4 clustered histone 14 |
| H4 | HIST2H4B | H4C15 | HGNC:29607 | H4 clustered histone 15 |
| H4 | HIST4H4 | H4C16 | HGNC:20510 | H4 histone 16 |
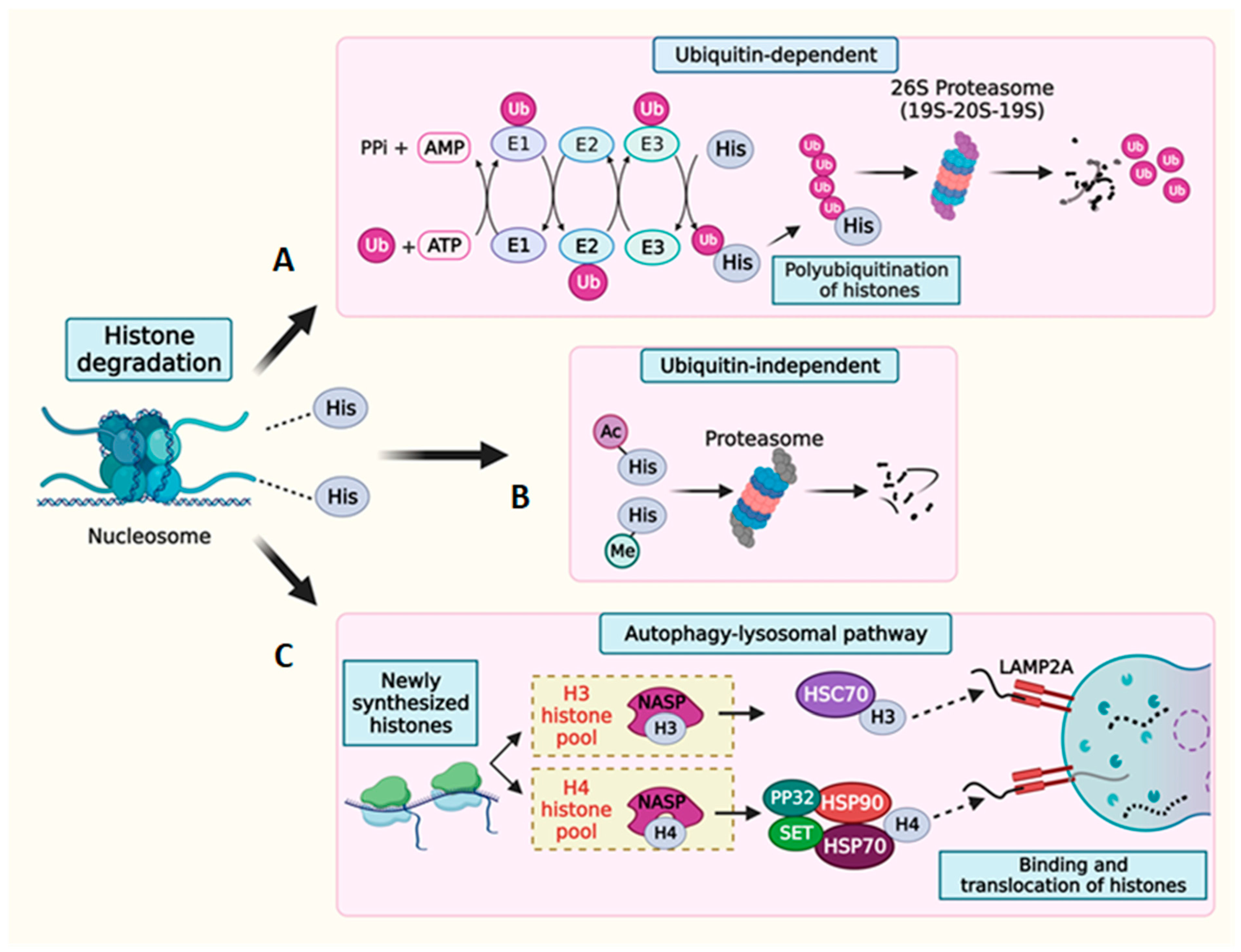
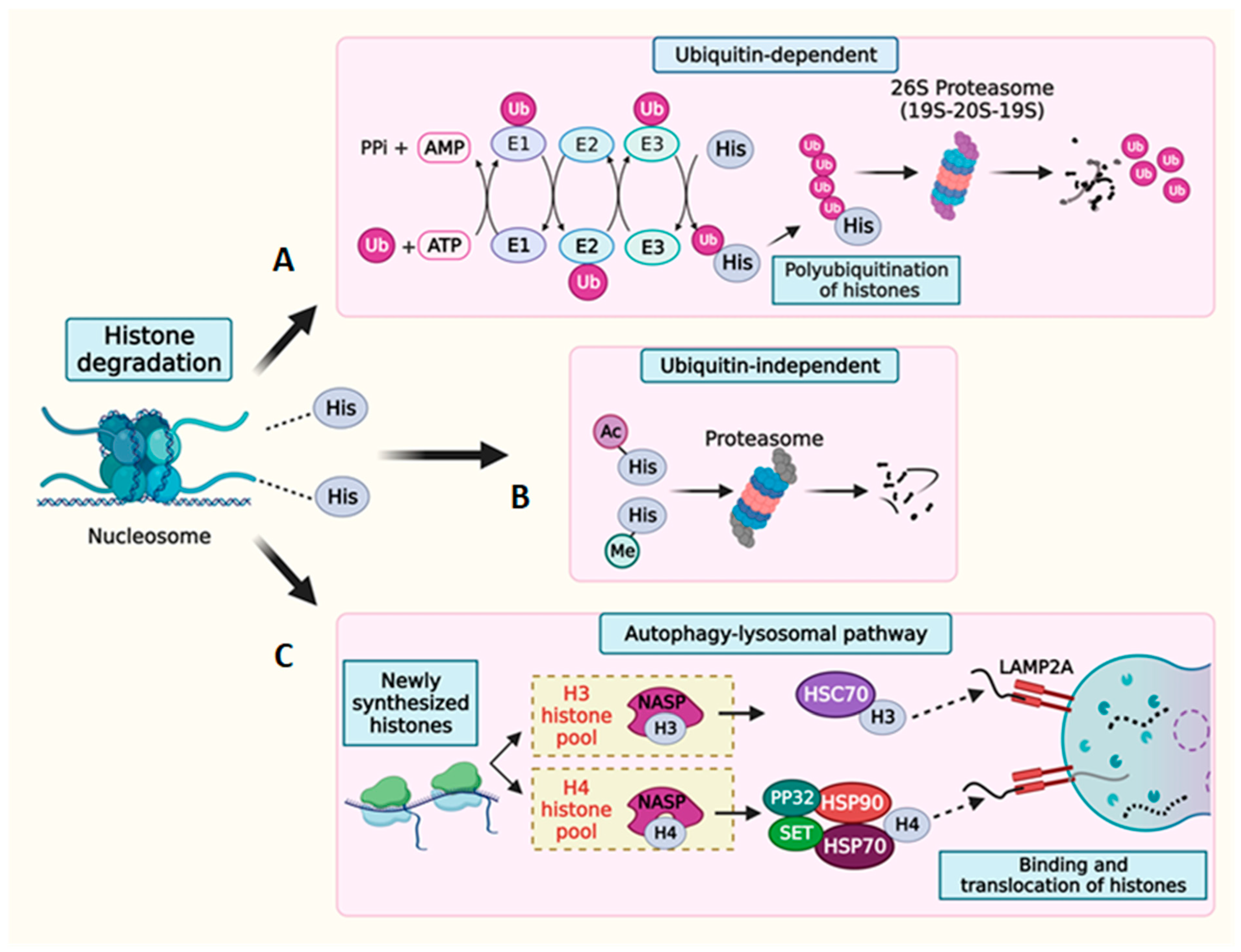
7. Multilevel Regulation of Histone Degradation
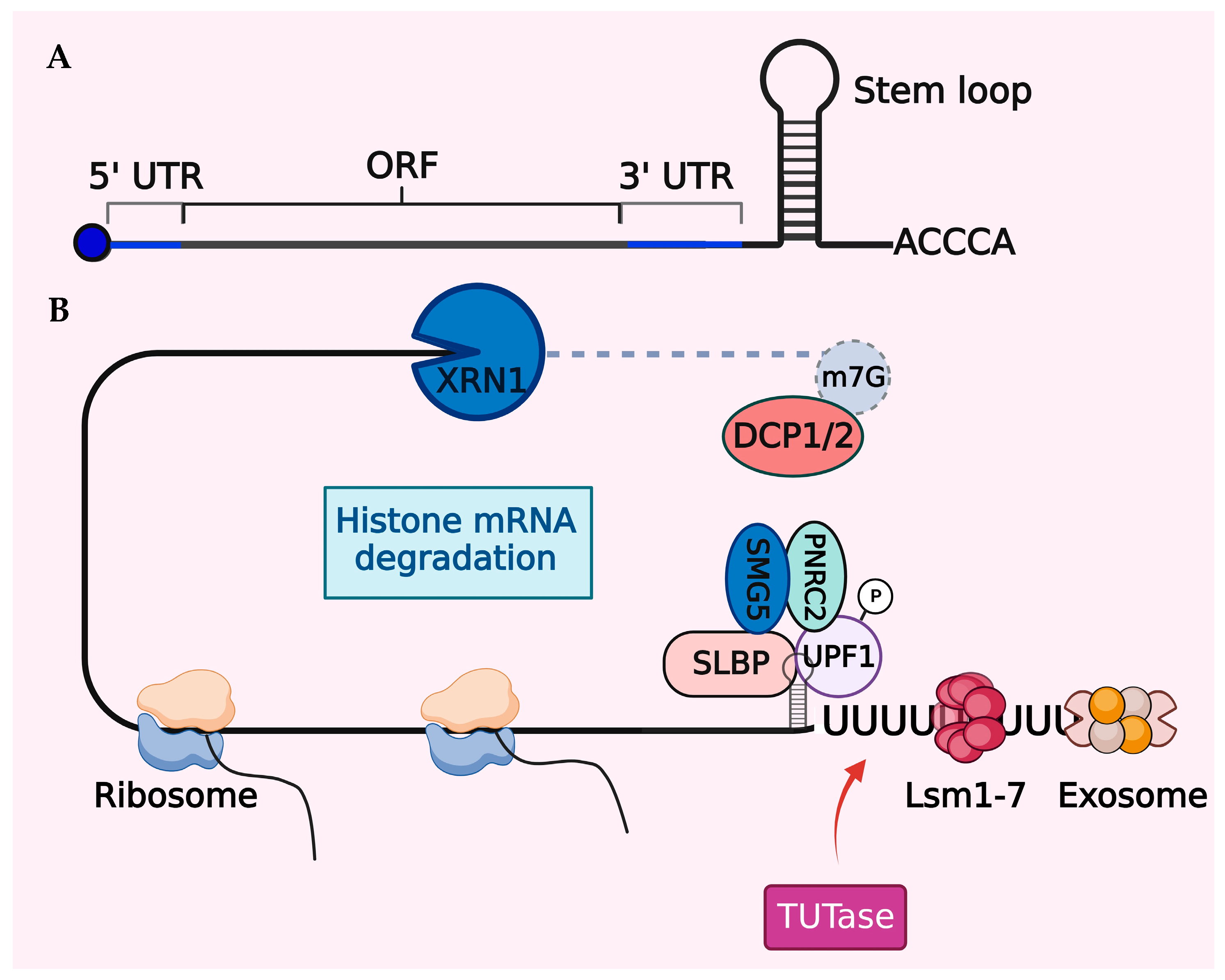
7.1. Histone mRNA Degradation
7.2. Histone Protein Degradation
8. Histone Complementation in Cells Experiencing Histone Loss
9. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- United Nations. World Population Ageing 2020 Highlights; United Nations Department of Economic and Social Affairs: New York, NY, USA, 2020. [Google Scholar]
- Jaul, E.; Barron, J. Age-Related Diseases and Clinical and Public Health Implications for the 85 Years Old and Over Population. Front. Public Health 2017, 5, 335. [Google Scholar] [CrossRef]
- Swenor, B.K.; Ehrlich, J.R. Ageing and vision loss: Looking to the future. Lancet Glob. Health 2021, 9, e385–e386. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodero, S.; Fernandez-Morera, J.L.; Menendez-Torre, E.; Calvanese, V.; Fernandez, A.F.; Fraga, M.F. Aging genetics and aging. Aging Dis. 2011, 2, 186–195. [Google Scholar] [PubMed]
- Wheeler, H.E.; Kim, S.K. Genetics and genomics of human ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- van der Rijt, S.; Molenaars, M.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Integrating the Hallmarks of Aging Throughout the Tree of Life: A Focus on Mitochondrial Dysfunction. Front. Cell Dev. Biol. 2020, 8, 594416. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Ahuir, A.; Manzanares-Estreder, S.; Proft, M. Pro- and Antioxidant Functions of the Peroxisome-Mitochondria Connection and Its Impact on Aging and Disease. Oxid. Med. Cell Longev. 2017, 2017, 9860841. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O.; Van Veldhoven, P.P. Aging, age-related diseases and peroxisomes. Subcell. Biochem. 2013, 69, 45–65. [Google Scholar] [CrossRef]
- Terlecky, S.R.; Koepke, J.I.; Walton, P.A. Peroxisomes and aging. Biochim. Biophys. Acta 2006, 1763, 1749–1754. [Google Scholar] [CrossRef]
- Vigne, S.; Pot, C. Implication of Oxysterols and Phytosterols in Aging and Human Diseases. Adv. Exp. Med. Biol. 2024, 1440, 231–260. [Google Scholar] [CrossRef]
- Zarrouk, A.; Vejux, A.; Mackrill, J.; O’Callaghan, Y.; Hammami, M.; O’Brien, N.; Lizard, G. Involvement of oxysterols in age-related diseases and ageing processes. Ageing Res. Rev. 2014, 18, 148–162. [Google Scholar] [CrossRef]
- Spinedi, M.; Clark, C.; Zullo, L.; Kerksiek, A.; Pistis, G.; Castelao, E.; von Gunten, A.; Preisig, M.; Lütjohann, D.; Popp, J. Cholesterol-metabolism, plant sterols, and long-term cognitive decline in older people—Effects of sex and APOEe4. iScience 2024, 27, 109013. [Google Scholar] [CrossRef]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264. [Google Scholar] [CrossRef]
- Mogilenko, D.A.; Shpynov, O.; Andhey, P.S.; Arthur, L.; Swain, A.; Esaulova, E.; Brioschi, S.; Shchukina, I.; Kerndl, M.; Bambouskova, M.; et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK+ CD8+ T Cells as Conserved Hallmark of Inflammaging. Immunity 2021, 54, 99–115.e112. [Google Scholar] [CrossRef] [PubMed]
- Bosco, N.; Noti, M. The aging gut microbiome and its impact on host immunity. Genes. Immun. 2021, 22, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Szilard, L. On the Nature of the Aging Process. Proc. Natl. Acad. Sci. USA 1959, 45, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Failla, G. The aging process and cancerogenesis. Ann. N. Y. Acad. Sci. 1958, 71, 1124–1140. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Charlesworth, B. A genetic analysis of senescence in Drosophila. Nature 1994, 367, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L. Evolutionary theories of ageing applied to long-lived organisms. Exp. Gerontol. 2001, 36, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef]
- Yang, J.H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e327. [Google Scholar] [CrossRef]
- De Majo, F.; Martens, L.; Hegenbarth, J.C.; Ruhle, F.; Hamczyk, M.R.; Nevado, R.M.; Andres, V.; Hilbold, E.; Bar, C.; Thum, T.; et al. Genomic instability in the naturally and prematurely aged myocardium. Proc. Natl. Acad. Sci. USA 2021, 118, e2022974118. [Google Scholar] [CrossRef]
- Kaya, A.; Lobanov, A.V.; Gladyshev, V.N. Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae. Aging Cell 2015, 14, 366–371. [Google Scholar] [CrossRef]
