
Neural Circuitry Polarization in the Spinal Dorsal Horn (SDH): A Novel Form of Dysregulated Circuitry Plasticity during Pain Pathogenesis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
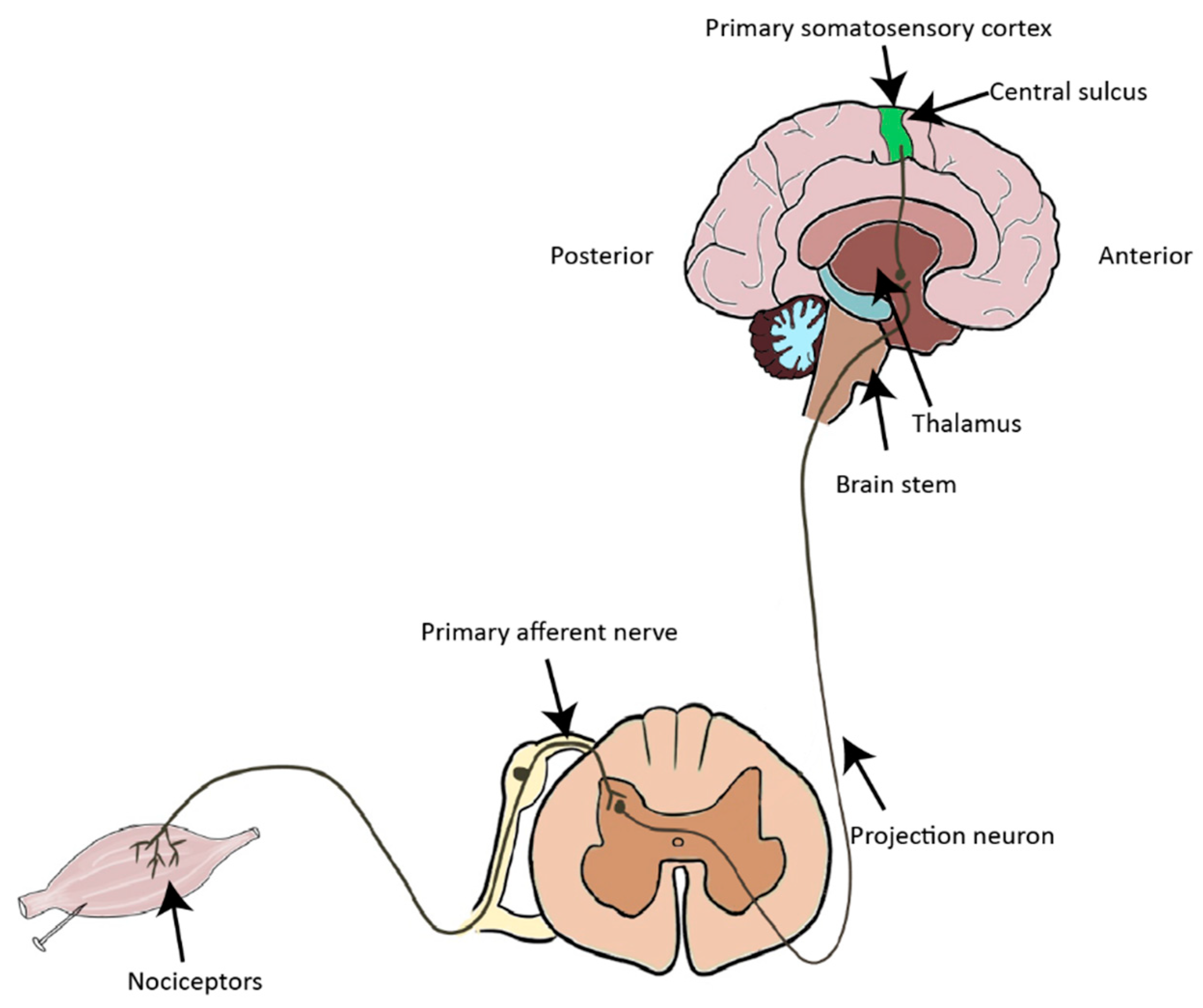
1. Introduction to the Nociceptive Pathway
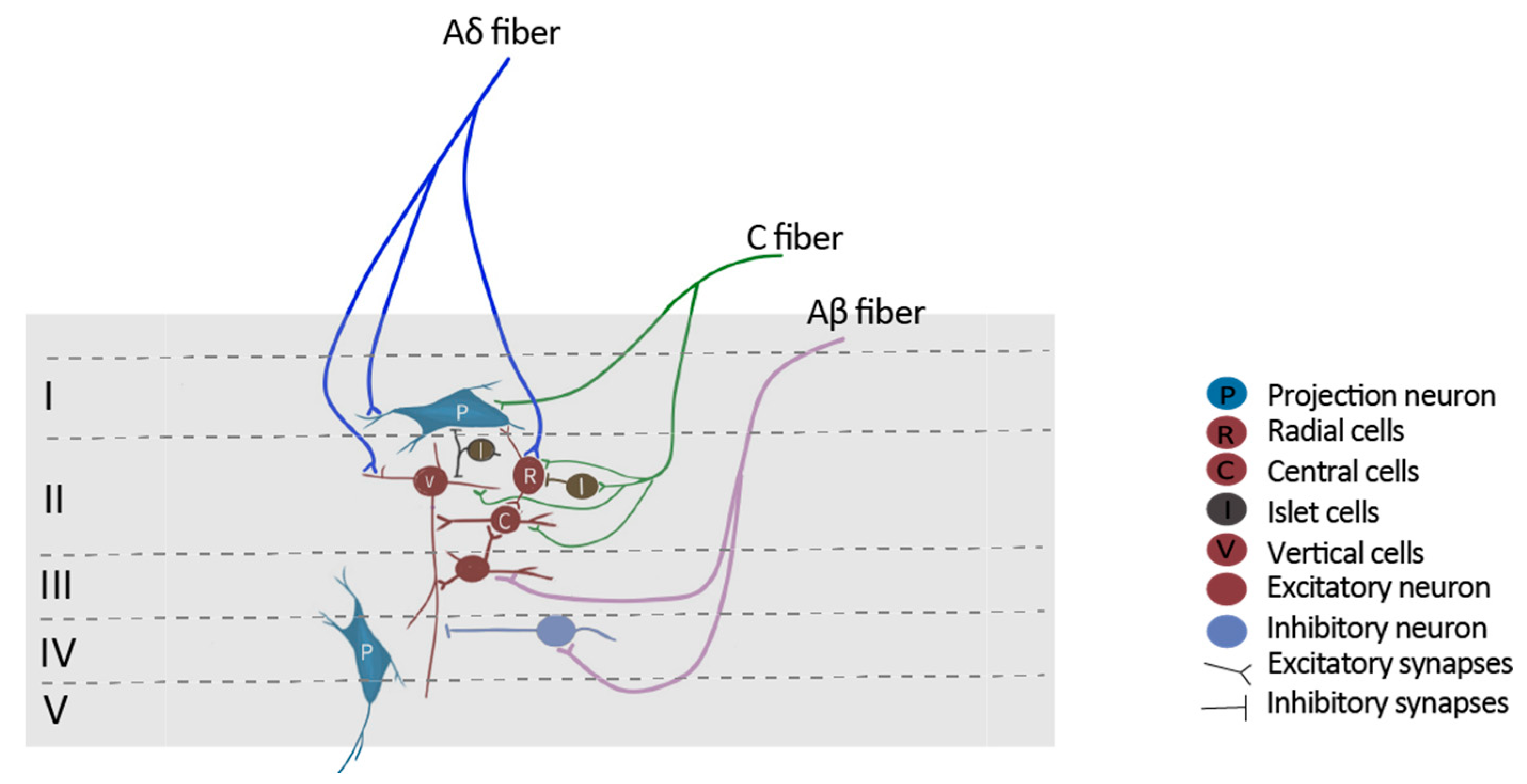
2. Principal Synaptic Receptors in Pain Neural Circuits
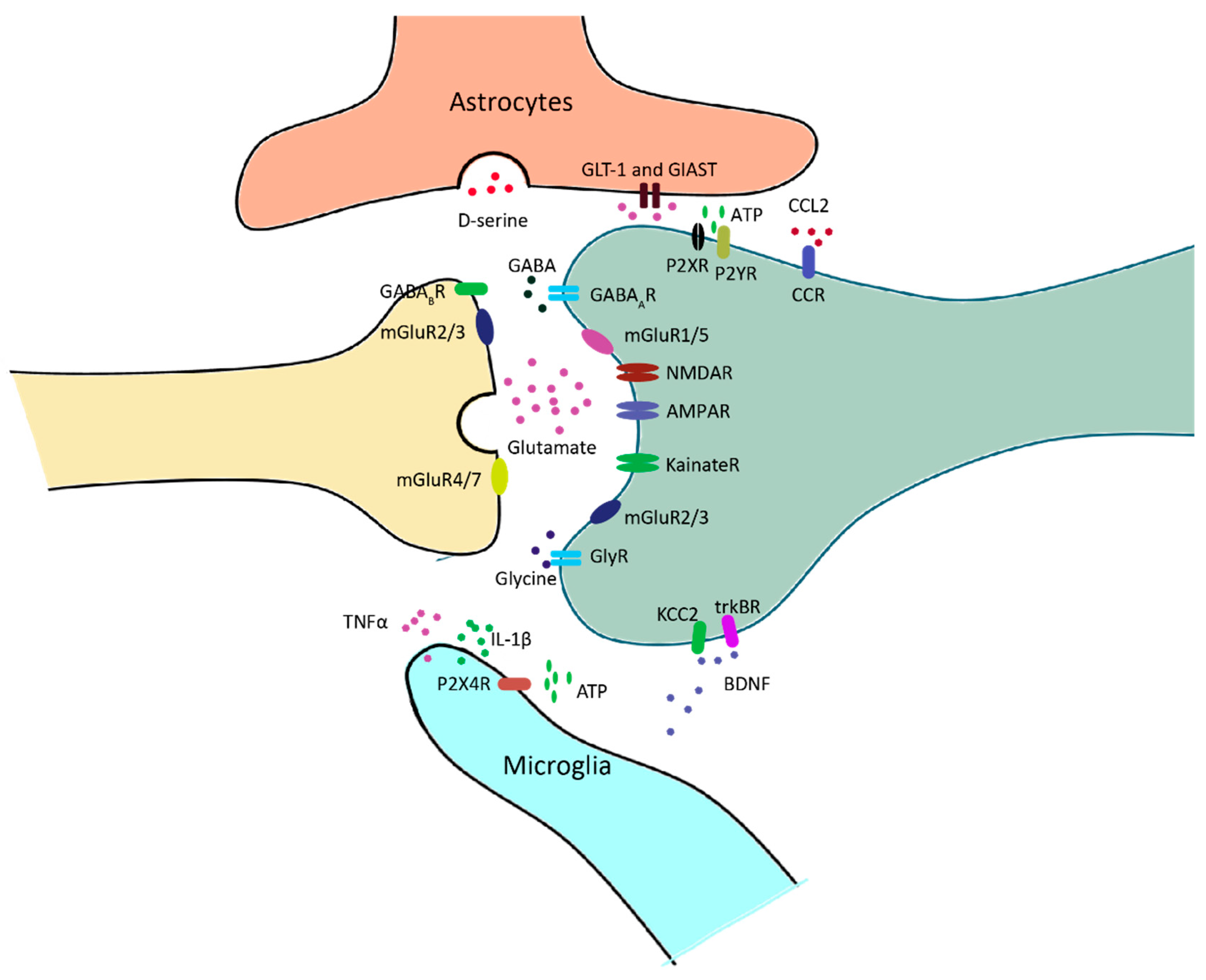
3. Glial Cells in Pain Neural Circuits
4. Neural Circuitry Plasticity in the SDH during Pain Pathogenesis
4.1. Central Sensitization
4.2. Short-Term Synaptic Plasticity
4.3. Long-Term Synaptic Plasticity
4.4. Homeostatic Plasticity/Synaptic Scaling
4.5. Excitation/Inhibition Balance
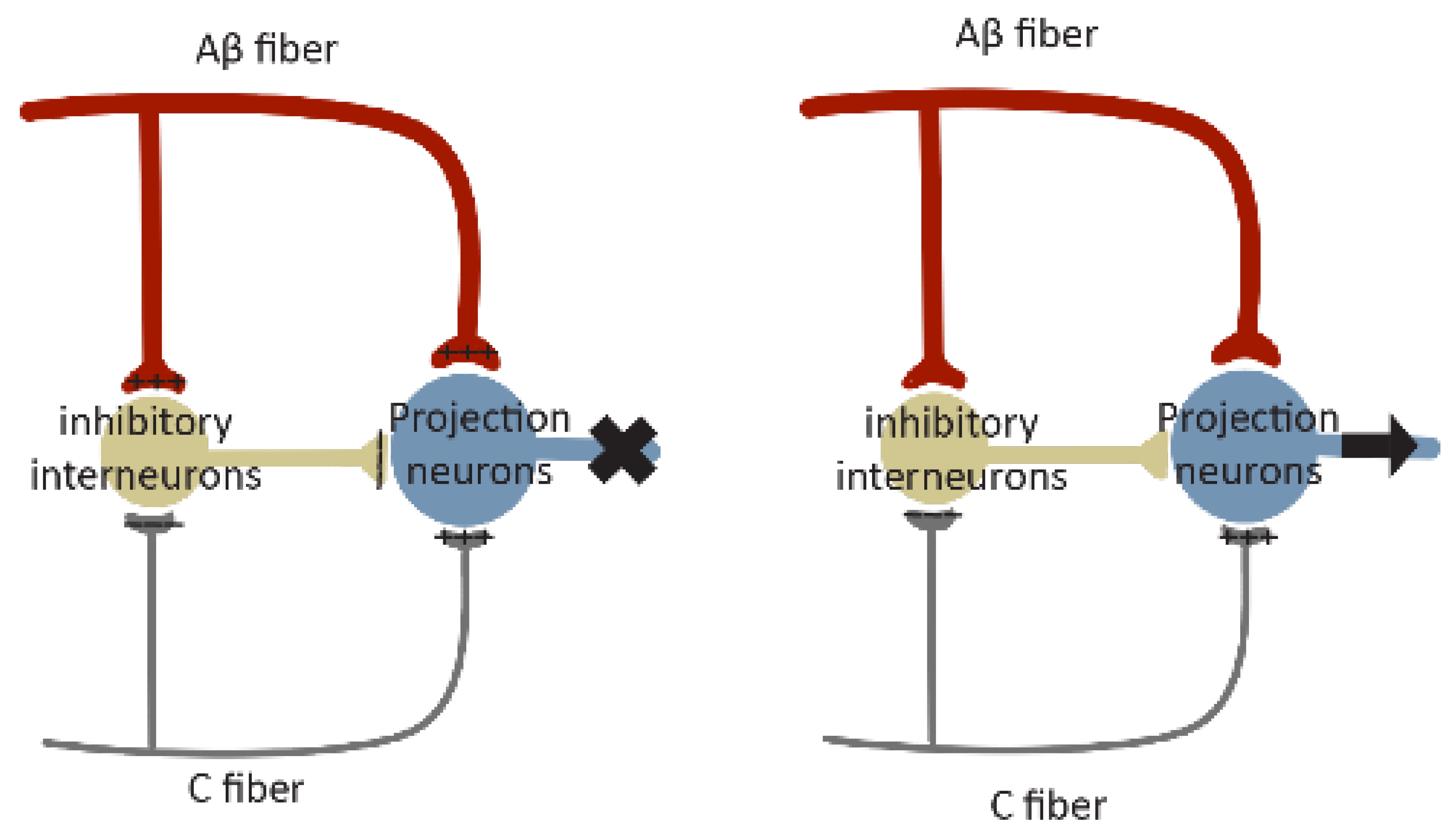
5. Neural Circuit Polarization (NCP): A Novel Form of Neural Circuitry Plasticity in Pain Pathogenesis
6. Comparison of NCP with Other Types of Circuitry Plasticity in Chronic Pain Models
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Belmonte, C.; Viana, F. Molecular and cellular limits to somatosensory specificity. Mol. Pain 2008, 4, 14. [Google Scholar] [CrossRef]
- Pope, J.E.; Deer, T.R.; Kramer, J. A systematic review: Current and future directions of dorsal root ganglion therapeutics to treat chronic pain. Pain Med. 2013, 14, 1477–1496. [Google Scholar] [CrossRef]
- Jessell, T.M.; Kandel, E.R.; Schwartz, J.H. Principles of Neural Science; McGraw-Hill: New York, NY, USA, 2000; Volume 4, pp. 1227–1246. [Google Scholar]
- Todd, A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010, 11, 823–836. [Google Scholar] [CrossRef]
- Todd, A.J.; Puskar, Z.; Spike, R.C.; Hughes, C.; Watt, C.; Forrest, L. Projection neurons in lamina I of rat spinal cord with the neurokinin 1 receptor are selectively innervated by substance p-containing afferents and respond to noxious stimulation. J. Neurosci. 2002, 22, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, P.W.; Lawson, S.N. Cell type and conduction velocity of rat primary sensory neurons with substance P-like immunoreactivity. Neuroscience 1989, 28, 745–753. [Google Scholar] [CrossRef]
- Lawson, S.N.; Perry, M.J.; Prabhakar, E.; McCarthy, P.W. Primary sensory neurones: Neurofilament, neuropeptides, and conduction velocity. Brain. Res. Bull. 1993, 30, 239–243. [Google Scholar] [CrossRef]
- Naim, M.; Spike, R.C.; Watt, C.; Shehab, S.A.; Todd, A.J. Cells in laminae III and IV of the rat spinal cord that possess the neurokinin-1 receptor and have dorsally directed dendrites receive a major synaptic input from tachykinin-containing primary afferents. J. Neurosci. 1997, 17, 5536–5548. [Google Scholar] [CrossRef]
- Westlund, K.N. Chapter 9 The dorsal horn and hyperalgesia. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2006; Volume 81, pp. 103–125. [Google Scholar] [CrossRef]
- Schaible, H.G.; Grubb, B.D.; Neugebauer, V.; Oppmann, M. The Effects of NMDA Antagonists on Neuronal Activity in Cat Spinal Cord Evoked by Acute Inflammation in the Knee Joint. Eur. J. Neurosci. 1991, 3, 981–991. [Google Scholar] [CrossRef]
- Dougherty, P.M.; Palecek, J.; Paleckova, V.; Sorkin, L.S.; Willis, W.D. The role of NMDA and non-NMDA excitatory amino acid receptors in the excitation of primate spinothalamic tract neurons by mechanical, chemical, thermal, and electrical stimuli. J. Neurosci. 1992, 12, 3025–3041. [Google Scholar] [CrossRef]
- Ren, K.; Williams, G.M.; Hylden, J.L.; Ruda, M.A.; Dubner, R. The intrathecal administration of excitatory amino acid receptor antagonists selectively attenuated carrageenan-induced behavioral hyperalgesia in rats. Eur. J. Pharmacol. 1992, 219, 235–243. [Google Scholar] [CrossRef]
- Urban, L.; Dray, A. Synaptic activation of dorsal horn neurons by selective C-fibre excitation with capsaicin in the mouse spinal cord in vitro. Neuroscience 1992, 47, 693–702. [Google Scholar] [CrossRef]
- Polgar, E.; Watanabe, M.; Hartmann, B.; Grant, S.G.; Todd, A.J. Expression of AMPA receptor subunits at synapses in laminae I-III of the rodent spinal dorsal horn. Mol. Pain 2008, 4, 5. [Google Scholar] [CrossRef]
- Antal, M.; Fukazawa, Y.; Eordogh, M.; Muszil, D.; Molnar, E.; Itakura, M.; Takahashi, M.; Shigemoto, R. Numbers, densities, and colocalization of AMPA- and NMDA-type glutamate receptors at individual synapses in the superficial spinal dorsal horn of rats. J. Neurosci. 2008, 28, 9692–9701. [Google Scholar] [CrossRef]
- Nagy, G.G.; Al-Ayyan, M.; Andrew, D.; Fukaya, M.; Watanabe, M.; Todd, A.J. Widespread expression of the AMPA receptor GluR2 subunit at glutamatergic synapses in the rat spinal cord and phosphorylation of GluR1 in response to noxious stimulation revealed with an antigen-unmasking method. J. Neurosci. 2004, 24, 5766–5777. [Google Scholar] [CrossRef]
- Harris, J.A.; Corsi, M.; Quartaroli, M.; Arban, R.; Bentivoglio, M. Upregulation of spinal glutamate receptors in chronic pain. Neuroscience 1996, 74, 7–12. [Google Scholar] [CrossRef]
- Gangadharan, V.; Wang, R.; Ulzhofer, B.; Luo, C.; Bardoni, R.; Bali, K.K.; Agarwal, N.; Tegeder, I.; Hildebrandt, U.; Nagy, G.G.; et al. Peripheral calcium-permeable AMPA receptors regulate chronic inflammatory pain in mice. J. Clin. Investig. 2011, 121, 1608–1623. [Google Scholar] [CrossRef]
- Hartmann, B.; Ahmadi, S.; Heppenstall, P.A.; Lewin, G.R.; Schott, C.; Borchardt, T.; Seeburg, P.H.; Zeilhofer, H.U.; Sprengel, R.; Kuner, R. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron 2004, 44, 637–650. [Google Scholar] [CrossRef]
- Garry, E.M.; Fleetwood-Walker, S.M. A new view on how AMPA receptors and their interacting proteins mediate neuropathic pain. Pain 2004, 109, 210–213. [Google Scholar] [CrossRef]
- Larsson, M.; Broman, J. Translocation of GluR1-containing AMPA receptors to a spinal nociceptive synapse during acute noxious stimulation. J Neurosci. 2008, 28, 7084–7090. [Google Scholar] [CrossRef]
- Vikman, K.S.; Rycroft, B.K.; Christie, M.J. Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. J. Physiol. 2008, 586, 515–527. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed]
- Galan, A.; Laird, J.M.A.; Cervero, F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain 2004, 112, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Voitenko, N.; Petralia, R.S.; Guan, X.; Xu, J.T.; Steinberg, J.P.; Takamiya, K.; Sotnik, A.; Kopach, O.; Huganir, R.L.; et al. Persistent inflammation induces GluR2 internalization via NMDA receptor-triggered PKC activation in dorsal horn neurons. J. Neurosci. 2009, 29, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Goudet, C.; Magnaghi, V.; Landry, M.; Nagy, F.; Gereau RWt Pin, J.P. Metabotropic receptors for glutamate and GABA in pain. Brain Res. Rev. 2009, 60, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Paterlini, M.; Boifava, M.; Memo, M.; Spano, P. Metabotropic glutamate receptor mRNA expression in rat spinal cord. Neuroreport 1997, 8, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- Azkue, J.J.; Murga, M.; Fernandez-Capetillo, O.; Mateos, J.M.; Elezgarai, I.; Benitez, R.; Osorio, A.; Diez, J.; Puente, N.; Bilbao, A.; et al. Immunoreactivity for the group III metabotropic glutamate receptor subtype mGluR4a in the superficial laminae of the rat spinal dorsal horn. J. Comp. Neurol. 2001, 430, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Rustioni, A.; Valtschanoff, J.G. Metabotropic glutamate receptors in superficial laminae of the rat dorsal horn. J. Comp. Neurol. 1999, 410, 627–642. [Google Scholar] [CrossRef]
- Li, H.; Ohishi, H.; Kinoshita, A.; Shigemoto, R.; Nomura, S.; Mizuno, N. Localization of a metabotropic glutamate receptor, mGluR7, in axon terminals of presumed nociceptive, primary afferent fibers in the superficial layers of the spinal dorsal horn: An electron microscope study in the rat. Neurosci. Lett. 1997, 223, 153–156. [Google Scholar] [CrossRef]
- Ohishi, H.; Akazawa, C.; Shigemoto, R.; Nakanishi, S.; Mizuno, N. Distributions of the mRNAs for L-2-amino-4-phosphonobutyrate-sensitive metabotropic glutamate receptors, mGluR4 and mGluR7, in the rat brain. J. Comp. Neurol. 1995, 360, 555–570. [Google Scholar] [CrossRef]
- Ohishi, H.; Nomura, S.; Ding, Y.Q.; Shigemoto, R.; Wada, E.; Kinoshita, A.; Li, J.L.; Neki, A.; Nakanishi, S.; Mizuno, N. Presynaptic localization of a metabotropic glutamate receptor, mGluR7, in the primary afferent neurons: An immunohistochemical study in the rat. Neurosci. Lett. 1995, 202, 85–88. [Google Scholar] [CrossRef]
- Berthele, A.; Boxall, S.J.; Urban, A.; Anneser, J.M.; Zieglgansberger, W.; Urban, L.; Tolle, T.R. Distribution and developmental changes in metabotropic glutamate receptor messenger RNA expression in the rat lumbar spinal cord. Dev. Brain Res. 1999, 112, 39–53. [Google Scholar] [CrossRef]
- Bhave, G.; Karim, F.; Carlton, S.M.; Gereau Iv, R.W. Peripheral group I metabotropic glutamate receptors modulate nociception in mice. Nat. Neurosci. 2001, 4, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Carlton, S.M.; Hargett, G.L. Colocalization of metabotropic glutamate receptors in rat dorsal root ganglion cells. J. Comp. Neurol. 2007, 501, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, V.; Lucke, T.; Grubb, B.; Schaible, H.G. The involvement of N-methyl-D-aspartate (NMDA) and non-NMDA receptors in the responsiveness of rat spinal neurons with input from the chronically inflamed ankle. Neurosci. Lett. 1994, 170, 237–240. [Google Scholar] [CrossRef]