- Narayanan, L.; Fritzell, J.A.; Baker, S.M.; Liskay, R.M.; Glazer, P.M. Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2. Proc. Natl. Acad. Sci. USA 1997, 94, 3122–3127. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Gotta, M.; Sinclair, D.A.; Mills, K.; McNabb, D.S.; Murthy, M.; Pak, S.M.; Laroche, T.; Gasser, S.M.; Guarente, L. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 1997, 89, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.A.; Mills, K.; Guarente, L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science 1997, 277, 1313–1316. [Google Scholar] [CrossRef]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 2019, 26, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.K.M.; Pugh, B.F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 2017, 18, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Gurard-Levin, Z.A.; Quivy, J.P.; Almouzni, G. Histone chaperones: Assisting histone traffic and nucleosome dynamics. Annu. Rev. Biochem. 2014, 83, 487–517. [Google Scholar] [CrossRef]
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9. [Google Scholar] [CrossRef]
- Allahverdi, A.; Yang, R.; Korolev, N.; Fan, Y.; Davey, C.A.; Liu, C.F.; Nordenskiold, L. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 2011, 39, 1680–1691. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Swygert, S.G.; Peterson, C.L. Chromatin dynamics: Interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta 2014, 1839, 728–736. [Google Scholar] [CrossRef]
- McCauley, B.S.; Dang, W. Histone methylation and aging: Lessons learned from model systems. Biochim. Biophys. Acta 2014, 1839, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, Q.; Xie, L. Histone Modifications in Aging: The Underlying Mechanisms and Implications. Curr. Stem Cell Res. Ther. 2018, 13, 125–135. [Google Scholar] [CrossRef]
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Bainor, A.J.; David, G. Chapter 7—The Dynamics of Histone Modifications During Aging. In Epigenomics in Health and Disease; Fraga, M.F., Fernández, A.F., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 145–162. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Fan, X.G.; Tang, D. Release and activity of histone in diseases. Cell Death Dis. 2014, 5, e1370. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Chang, M.; Kim, U.J.; Grunstein, M. Histone H2B repression causes cell-cycle-specific arrest in yeast: Effects on chromosomal segregation, replication, and transcription. Cell 1987, 48, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.J.; Han, M.; Kayne, P.; Grunstein, M. Effects of histone H4 depletion on the cell cycle and transcription of Saccharomyces cerevisiae. EMBO J. 1988, 7, 2211–2219. [Google Scholar] [CrossRef]
- Gossett, A.J.; Lieb, J.D. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet. 2012, 8, e1002771. [Google Scholar] [CrossRef]
- Wyrick, J.J.; Holstege, F.C.; Jennings, E.G.; Causton, H.C.; Shore, D.; Grunstein, M.; Lander, E.S.; Young, R.A. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 1999, 402, 418–421. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes. Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef]
- Ni, Z.; Ebata, A.; Alipanahiramandi, E.; Lee, S.S. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 2012, 11, 315–325. [Google Scholar] [CrossRef]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef]
- Celona, B.; Weiner, A.; Di Felice, F.; Mancuso, F.M.; Cesarini, E.; Rossi, R.L.; Gregory, L.; Baban, D.; Rossetti, G.; Grianti, P.; et al. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 2011, 9, e1001086. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.M.; Ryvkin, P.; Wanat, J.J.; Donahue, G.; Ricketts, M.D.; Barrett, S.P.; Waters, H.J.; Song, S.; Chavez, A.; Abdallah, K.O.; et al. Rap1 relocalization contributes to the chromatin-mediated gene expression profile and pace of cell senescence. Genes Dev. 2013, 27, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Janssens, G.E.; Veenhoff, L.M. Evidence for the hallmarks of human aging in replicatively aging yeast. Microb. Cell 2016, 3, 263–274. [Google Scholar] [CrossRef]
- Denoth Lippuner, A.; Julou, T.; Barral, Y. Budding yeast as a model organism to study the effects of age. FEMS Microbiol. Rev. 2014, 38, 300–325. [Google Scholar] [CrossRef]
- Mitsui, Y.; Sakagami, H.; Murota, S.; Yamada, M. Age-related decline in histone H1 fraction in human diploid fibroblast cultures. Exp. Cell Res. 1980, 126, 289–298. [Google Scholar] [CrossRef]
- Houde, M.; Shmookler Reis, R.J.; Goldstein, S. Proportions of H1 histone subspecies in human fibroblasts shift during density-dependent growth arrest independent of replicative senescence. Exp. Cell Res. 1989, 184, 256–261. [Google Scholar] [CrossRef]
- Ishimi, Y.; Kojima, M.; Takeuchi, F.; Miyamoto, T.; Yamada, M.; Hanaoka, F. Changes in chromatin structure during aging of human skin fibroblasts. Exp. Cell Res. 1987, 169, 458–467. [Google Scholar] [CrossRef]
- Macieira-Coelho, A. Chromatin reorganization during senescence of proliferating cells. Mutat. Res. 1991, 256, 81–104. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef]