- Fundytus, M.E.; Osborne, M.G.; Henry, J.L.; Coderre, T.J.; Dray, A. Antisense oligonucleotide knockdown of mGluR1 alleviates hyperalgesia and allodynia associated with chronic inflammation. Pharmacol. Biochem. Behav. 2002, 73, 401–410. [Google Scholar] [CrossRef]
- Fundytus, M.E.; Yashpal, K.; Chabot, J.G.; Osborne, M.G.; Lefebvre, C.D.; Dray, A.; Henry, J.L.; Coderre, T.J. Knockdown of spinal metabotropic glutamate receptor 1 (mGluR(1)) alleviates pain and restores opioid efficacy after nerve injury in rats. Br. J. Pharmacol. 2001, 132, 354–367. [Google Scholar] [CrossRef]
- Fundytus, M.E.; Fisher, K.; Dray, A.; Henry, J.L.; Coderre, T.J. In vivo antinociceptive activity of anti-rat mGluR1 and mGluR5 antibodies in rats. Neuroreport 1998, 9, 731–735. [Google Scholar] [CrossRef]
- Chery, N.; De Koninck, Y. GABA(B) receptors are the first target of released GABA at lamina I inhibitory synapses in the adult rat spinal cord. J. Neurophysiol. 2000, 84, 1006–1011. [Google Scholar] [CrossRef]
- Salio, C.; Merighi, A.; Bardoni, R. GABA(B) receptors-mediated tonic inhibition of glutamate release from Abeta fibers in rat laminae III/IV of the spinal cord dorsal horn. Mol. Pain 2017, 13, 1744806917710041. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.A.; Kohno, T.; Karchewski, L.A.; Scholz, J.; Baba, H.; Woolf, C.J. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J. Neurosci. 2002, 22, 6724–6731. [Google Scholar] [CrossRef] [PubMed]
- Polgar, E.; Todd, A.J. Tactile allodynia can occur in the spared nerve injury model in the rat without selective loss of GABA or GABA(A) receptors from synapses in laminae I-II of the ipsilateral spinal dorsal horn. Neuroscience 2008, 156, 193–202. [Google Scholar] [CrossRef]
- Chen, S.R.; Zhu, L.; Chen, H.; Wen, L.; Laumet, G.; Pan, H.L. Increased spinal cord Na(+)-K(+)-2Cl(−) cotransporter-1 (NKCC1) activity contributes to impairment of synaptic inhibition in paclitaxel-induced neuropathic pain. J. Biol. Chem. 2014, 289, 31111–31120. [Google Scholar] [CrossRef]
- Modol, L.; Cobianchi, S.; Navarro, X. Prevention of NKCC1 phosphorylation avoids downregulation of KCC2 in central sensory pathways and reduces neuropathic pain after peripheral nerve injury. Pain 2014, 155, 1577–1590. [Google Scholar] [CrossRef]
- Grudzinska, J.; Schemm, R.; Haeger, S.; Nicke, A.; Schmalzing, G.; Betz, H.; Laube, B. The beta subunit determines the ligand binding properties of synaptic glycine receptors. Neuron 2005, 45, 727–739. [Google Scholar] [CrossRef]
- Zeilhofer, H.U.; Werynska, K.; Gingras, J.; Yevenes, G.E. Glycine Receptors in Spinal Nociceptive Control—An Update. Biomolecules 2021, 11, 846. [Google Scholar] [CrossRef]
- Ji, R.R.; Donnelly, C.R.; Nedergaard, M. Astrocytes in chronic pain and itch. Nat. Rev. Neurosci. 2019, 20, 667–685. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, Y.Q. Spinal glial activation contributes to pathological pain states. Neurosci. Biobehav. Rev. 2008, 32, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bae, C.; Gelman, B.B.; Chung, J.M.; Tang, S.J. A neuron-to-astrocyte Wnt5a signal governs astrogliosis during HIV-associated pain pathogenesis. Brain 2022, 145, 4108–4123. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bae, C.; Liu, B.; Zhang, Y.M.; Zhou, X.; Zhang, D.; Zhou, C.; DiBua, A.; Schutz, L.; Kaczocha, M.; et al. Development of opioid-induced hyperalgesia depends on reactive astrocytes controlled by Wnt5a signaling. Mol. Psychiatry 2023, 28, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis. Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef]
- Beggs, S.; Salter, M.W. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav. Immun. 2007, 21, 624–633. [Google Scholar] [CrossRef]
- Watkins, L.R.; Milligan, E.D.; Maier, S.F. Glial activation: A driving force for pathological pain. Trends Neurosci. 2001, 24, 450–455. [Google Scholar] [CrossRef]
- Calvo, M.; Bennett, D.L. The mechanisms of microgliosis and pain following peripheral nerve injury. Exp. Neurol. 2012, 234, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, S.; Shi, X.Q.; Zhang, J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain 2008, 135, 37–47. [Google Scholar] [CrossRef]
- Liu, L.; Rudin, M.; Kozlova, E.N. Glial cell proliferation in the spinal cord after dorsal rhizotomy or sciatic nerve transection in the adult rat. Exp. Brain Res. 2000, 131, 64–73. [Google Scholar] [CrossRef]
- Svensson, C.I.; Schafers, M.; Jones, T.L.; Powell, H.; Sorkin, L.S. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci. Lett. 2005, 379, 209–213. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, J.A.; Yezierski, R.P. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 2001, 90, 1–6. [Google Scholar] [CrossRef]
- Ru, W.; Liu, X.; Bae, C.; Shi, Y.; Walikonis, R.; Mo Chung, J.; Tang, S.J. Microglia Mediate HIV-1 gp120-Induced Synaptic Degeneration in Spinal Pain Neural Circuits. J. Neurosci. 2019, 39, 8408–8421. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Lu, N.; Xu, Z.Z.; Liu, T.; Serhan, C.N.; Ji, R.R. Resolving TRPV1- and TNF-alpha-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J. Neurosci. 2011, 31, 15072–15085. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.I.; Svensson, C.I.; Koehrn, F.J.; Bhuskute, A.; Sorkin, L.S. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain 2010, 149, 243–253. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Zhang, L.; Cheng, J.K.; Ji, R.R. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 2008, 28, 5189–5194. [Google Scholar] [CrossRef]
- Zhang, R.X.; Liu, B.; Li, A.; Wang, L.; Ren, K.; Qiao, J.T.; Berman, B.M.; Lao, L. Interleukin 1beta facilitates bone cancer pain in rats by enhancing NMDA receptor NR-1 subunit phosphorylation. Neuroscience 2008, 154, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.X.; Li, A.; Liu, B.; Wang, L.; Ren, K.; Zhang, H.; Berman, B.M.; Lao, L. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain 2008, 135, 232–239. [Google Scholar] [CrossRef]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783. [Google Scholar] [CrossRef]
- Trang, T.; Beggs, S.; Wan, X.; Salter, M.W. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J. Neurosci. 2009, 29, 3518–3528. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.W.; Bennett, D.L.; Kerr, B.J.; Bradbury, E.J.; McMahon, S.B. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc. Natl. Acad. Sci. USA 1999, 96, 7714–7718. [Google Scholar] [CrossRef]
- Coull, J.A.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Li, H.; Thomas-Crusells, J.; Lahtinen, H.; Viitanen, T.; Nanobashvili, A.; Kokaia, Z.; Airaksinen, M.S.; Voipio, J.; Kaila, K.; et al. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J. Cell Biol. 2002, 159, 747–752. [Google Scholar] [CrossRef]
- Coull, J.A.; Boudreau, D.; Bachand, K.; Prescott, S.A.; Nault, F.; Sik, A.; De Koninck, P.; De Koninck, Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 2003, 424, 938–942. [Google Scholar] [CrossRef]
- Prescott, S.A.; Sejnowski, T.J.; De Koninck, Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: Towards a biophysical basis for neuropathic pain. Mol. Pain 2006, 2, 32. [Google Scholar] [CrossRef]
- Lever, I.; Cunningham, J.; Grist, J.; Yip, P.K.; Malcangio, M. Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur. J. Neurosci. 2003, 18, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Tuve, A.; Malcangio, M.; Ebersberger, A.; Mazario, J.; Schaible, H.G. Effect of brain-derived neurotrophic factor on the release of substance P from rat spinal cord. Neuroreport 2001, 12, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; Cunningham, J.; Patel, J.; Grist, J.; Gavazzi, I.; Lever, I.J.; Malcangio, M. BDNF modulates sensory neuron synaptic activity by a facilitation of GABA transmission in the dorsal horn. Mol. Cell Neurosci. 2002, 21, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.J. Reactive astrocytes in pain neural circuit pathogenesis. Curr. Opin. Neurobiol. 2022, 75, 102584. [Google Scholar] [CrossRef] [PubMed]
- Garrison, C.J.; Dougherty, P.M.; Kajander, K.C.; Carlton, S.M. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991, 565, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Garrison, C.J.; Dougherty, P.M.; Carlton, S.M. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp. Neurol. 1994, 129, 237–243. [Google Scholar] [CrossRef]
- Nesic, O.; Lee, J.; Johnson, K.M.; Ye, Z.; Xu, G.Y.; Unabia, G.C.; Wood, T.G.; McAdoo, D.J.; Westlund, K.N.; Hulsebosch, C.E.; et al. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J. Neurochem. 2005, 95, 998–1014. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, V.; Tanga, F.Y.; DeLeo, J.A. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur. J. Neurosci. 2004, 20, 467–473. [Google Scholar] [CrossRef]
- Sun, S.; Cao, H.; Han, M.; Li, T.T.; Pan, H.L.; Zhao, Z.Q.; Zhang, Y.Q. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain 2007, 129, 64–75. [Google Scholar] [CrossRef]
- Tsuda, M.; Kohro, Y.; Yano, T.; Tsujikawa, T.; Kitano, J.; Tozaki-Saitoh, H.; Koyanagi, S.; Ohdo, S.; Ji, R.R.; Salter, M.W.; et al. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain 2011, 134, 1127–1139. [Google Scholar] [CrossRef]