- Kim, C.; Jin, J.; Ye, Z.; Jadhav, R.R.; Gustafson, C.E.; Hu, B.; Cao, W.; Tian, L.; Weyand, C.M.; Goronzy, J.J. Histone deficiency and accelerated replication stress in T cell aging. J. Clin. Investig. 2021, 131, e143632. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Dubey, R.; Jung, K.S.; Prajapati, S.; Tian, W.; Kleinman, M.E. Age-related global histone loss and altered histone acetylation in retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2023, 64, 4444. [Google Scholar]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: The regulation and intervention. Signal Transduct. Target. Ther. 2021, 6, 245. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence-Current Concepts and Open Lessons. Cells 2019, 8, 1446. [Google Scholar] [CrossRef] [PubMed]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Schwartz, S.; Kirkland, J.L.; Oshima, J. Key elements of cellular senescence involve transcriptional repression of mitotic and DNA repair genes through the p53-p16/RB-E2F-DREAM complex. Aging 2023, 15, 4012–4034. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Domen, A.; Deben, C.; Verswyvel, J.; Flieswasser, T.; Prenen, H.; Peeters, M.; Lardon, F.; Wouters, A. Cellular senescence in cancer: Clinical detection and prognostic implications. J. Exp. Clin. Cancer Res. 2022, 41, 360. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Amatori, S.; Tavolaro, S.; Gambardella, S.; Fanelli, M. The dark side of histones: Genomic organization and role of oncohistones in cancer. Clin. Epigenetics 2021, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Gautrey, H.E.; van Otterdijk, S.D.; Cordell, H.J.; Newcastle 85+ Study Core, T.; Mathers, J.C.; Strathdee, G. DNA methylation abnormalities at gene promoters are extensive and variable in the elderly and phenocopy cancer cells. FASEB J. 2014, 28, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.; Moore, I.K.; Fondufe-Mittendorf, Y.; Gossett, A.J.; Tillo, D.; Field, Y.; LeProust, E.M.; Hughes, T.R.; Lieb, J.D.; Widom, J.; et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009, 458, 362–366. [Google Scholar] [CrossRef]
- Bochkis, I.M.; Przybylski, D.; Chen, J.; Regev, A. Changes in nucleosome occupancy associated with metabolic alterations in aged mammalian liver. Cell Rep. 2014, 9, 996–1006. [Google Scholar] [CrossRef]
- Wheaton, K.; Riabowol, K. Protein kinase C delta blocks immediate-early gene expression in senescent cells by inactivating serum response factor. Mol. Cell Biol. 2004, 24, 7298–7311. [Google Scholar] [CrossRef]
- Ding, W.; Gao, S.; Scott, R.E. Senescence represses the nuclear localization of the serum response factor and differentiation regulates its nuclear localization with lineage specificity. J. Cell Sci. 2001, 114, 1011–1018. [Google Scholar] [CrossRef]
- Chen, Y.; Bravo, J.I.; Son, J.M.; Lee, C.; Benayoun, B.A. Remodeling of the H3 nucleosomal landscape during mouse aging. Transl. Med. Aging 2020, 4, 22–31. [Google Scholar] [CrossRef]
- Funayama, R.; Saito, M.; Tanobe, H.; Ishikawa, F. Loss of linker histone H1 in cellular senescence. J. Cell Biol. 2006, 175, 869–880. [Google Scholar] [CrossRef]
- Ucar, D.; Marquez, E.J.; Chung, C.H.; Marches, R.; Rossi, R.J.; Uyar, A.; Wu, T.C.; George, J.; Stitzel, M.L.; Palucka, A.K.; et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J. Exp. Med. 2017, 214, 3123–3144. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, H.; Liu, J.; Hu, Q.; Zhang, S.; Zhang, N.; Liu, L.; Dai, Y.; Cao, D.; Li, X.; et al. Arginine hypomethylation-mediated proteasomal degradation of histone H4-an early biomarker of cellular senescence. Cell Death Differ. 2020, 27, 2697–2709. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Seeber, A.; Singh, V.; Thierry, R.; Sack, R.; Amitai, A.; Kryzhanovska, M.; Eglinger, J.; Holcman, D.; Owen-Hughes, T.; et al. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat. Struct. Mol. Biol. 2017, 24, 99–107. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Bar, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5, F1000 Faculty Rev-89. [Google Scholar] [CrossRef]
- Gottschling, D.E.; Aparicio, O.M.; Billington, B.L.; Zakian, V.A. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell 1990, 63, 751–762. [Google Scholar] [CrossRef]
- Andrulis, E.D.; Neiman, A.M.; Zappulla, D.C.; Sternglanz, R. Perinuclear localization of chromatin facilitates transcriptional silencing. Nature 1998, 394, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Galy, V.; Olivo-Marin, J.C.; Scherthan, H.; Doye, V.; Rascalou, N.; Nehrbass, U. Nuclear pore complexes in the organization of silent telomeric chromatin. Nature 2000, 403, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Weierich, C.; Brero, A.; Stein, S.; von Hase, J.; Cremer, C.; Cremer, T.; Solovei, I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res. 2003, 11, 485–502. [Google Scholar] [CrossRef]