- Kim, D.S.; Figueroa, K.W.; Li, K.W.; Boroujerdi, A.; Yolo, T.; Luo, Z.D. Profiling of dynamically changed gene expression in dorsal root ganglia post peripheral nerve injury and a critical role of injury-induced glial fibrillary acidic protein in maintenance of pain behaviors [corrected]. Pain 2009, 143, 114–122. [Google Scholar] [CrossRef]
- Shi, Y.; Gelman, B.B.; Lisinicchia, J.G.; Tang, S.J. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J. Neurosci. 2012, 32, 10833–10840. [Google Scholar] [CrossRef]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.S.; McCarthy, K.D. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R Soc. Lond B Biol. Sci. 2014, 369, 20130595. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.Y.; Li, Z.; Dostrovsky, J.O.; Hu, J.W.; Sessle, B.J. Glutamine uptake contributes to central sensitization in the medullary dorsal horn. Neuroreport 2008, 19, 1151–1154. [Google Scholar] [CrossRef]
- Xin, W.J.; Weng, H.R.; Dougherty, P.M. Plasticity in expression of the glutamate transporters GLT-1 and GLAST in spinal dorsal horn glial cells following partial sciatic nerve ligation. Mol. Pain. 2009, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Lim, G.; Mao, J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J. Neurosci. 2003, 23, 2899–2910. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.D.; Wang, H.; Zhang, Y.Q.; Zhao, Z.Q. Distinct effects of D-serine on spinal nociceptive responses in normal and carrageenan-injected rats. Biochem. Biophys. Res. Commun. 2006, 343, 401–406. [Google Scholar] [CrossRef]
- Gu, J.G.; MacDermott, A.B. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature 1997, 389, 749–753. [Google Scholar] [CrossRef]
- Rodrigues, R.J.; Almeida, T.; Richardson, P.J.; Oliveira, C.R.; Cunha, R.A. Dual presynaptic control by ATP of glutamate release via facilitatory P2X1, P2X2/3, and P2X3 and inhibitory P2Y1, P2Y2, and/or P2Y4 receptors in the rat hippocampus. J. Neurosci. 2005, 25, 6286–6295. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Fernandez, V.; Andrew, R.D.; Barajas-Lopez, C. ATP inhibits glutamate synaptic release by acting at P2Y receptors in pyramidal neurons of hippocampal slices. J. Pharmacol. Exp. Ther. 2000, 293, 172–179. [Google Scholar] [PubMed]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.L.; Van Eldik, L.J. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes [corrected]. Brain Res. 2009, 1287, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Thacker, M.A.; Clark, A.K.; Bishop, T.; Grist, J.; Yip, P.K.; Moon, L.D.; Thompson, S.W.; Marchand, F.; McMahon, S.B. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur. J. Pain. 2009, 13, 263–272. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, X.Q.; Echeverry, S.; Mogil, J.S.; De Koninck, Y.; Rivest, S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 2007, 27, 12396–12406. [Google Scholar] [CrossRef]
- Gosselin, R.D.; Varela, C.; Banisadr, G.; Mechighel, P.; Rostene, W.; Kitabgi, P.; Melik-Parsadaniantz, S. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J. Neurochem. 2005, 95, 1023–1034. [Google Scholar] [CrossRef]
- Raghavendra, V.; Tanga, F.; DeLeo, J.A. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J. Pharmacol. Exp. Ther. 2003, 306, 624–630. [Google Scholar] [CrossRef]
- Chen, G.; Park, C.K.; Xie, R.G.; Berta, T.; Nedergaard, M.; Ji, R.R. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 2014, 137, 2193–2209. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.R. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154 (Suppl. S1), S10–S28. [Google Scholar] [CrossRef]
- Zhang, J.; De Koninck, Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J. Neurochem. 2006, 97, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Gwak, Y.S.; Kang, J.; Unabia, G.C.; Hulsebosch, C.E. Spatial and temporal activation of spinal glial cells: Role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp. Neurol. 2012, 234, 362–372. [Google Scholar] [CrossRef]
- Yousefpour, N.; Locke, S.; Deamond, H.; Wang, C.; Marques, L.; St-Louis, M.; Ouellette, J.; Khoutorsky, A.; De Koninck, Y.; Ribeiro-da-Silva, A. Time-dependent and selective microglia-mediated removal of spinal synapses in neuropathic pain. Cell Rep. 2023, 42, 112010. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, B.L.; Yang, Q.; Zhou, X.; Tang, S.J. Microglial ablation does not affect opioid-induced hyperalgesia in rodents. Pain 2022, 163, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Sorge, R.E.; Mapplebeck, J.C.S.; Rosen, S.; Beggs, S.; Taves, S.; Alexander, J.K.; Martin, L.J.; Austin, J.-S.; Sotocinal, S.G.; Chen, D.; et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 2015, 18, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J. Evidence for a central component of post-injury pain hypersensitivity. Nature 1983, 306, 686–688. [Google Scholar] [CrossRef]