- Raz, V.; Vermolen, B.J.; Garini, Y.; Onderwater, J.J.; Mommaas-Kienhuis, M.A.; Koster, A.J.; Young, I.T.; Tanke, H.; Dirks, R.W. The nuclear lamina promotes telomere aggregation and centromere peripheral localization during senescence of human mesenchymal stem cells. J. Cell Sci. 2008, 121, 4018–4028. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nunez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Kreiling, J.A.; Tamamori-Adachi, M.; Sexton, A.N.; Jeyapalan, J.C.; Munoz-Najar, U.; Peterson, A.L.; Manivannan, J.; Rogers, E.S.; Pchelintsev, N.A.; Adams, P.D.; et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 2011, 10, 292–304. [Google Scholar] [CrossRef]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef]
- Kozak, M.L.; Chavez, A.; Dang, W.; Berger, S.L.; Ashok, A.; Guo, X.; Johnson, F.B. Inactivation of the Sas2 histone acetyltransferase delays senescence driven by telomere dysfunction. EMBO J. 2010, 29, 158–170. [Google Scholar] [CrossRef]
- Smeal, T.; Claus, J.; Kennedy, B.; Cole, F.; Guarente, L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 1996, 84, 633–642. [Google Scholar] [CrossRef]
- Harley, C.B. Telomere loss: Mitotic clock or genetic time bomb? Mutat. Res. 1991, 256, 271–282. [Google Scholar] [CrossRef]
- Munro, J.; Steeghs, K.; Morrison, V.; Ireland, H.; Parkinson, E.K. Human fibroblast replicative senescence can occur in the absence of extensive cell division and short telomeres. Oncogene 2001, 20, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Marthandan, S.; Priebe, S.; Hemmerich, P.; Klement, K.; Diekmann, S. Long-term quiescent fibroblast cells transit into senescence. PLoS ONE 2014, 9, e115597. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Victor, P.; Gutarra, S.; Garcia-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardi, M.; Ballestar, E.; Gonzalez, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Tumpel, S.; Rudolph, K.L. Quiescence: Good and Bad of Stem Cell Aging. Trends Cell Biol. 2019, 29, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Wang, T.; Lin, X.W.; Denlinger, D.L.; Xu, W.H. Reactive oxygen species extend insect life span using components of the insulin-signaling pathway. Proc. Natl. Acad. Sci. USA 2017, 114, E7832–E7840. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Lee, E.; Lee, J.; Oh, S.; Kim, S. Down-regulation of asymmetric arginine methylation during replicative and H2O2-induced premature senescence in WI-38 human diploid fibroblasts. J. Biochem. 2008, 144, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.X.; Ma, S.; Han, X.; Luo, Z.Y.; Zhu, Q.Q.; Chiba, T.; Xie, W.; Lin, K.; Qiu, X.B. Proteasome activator PA200 maintains stability of histone marks during transcription and aging. Theranostics 2021, 11, 1458–1472. [Google Scholar] [CrossRef]
- Yi, S.J.; Kim, K. New Insights into the Role of Histone Changes in Aging. Int. J. Mol. Sci. 2020, 21, 8241. [Google Scholar] [CrossRef]
- Maze, I.; Noh, K.M.; Soshnev, A.A.; Allis, C.D. Every amino acid matters: Essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014, 15, 259–271. [Google Scholar] [CrossRef]
- Akiyama, T.; Suzuki, O.; Matsuda, J.; Aoki, F. Dynamic replacement of histone H3 variants reprograms epigenetic marks in early mouse embryos. PLoS Genet. 2011, 7, e1002279. [Google Scholar] [CrossRef] [PubMed]
- Santenard, A.; Ziegler-Birling, C.; Koch, M.; Tora, L.; Bannister, A.J.; Torres-Padilla, M.E. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat. Cell Biol. 2010, 12, 853–862. [Google Scholar] [CrossRef]
- Pina, B.; Suau, P. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev. Biol. 1987, 123, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.W.; Shibata, Y.; Starmer, J.; Yee, D.; Magnuson, T. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 2015, 29, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Nashun, B.; Hill, P.W.; Smallwood, S.A.; Dharmalingam, G.; Amouroux, R.; Clark, S.J.; Sharma, V.; Ndjetehe, E.; Pelczar, P.; Festenstein, R.J.; et al. Continuous Histone Replacement by Hira Is Essential for Normal Transcriptional Regulation and De Novo DNA Methylation during Mouse Oogenesis. Mol. Cell 2015, 60, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Szenker, E.; Lacoste, N.; Almouzni, G. A developmental requirement for HIRA-dependent H3.3 deposition revealed at gastrulation in Xenopus. Cell Rep. 2012, 1, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, M.D.; Frederick, B.; Hoff, H.; Tang, Y.; Schultz, D.C.; Singh Rai, T.; Grazia Vizioli, M.; Adams, P.D.; Marmorstein, R. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat. Commun. 2015, 6, 7711. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef]
- Elsaesser, S.J.; Allis, C.D. HIRA and Daxx constitute two independent histone H3.3-containing predeposition complexes. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ray-Gallet, D.; Woolfe, A.; Vassias, I.; Pellentz, C.; Lacoste, N.; Puri, A.; Schultz, D.C.; Pchelintsev, N.A.; Adams, P.D.; Jansen, L.E.; et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 2011, 44, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Wenderski, W.; Noh, K.M.; Bagot, R.C.; Tzavaras, N.; Purushothaman, I.; Elsasser, S.J.; Guo, Y.; Ionete, C.; Hurd, Y.L.; et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron 2015, 87, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Tvardovskiy, A.; Schwammle, V.; Kempf, S.J.; Rogowska-Wrzesinska, A.; Jensen, O.N. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 2017, 45, 9272–9289. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Occean, J.R.; Melters, D.P.; Shi, C.; Wang, L.; Stransky, S.; Doyle, M.E.; Cui, C.Y.; Delannoy, M.; Fan, J.; et al. A hyper-quiescent chromatin state formed during aging is reversed by regeneration. Mol. Cell 2023, 83, 1659–1676. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Young, A.R.; Wang, Z.; Wu, H.A.; Panda, T.; Kou, Y.; Kapoor, A.; Hasson, D.; Mills, N.R.; Ma’ayan, A.; et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014, 5, 5210. [Google Scholar] [CrossRef]
- Oberdoerffer, P.; Miller, K.M. Histone H2A variants: Diversifying chromatin to ensure genome integrity. Semin. Cell Dev. Biol. 2023, 135, 59–72. [Google Scholar] [CrossRef]
- Stefanelli, G.; Azam, A.B.; Walters, B.J.; Brimble, M.A.; Gettens, C.P.; Bouchard-Cannon, P.; Cheng, H.M.; Davidoff, A.M.; Narkaj, K.; Day, J.J.; et al. Learning and Age-Related Changes in Genome-wide H2A.Z Binding in the Mouse Hippocampus. Cell Rep. 2018, 22, 1124–1131. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- Contrepois, K.; Mann, C.; Fenaille, F. H2B Type 1-K Accumulates in Senescent Fibroblasts with Persistent DNA Damage along with Methylated and Phosphorylated Forms of HMGA1. Proteomes 2021, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Isermann, A.; Mann, C.; Rube, C.E. Histone Variant H2A.J Marks Persistent DNA Damage and Triggers the Secretory Phenotype in Radiation-Induced Senescence. Int. J. Mol. Sci. 2020, 21, 9130. [Google Scholar] [CrossRef] [PubMed]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [PubMed]
- Rube, C.E.; Baumert, C.; Schuler, N.; Isermann, A.; Schmal, Z.; Glanemann, M.; Mann, C.; Scherthan, H. Human skin aging is associated with increased expression of the histone variant H2A.J in the epidermis. NPJ Aging Mech. Dis. 2021, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Happel, N.; Doenecke, D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene 2009, 431, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ponte, I.; Romero, D.; Yero, D.; Suau, P.; Roque, A. Complex Evolutionary History of the Mammalian Histone H1.1-H1.5 Gene Family. Mol. Biol. Evol. 2017, 34, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Sekeri-Pataryas, K.E.; Sourlingas, T.G. The differentiation-associated linker histone, H1.0, during the in vitro aging and senescence of human diploid fibroblasts. Ann. N. Y. Acad. Sci. 2007, 1100, 361–367. [Google Scholar] [CrossRef]
- Nishibuchi, I.; Suzuki, H.; Kinomura, A.; Sun, J.; Liu, N.A.; Horikoshi, Y.; Shima, H.; Kusakabe, M.; Harata, M.; Fukagawa, T.; et al. Reorganization of damaged chromatin by the exchange of histone variant H2A.Z-2. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 736–744. [Google Scholar] [CrossRef]
- Faast, R.; Thonglairoam, V.; Schulz, T.C.; Beall, J.; Wells, J.R.; Taylor, H.; Matthaei, K.; Rathjen, P.D.; Tremethick, D.J.; Lyons, I. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 2001, 11, 1183–1187. [Google Scholar] [CrossRef]
- van Daal, A.; Elgin, S.C. A histone variant, H2AvD, is essential in Drosophila melanogaster. Mol. Biol. Cell 1992, 3, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.D.; Gorovsky, M.A. Histone H2A.Z has a conserved function that is distinct from that of the major H2A sequence variants. Nucleic Acids Res. 2000, 28, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, B.; Gorovsky, M.A. Essential and nonessential histone H2A variants in Tetrahymena thermophila. Mol. Cell Biol. 1996, 16, 4305–4311. [Google Scholar] [CrossRef] [PubMed]
- Rai, T.S.; Cole, J.J.; Nelson, D.M.; Dikovskaya, D.; Faller, W.J.; Vizioli, M.G.; Hewitt, R.N.; Anannya, O.; McBryan, T.; Manoharan, I.; et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014, 28, 2712–2725. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone variants--ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Bonisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef]
- Seal, R.L.; Denny, P.; Bruford, E.A.; Gribkova, A.K.; Landsman, D.; Marzluff, W.F.; McAndrews, M.; Panchenko, A.R.; Shaytan, A.K.; Talbert, P.B. A standardized nomenclature for mammalian histone genes. Epigenetics Chromatin 2022, 15, 34. [Google Scholar] [CrossRef]