- Mannion, R.J.; Costigan, M.; Decosterd, I.; Amaya, F.; Ma, Q.P.; Holstege, J.C.; Ji, R.R.; Acheson, A.; Lindsay, R.M.; Wilkinson, G.A.; et al. Neurotrophins: Peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc. Natl. Acad. Sci. USA 1999, 96, 9385–9390. [Google Scholar] [CrossRef]
- Neumann, S.; Doubell, T.P.; Leslie, T.; Woolf, C.J. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature 1996, 384, 360–364. [Google Scholar] [CrossRef]
- Baba, H.; Doubell, T.P.; Woolf, C.J. Peripheral inflammation facilitates Abeta fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J. Neurosci. 1999, 19, 859–867. [Google Scholar] [CrossRef]
- Zieglgansberger, W. Substance P and pain chronicity. Cell Tissue Res. 2019, 375, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Matayoshi, S.; Jiang, N.; Katafuchi, T.; Koga, K.; Furue, H.; Yasaka, T.; Nakatsuka, T.; Zhou, X.F.; Kawasaki, Y.; Tanaka, N.; et al. Actions of brain-derived neurotrophic factor on spinal nociceptive transmission during inflammation in the rat. J. Physiol. 2005, 569, 685–695. [Google Scholar] [CrossRef]
- Lekan, H.A.; Carlton, S.M.; Coggeshall, R.E. Sprouting of A beta fibers into lamina II of the rat dorsal horn in peripheral neuropathy. Neurosci. Lett. 1996, 208, 147–150. [Google Scholar] [CrossRef]
- Mannion, R.J.; Doubell, T.P.; Coggeshall, R.E.; Woolf, C.J. Collateral sprouting of uninjured primary afferent A-fibers into the superficial dorsal horn of the adult rat spinal cord after topical capsaicin treatment to the sciatic nerve. J. Neurosci. 1996, 16, 5189–5195. [Google Scholar] [CrossRef] [PubMed]
- Shortland, P.; Kinman, E.; Molander, C. Sprouting of A-fibre primary afferents into lamina II in two rat models of neuropathic pain. Eur. J. Pain 1997, 1, 215–227. [Google Scholar] [CrossRef]
- Woolf, C.J.; Shortland, P.; Coggeshall, R.E. Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature 1992, 355, 75–78. [Google Scholar] [CrossRef]
- Feng, T. Studies on the neuromuscular junction XXVI. The changes of the end-plate potential during and after prolonged stimulation. Chin. J. Physiol. 1941, 16, 341–372. [Google Scholar]
- Eccles, J.C.; O’Connor, W.J. Abortive impulses at the neuro-muscular junction. J Physiol 1941, 100, 318–328. [Google Scholar] [CrossRef]
- Stevens, C.F.; Wang, Y. Facilitation and depression at single central synapses. Neuron 1995, 14, 795–802. [Google Scholar] [CrossRef]
- Abbott, L.F.; Varela, J.A.; Sen, K.; Nelson, S.B. Synaptic depression and cortical gain control. Science 1997, 275, 220–224. [Google Scholar] [CrossRef]
- Tsodyks, M.V.; Markram, H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. USA 1997, 94, 719–723. [Google Scholar] [CrossRef]
- Chen, C.; Blitz, D.M.; Regehr, W.G. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron 2002, 33, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Aslam, M.; Gollisch, T.; Allen, K.; von Engelhardt, J. CKAMP44 modulates integration of visual inputs in the lateral geniculate nucleus. Nat. Commun. 2018, 9, 261. [Google Scholar] [CrossRef]
- LaMotte, R.H.; Campbell, J.N. Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgments of thermal pain. J. Neurophysiol. 1978, 41, 509–528. [Google Scholar] [CrossRef]
- Greffrath, W.; Nemenov, M.I.; Schwarz, S.; Baumgartner, U.; Vogel, H.; Arendt-Nielsen, L.; Treede, R.D. Inward currents in primary nociceptive neurons of the rat and pain sensations in humans elicited by infrared diode laser pulses. Pain 2002, 99, 145–155. [Google Scholar] [CrossRef]
- Peng, Y.B.; Ringkamp, M.; Meyer, R.A.; Campbell, J.N. Fatigue and paradoxical enhancement of heat response in C-fiber nociceptors from cross-modal excitation. J. Neurosci. 2003, 23, 4766–4774. [Google Scholar] [CrossRef] [PubMed]
- Shyu, B.C.; Vogt, B.A. Short-term synaptic plasticity in the nociceptive thalamic-anterior cingulate pathway. Mol. Pain 2009, 5, 51. [Google Scholar] [CrossRef]
- Luo, C.; Kuner, T.; Kuner, R. Synaptic plasticity in pathological pain. Trends Neurosci. 2014, 37, 343–355. [Google Scholar] [CrossRef]
- Bliss, T.V.; Lomo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Zhou, L.-J.; Peng, J.; Xu, Y.-N.; Zeng, W.-J.; Zhang, J.; Wei, X.; Mai, C.-L.; Lin, Z.-J.; Liu, Y.; Murugan, M.; et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. 2019, 27, 3844–3859.e6. [Google Scholar] [CrossRef]