- Osley, M.A. The regulation of histone synthesis in the cell cycle. Annu. Rev. Biochem. 1991, 60, 827–861. [Google Scholar] [CrossRef]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef]
- Gunesdogan, U.; Jackle, H.; Herzig, A. Histone supply regulates S phase timing and cell cycle progression. eLife 2014, 3, e02443. [Google Scholar] [CrossRef]
- Armstrong, C.; Spencer, S.L. Replication-dependent histone biosynthesis is coupled to cell-cycle commitment. Proc. Natl. Acad. Sci. USA 2021, 118, e2100178118. [Google Scholar] [CrossRef] [PubMed]
- Hogan, A.K.; Foltz, D.R. Reduce, Retain, Recycle: Mechanisms for Promoting Histone Protein Degradation versus Stability and Retention. Mol. Cell Biol. 2021, 41, e0000721. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Huang, J.; Chen, W.; Tang, J.; Xu, C.; Yu, Q.; Cheng, Y.; Ma, L.; Yu, X.; Li, S. Regulation of DNA replication-coupled histone gene expression. Oncotarget 2017, 8, 95005–95022. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Koreski, K.P. Birth and Death of Histone mRNAs. Trends Genet. 2017, 33, 745–759. [Google Scholar] [CrossRef]
- Ewen, M.E. Where the cell cycle and histones meet. Genes Dev. 2000, 14, 2265–2270. [Google Scholar] [CrossRef]
- Armstrong, C.; Passanisi, V.J.; Ashraf, H.M.; Spencer, S.L. Cyclin E/CDK2 and feedback from soluble histone protein regulate the S phase burst of histone biosynthesis. Cell Rep. 2023, 42, 112768. [Google Scholar] [CrossRef]
- Harris, M.E.; Bohni, R.; Schneiderman, M.H.; Ramamurthy, L.; Schumperli, D.; Marzluff, W.F. Regulation of histone mRNA in the unperturbed cell cycle: Evidence suggesting control at two posttranscriptional steps. Mol. Cell Biol. 1991, 11, 2416–2424. [Google Scholar] [CrossRef]
- Dominski, Z.; Zheng, L.X.; Sanchez, R.; Marzluff, W.F. Stem-loop binding protein facilitates 3′-end formation by stabilizing U7 snRNP binding to histone pre-mRNA. Mol. Cell Biol. 1999, 19, 3561–3570. [Google Scholar] [CrossRef]
- Sanchez, R.; Marzluff, W.F. The stem-loop binding protein is required for efficient translation of histone mRNA in vivo and in vitro. Mol. Cell Biol. 2002, 22, 7093–7104. [Google Scholar] [CrossRef] [PubMed]
- Peltz, S.W.; Ross, J. Autogenous regulation of histone mRNA decay by histone proteins in a cell-free system. Mol. Cell Biol. 1987, 7, 4345–4356. [Google Scholar] [CrossRef] [PubMed]
- Kaygun, H.; Marzluff, W.F. Translation termination is involved in histone mRNA degradation when DNA replication is inhibited. Mol. Cell Biol. 2005, 25, 6879–6888. [Google Scholar] [CrossRef]
- Bao, H.; Carraro, M.; Flury, V.; Liu, Y.; Luo, M.; Chen, L.; Groth, A.; Huang, H. NASP maintains histone H3-H4 homeostasis through two distinct H3 binding modes. Nucleic Acids Res. 2022, 50, 5349–5368. [Google Scholar] [CrossRef]
- Finn, R.M.; Browne, K.; Hodgson, K.C.; Ausio, J. sNASP, a histone H1-specific eukaryotic chaperone dimer that facilitates chromatin assembly. Biophys. J. 2008, 95, 1314–1325. [Google Scholar] [CrossRef]
- Mullen, T.E.; Marzluff, W.F. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes. Dev. 2008, 22, 50–65. [Google Scholar] [CrossRef]
- Meaux, S.A.; Holmquist, C.E.; Marzluff, W.F. Role of oligouridylation in normal metabolism and regulated degradation of mammalian histone mRNAs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20180170. [Google Scholar] [CrossRef]
- Kim, Y.K.; Maquat, L.E. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 2019, 25, 407–422. [Google Scholar] [CrossRef]
- Choe, J.; Ahn, S.H.; Kim, Y.K. The mRNP remodeling mediated by UPF1 promotes rapid degradation of replication-dependent histone mRNA. Nucleic Acids Res. 2014, 42, 9334–9349. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.K.; Meaux, S.; Welch, J.D.; Bigler, R.; Miliani de Marval, P.L.; Su, W.; Rhoads, R.E.; Prins, J.F.; Marzluff, W.F. Deep sequencing shows multiple oligouridylations are required for 3′ to 5′ degradation of histone mRNAs on polyribosomes. Mol. Cell 2014, 53, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Hoefig, K.P.; Rath, N.; Heinz, G.A.; Wolf, C.; Dameris, J.; Schepers, A.; Kremmer, E.; Ansel, K.M.; Heissmeyer, V. Eri1 degrades the stem-loop of oligouridylated histone mRNAs to induce replication-dependent decay. Nat. Struct. Mol. Biol. 2013, 20, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ha, M.; Chang, H.; Kwon, S.C.; Simanshu, D.K.; Patel, D.J.; Kim, V.N. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 2014, 159, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef]
- Commerford, S.L.; Carsten, A.L.; Cronkite, E.P. Histone turnover within nonproliferating cells. Proc. Natl. Acad. Sci. USA 1982, 79, 1163–1165. [Google Scholar] [CrossRef]
- Pina, B.; Martinez, P.; Simon, L.; Suau, P. Differential kinetics of histone H1(0) accumulation in neuronal and glial cells from rat cerebral cortex during postnatal development. Biochem. Biophys. Res. Commun. 1984, 123, 697–702. [Google Scholar] [CrossRef]
- Shmueli, M.D.; Sheban, D.; Eisenberg-Lerner, A.; Merbl, Y. Histone degradation by the proteasome regulates chromatin and cellular plasticity. FEBS J. 2022, 289, 3304–3316. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Orian, A.; Schwartz, A.L. Ubiquitin-mediated proteolysis: Biological regulation via destruction. Bioessays 2000, 22, 442–451. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989, 243, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Arrieta, A.; Vondriska, T.M. Nucleosome proteostasis and histone turnover. Front. Mol. Biosci. 2022, 9, 990006. [Google Scholar] [CrossRef]
- de Poot, S.A.H.; Tian, G.; Finley, D. Meddling with Fate: The Proteasomal Deubiquitinating Enzymes. J. Mol. Biol. 2017, 429, 3525–3545. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.X.; Pang, Y.; Liu, C.H.; Haratake, K.; Du, B.Y.; Ji, D.Y.; Wang, G.F.; Zhu, Q.Q.; Song, W.; Yu, Y.; et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 2013, 153, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Hormazabal, J.; Saavedra, F.; Espinoza-Arratia, C.; Martinez, N.W.; Cruces, T.; Alfaro, I.E.; Loyola, A. Chaperone mediated autophagy contributes to the newly synthesized histones H3 and H4 quality control. Nucleic Acids Res. 2022, 50, 1875–1887. [Google Scholar] [CrossRef] [PubMed]
- Tirgar, R.; Davies, J.P.; Plate, L.; Nordman, J.T. The histone chaperone NASP maintains H3-H4 reservoirs in the early Drosophila embryo. PLoS Genet. 2023, 19, e1010682. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Liang, D.; Gajjalaiahvari, U.R.; Kabbaj, M.H.; Paik, J.; Gunjan, A. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 2010, 9, 4236–4244. [Google Scholar] [CrossRef] [PubMed]
- Maya Miles, D.; Penate, X.; Sanmartin Olmo, T.; Jourquin, F.; Munoz Centeno, M.C.; Mendoza, M.; Simon, M.N.; Chavez, S.; Geli, V. High levels of histones promote whole-genome-duplications and trigger a Swe1(WEE1)-dependent phosphorylation of Cdc28(CDK1). eLife 2018, 7, e35337. [Google Scholar] [CrossRef] [PubMed]
- Au, W.C.; Crisp, M.J.; DeLuca, S.Z.; Rando, O.J.; Basrai, M.A. Altered dosage and mislocalization of histone H3 and Cse4p lead to chromosome loss in Saccharomyces cerevisiae. Genetics 2008, 179, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.G.; Mellone, B.G.; Partridge, J.F.; Richardson, W.; Hamilton, G.L.; Allshire, R.C.; Pidoux, A.L. Plasticity of fission yeast CENP-A chromatin driven by relative levels of histone H3 and H4. PLoS Genet. 2007, 3, e121. [Google Scholar] [CrossRef]
- Yu, R.; Sun, L.; Sun, Y.; Han, X.; Qin, L.; Dang, W. Cellular response to moderate chromatin architectural defects promotes longevity. Sci. Adv. 2019, 5, eaav1165. [Google Scholar] [CrossRef]
- Meeks-Wagner, D.; Hartwell, L.H. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell 1986, 44, 43–52. [Google Scholar] [CrossRef]
- Morillo-Huesca, M.; Maya, D.; Munoz-Centeno, M.C.; Singh, R.K.; Oreal, V.; Reddy, G.U.; Liang, D.; Geli, V.; Gunjan, A.; Chavez, S. FACT prevents the accumulation of free histones evicted from transcribed chromatin and a subsequent cell cycle delay in G1. PLoS Genet. 2010, 6, e1000964. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.M.; Stromme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017, 18, 141–158. [Google Scholar] [CrossRef]
- Ghule, P.N.; Xie, R.L.; Medina, R.; Colby, J.L.; Jones, S.N.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Fidelity of histone gene regulation is obligatory for genome replication and stability. Mol. Cell Biol. 2014, 34, 2650–2659. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; McKillop-Smith, S.; Muller, B. The human histone gene expression regulator HBP/SLBP is required for histone and DNA synthesis, cell cycle progression and cell proliferation in mitotic cells. J. Cell Sci. 2004, 117, 6043–6051. [Google Scholar] [CrossRef] [PubMed]
- Barcaroli, D.; Bongiorno-Borbone, L.; Terrinoni, A.; Hofmann, T.G.; Rossi, M.; Knight, R.A.; Matera, A.G.; Melino, G.; De Laurenzi, V. FLASH is required for histone transcription and S-phase progression. Proc. Natl. Acad. Sci. USA 2006, 103, 14808–14812. [Google Scholar] [CrossRef]
- Yoo, Y.; Park, S.Y.; Jo, E.B.; Choi, M.; Lee, K.W.; Hong, D.; Lee, S.; Lee, C.R.; Lee, Y.; Um, J.Y.; et al. Overexpression of Replication-Dependent Histone Signifies a Subset of Dedifferentiated Liposarcoma with Increased Aggressiveness. Cancers 2021, 13, 3122. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.H. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef]
- Lu, Y.X.; Regan, J.C.; Esser, J.; Drews, L.F.; Weinseis, T.; Stinn, J.; Hahn, O.; Miller, R.A.; Gronke, S.; Partridge, L. A TORC1-histone axis regulates chromatin organisation and non-canonical induction of autophagy to ameliorate ageing. eLife 2021, 10, e62233. [Google Scholar] [CrossRef]