- Ikeda, H.; Stark, J.; Fischer, H.; Wagner, M.; Drdla, R.; Jager, T.; Sandkuhler, J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science 2006, 312, 1659–1662. [Google Scholar] [CrossRef]
- Azkue, J.J.; Liu, X.G.; Zimmermann, M.; Sandkuhler, J. Induction of long-term potentiation of C fibre-evoked spinal field potentials requires recruitment of group I, but not group II/III metabotropic glutamate receptors. Pain 2003, 106, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C.; Zhang, Y.Q.; Zhao, Z.Q. Involvement of nitric oxide in long-term potentiation of spinal nociceptive responses in rats. Neuroreport 2005, 16, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Magerl, W.; Hopf, H.C.; Sandkuhler, J.; Treede, R.D. Perceptual correlates of nociceptive long-term potentiation and long-term depression in humans. J. Neurosci. 2004, 24, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Li, P.; Zhuo, M. Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J. Neurosci. 1999, 19, 9346–9354. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Liu, M.G.; Chen, T.; Ko, H.G.; Baek, G.C.; Lee, H.R.; Lee, K.; Collingridge, G.L.; Kaang, B.K.; Zhuo, M. Plasticity of metabotropic glutamate receptor-dependent long-term depression in the anterior cingulate cortex after amputation. J. Neurosci. 2012, 32, 11318–11329. [Google Scholar] [CrossRef] [PubMed]
- Rottmann, S.; Jung, K.; Ellrich, J. Electrical low-frequency stimulation induces homotopic long-term depression of nociception and pain from hand in man. Clin. Neurophysiol. 2008, 119, 1895–1904. [Google Scholar] [CrossRef]
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef] [PubMed]
- Del Rosario, J.S.; McIlvried, L.A.; Pullen, M.Y.; Wangzhou, A.; Sheahan, T.D.; Shepherd, A.J.; Slivicki, R.A.; Price, T.J.; Copits, B.A.; Bertels, Z. Sustained Depolarization Induces Homeostatic Plasticity in Mouse and Human Sensory Neurons. J. Pain 2022, 23, 6. [Google Scholar] [CrossRef]
- Aguilar, J.; Humanes-Valera, D.; Alonso-Calvino, E.; Yague, J.G.; Moxon, K.A.; Oliviero, A.; Foffani, G. Spinal cord injury immediately changes the state of the brain. J. Neurosci. 2010, 30, 7528–7537. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.H.; Kim, D.S.; Cho, Z.H.; Sohn, J.H.; Chung, M.A.; Lee, H.J.; Nam, T.S.; Lee, B.H. Modification of cortical excitability in neuropathic rats: A voltage-sensitive dye study. Neurosci. Lett. 2009, 464, 117–121. [Google Scholar] [CrossRef]
- Boord, P.; Siddall, P.J.; Tran, Y.; Herbert, D.; Middleton, J.; Craig, A. Electroencephalographic slowing and reduced reactivity in neuropathic pain following spinal cord injury. Spinal Cord 2008, 46, 118–123. [Google Scholar] [CrossRef]
- Xiong, W.; Ping, X.; Ripsch, M.S.; Chavez, G.S.C.; Hannon, H.E.; Jiang, K.; Bao, C.; Jadhav, V.; Chen, L.; Chai, Z.; et al. Enhancing excitatory activity of somatosensory cortex alleviates neuropathic pain through regulating homeostatic plasticity. Sci. Rep. 2017, 7, 12743. [Google Scholar] [CrossRef]
- Galhardoni, R.; Correia, G.S.; Araujo, H.; Yeng, L.T.; Fernandes, D.T.; Kaziyama, H.H.; Marcolin, M.A.; Bouhassira, D.; Teixeira, M.J.; de Andrade, D.C. Repetitive transcranial magnetic stimulation in chronic pain: A review of the literature. Arch. Phys. Med. Rehabil. 2015, 96, S156–S172. [Google Scholar] [CrossRef]
- Fregni, F.; Boggio, P.S.; Lima, M.C.; Ferreira, M.J.; Wagner, T.; Rigonatti, S.P.; Castro, A.W.; Souza, D.R.; Riberto, M.; Freedman, S.D.; et al. A sham-controlled, phase II trial of transcranial direct current stimulation for the treatment of central pain in traumatic spinal cord injury. Pain 2006, 122, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Scherrer, G.; Chen, L. Spinal cord retinoic acid receptor signaling gates mechanical hypersensitivity in neuropathic pain. Neuron 2022, 110, 4108–4124 e6. [Google Scholar] [CrossRef] [PubMed]
- Melzack, R.; Wall, P.D. Pain mechanisms: A new theory. Science 1965, 150, 971–979. [Google Scholar] [CrossRef]
- Antal, M.; Petko, M.; Polgar, E.; Heizmann, C.W.; Storm-Mathisen, J. Direct evidence of an extensive GABAergic innervation of the spinal dorsal horn by fibres descending from the rostral ventromedial medulla. Neuroscience 1996, 73, 509–518. [Google Scholar] [CrossRef]
- Harvey, R.J.; Depner, U.B.; Wässle, H.; Ahmadi, S.; Heindl, C.; Reinold, H.; Smart, T.G.; Schütz, B.; Abo-Salem, O.M.; Zimmer, A.; et al. GlyR alpha3, an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science 2004, 304, 884–887. [Google Scholar] [CrossRef]