- Mahmassani, Z.S.; McKenzie, A.I.; Petrocelli, J.J.; de Hart, N.M.; Reidy, P.T.; Fix, D.K.; Ferrara, P.J.; Funai, K.; Drummond, M.J. Short-term metformin ingestion by healthy older adults improves myoblast function. Am. J. Physiol. Cell Physiol. 2021, 320, C566–C576. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Aleksic, S.; Berger, D.M.; Sierra, F.; Kuchel, G.A.; Barzilai, N. Geroscience-guided repurposing of FDA-approved drugs to target aging: A proposed process and prioritization. Aging Cell 2022, 21, e13596. [Google Scholar] [CrossRef] [PubMed]
- Elliehausen, C.J.; Anderson, R.M.; Diffee, G.M.; Rhoads, T.W.; Lamming, D.W.; Hornberger, T.A.; Konopka, A.R. Geroprotector drugs and exercise: Friends or foes on healthy longevity? BMC Biol. 2023, 21, 287. [Google Scholar] [CrossRef] [PubMed]
- Juricic, P.; Lu, Y.X.; Leech, T.; Drews, L.F.; Paulitz, J.; Lu, J.; Nespital, T.; Azami, S.; Regan, J.C.; Funk, E.; et al. Long-lasting geroprotection from brief rapamycin treatment in early adulthood by persistently increased intestinal autophagy. Nat. Aging 2022, 2, 824–836. [Google Scholar] [CrossRef]
| Part A | Histone Reduction | Organism | Reason for Histone Alterations | Impact of Histone Loss | Reference |
|---|---|---|---|---|---|
| 1 | H2B | MHY103 | Conditional repression of H2B mRNA synthesis using a yeast strain, with single H2B gene fused with a repressible GAL10 promoter | Cell cycle arrest, disruption of chromatin structure, disruption of nuclear segregation | Han et al., 1987 [52] |
| 2 | H4 | UKY403 | Conditional repression of H4 in yeast strain, with single histone H4 gene under the control of GAL1 promoter | Cell cycle arrest at G2, disruption of chromatin structure and chromosomal segregation | Kim et al., 1988 [53] |
| 3 | H4 | S. cerevisiae UKY403 and MHY308 | Conditional repression of H4 in yeast strain, with single histone H4 gene under the control of GAL1 promoter | Preferential derepression of genes at telomeric heterochromatin | Wyrick et al., 1999 [55] |
| 4 | H4 | Yeast | Replicative aging | Decreased expression of histone deacetylase, Sir2, accompanied by an increase in H4K16 acetylation; transcriptional derepression at specific loci near telomeres | Dang et al., 2009 [58] |
| 5 | H3 and H2B | Yeast | Age-related histone loss | Increased transcription of histone genes with age, but depletion of histone proteins; histone occupancy is reduced by 50–75% in the aged population | Feser et al. 2010 [59] |
| 6 | H2A, H2B, H3, H4 | Yeast nhp6a/b double mutant | Yeast cells with the nhp6a/b double mutation; lack HMG-box proteins Nhp6a and Nhp6b; they demonstrate many senescence-related characteristics including reduced histone content | Does not affect nucleosome spacing, but rather, changes nucleosome occupancy with the loss typically being concentrated in nucleosome-poor regions of the chromatin; global upregulation in transcription due to increased DNA accessibility | Celona et al., 2011 [60] |
| 7 | H3 | YEF473A (WT) and DCB200.1 | Inducing the repression of H3 conditionally in a yeast mutant strain involved deleting one H3-encoding gene, HHT1, and placing HHT2 under the regulation of the GAL1 promoter | Specific sets of nucleosomes within undergo changes in their occupancy, and nucleosomes are typically lost at gene promoters; limited histone availability results in DNA-encoded preferred nucleosome occupancy and chromatin stability | Gossett et al. 2012 [54] |
| 8 | H2A, H2B, H3, H4 | Derivative strains of S. cerevisae BY4741/2 | Yeast mutant strain lacking telomerase in senescence relocalize Rap1 transcription factor to canonical histone promoters and downregulate their expression | Rap1 binds and represses histone genes in senescent cells; Rap1 and nucleosome occupancy exhibit an inverse correlation at many genomic loci | Platt et al., 2013 [61] |
| 9 | H2A, H2B, H3, H4 | Derivative strains of S. cerevisiae S288c (BY4741) | Replicative aging of yeast cells | Nucleosome positioning became less precise; transcriptional activation; derepression of genes that are transcriptionally silent due to loss of histones from the promoters; elevated levels of DNA strand breaks; mitochondrial DNA transfer; genomic instability is attributed to large-scale chromatin rearrangements including translocations and retrotransposition | Hu et al., 2014 [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubey, S.K.; Dubey, R.; Kleinman, M.E. Unraveling Histone Loss in Aging and Senescence. Cells 2024, 13, 320. https://doi.org/10.3390/cells13040320
Dubey SK, Dubey R, Kleinman ME. Unraveling Histone Loss in Aging and Senescence. Cells. 2024; 13(4):320. https://doi.org/10.3390/cells13040320
Chicago/Turabian StyleDubey, Sushil Kumar, Rashmi Dubey, and Mark Ellsworth Kleinman. 2024. "Unraveling Histone Loss in Aging and Senescence" Cells 13, no. 4: 320. https://doi.org/10.3390/cells13040320