- Scholz, J.; Broom, D.C.; Youn, D.H.; Mills, C.D.; Kohno, T.; Suter, M.R.; Moore, K.A.; Decosterd, I.; Coggeshall, R.E.; Woolf, C.J. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J. Neurosci. 2005, 25, 7317–7323. [Google Scholar] [CrossRef] [PubMed]
- Drew, G.M.; Siddall, P.J.; Duggan, A.W. Mechanical allodynia following contusion injury of the rat spinal cord is associated with loss of GABAergic inhibition in the dorsal horn. Pain 2004, 109, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T.L. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: Effects of modulatory receptor systems and excitatory amino acid antagonists. Pain 1989, 37, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Torsney, C.; MacDermott, A.B. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J. Neurosci. 2006, 26, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Malan, T.P.; Mata, H.P.; Porreca, F. Spinal GABA(A) and GABA(B) receptor pharmacology in a rat model of neuropathic pain. Anesthesiology 2002, 96, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.P.; Mezzatesta, J.; Nadeson, R.; Goodchild, C.S. Intrathecal midazolam II: Combination with intrathecal fentanyl for labor pain. Anesth. Analg. 2004, 98, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Ratte, S.; Prescott, S.A. Excitatory neurons are more disinhibited than inhibitory neurons by chloride dysregulation in the spinal dorsal horn. eLife 2019, 8, e49753. [Google Scholar] [CrossRef]
- Lu, V.B.; Biggs, J.E.; Stebbing, M.J.; Balasubramanyan, S.; Todd, K.G.; Lai, A.Y.; Colmers, W.F.; Dawbarn, D.; Ballanyi, K.; Smith, P.A. Brain-derived neurotrophic factor drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. J. Physiol. 2009, 587, 1013–1032. [Google Scholar] [CrossRef]
- Wang, J.; Duan, Y.; Zhang, T.; Huang, J.; Ren, Z.; Ye, J.; Wang, N.; Li, Y.; Chen, X.; Gao, P.; et al. Aberrant multimodal brain networks in patients with anti-NMDA receptor encephalitis. CNS. Neurosci. Ther. 2021, 27, 652–663. [Google Scholar] [CrossRef]
- Kumar, S.S.; Jin, X.; Buckmaster, P.S.; Huguenard, J.R. Recurrent circuits in layer II of medial entorhinal cortex in a model of temporal lobe epilepsy. J. Neurosci. 2007, 27, 1239–1246. [Google Scholar] [CrossRef]
- Dallerac, G.; Chever, O.; Rouach, N. How do astrocytes shape synaptic transmission? Insights from electrophysiology. Front. Cell Neurosci. 2013, 7, 159. [Google Scholar] [CrossRef]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef]
- Pannasch, U.; Vargova, L.; Reingruber, J.; Ezan, P.; Holcman, D.; Giaume, C.; Sykova, E.; Rouach, N. Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 8467–8472. [Google Scholar] [CrossRef] [PubMed]
- Covelo, A.; Araque, A. Neuronal activity determines distinct gliotransmitter release from a single astrocyte. eLife 2018, 7, e32237. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Araque, A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 2010, 68, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Gómez, R.; Mederos, S.; Covelo, A.; Ballesteros, J.J.; Schlosser, L.; Hernández-Vivanco, A.; Martín-Fernández, M.; Quintana, R.; Rayan, A.; et al. Activity-dependent switch of GABAergic inhibition into glutamatergic excitation in astrocyte-neuron networks. eLife 2016, 5, e20362. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gonzalo, M.; Navarrete, M.; Perea, G.; Covelo, A.; Martin-Fernandez, M.; Shigemoto, R.; Lujan, R.; Araque, A. Endocannabinoids Induce Lateral Long-Term Potentiation of Transmitter Release by Stimulation of Gliotransmission. Cereb. Cortex 2015, 25, 3699–3712. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Perea, G.; Fernandez de Sevilla, D.; Gomez-Gonzalo, M.; Nunez, A.; Martin, E.D.; Araque, A. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 2012, 10, e1001259. [Google Scholar] [CrossRef]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef]
- Kang, J.; Jiang, L.; Goldman, S.A.; Nedergaard, M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1998, 1, 683–692. [Google Scholar] [CrossRef]
- Liu, Q.S.; Xu, Q.; Arcuino, G.; Kang, J.; Nedergaard, M. Astrocyte-mediated activation of neuronal kainate receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 3172–3177. [Google Scholar] [CrossRef]
- Liu, Q.S.; Xu, Q.; Kang, J.; Nedergaard, M. Astrocyte activation of presynaptic metabotropic glutamate receptors modulates hippocampal inhibitory synaptic transmission. Neuron Glia Biol. 2004, 1, 307–316. [Google Scholar] [CrossRef]
- Varela-Nallar, L.; Alfaro, I.E.; Serrano, F.G.; Parodi, J.; Inestrosa, N.C. Wingless-type family member 5A (Wnt-5a) stimulates synaptic differentiation and function of glutamatergic synapses. Proc. Natl. Acad. Sci. USA 2010, 107, 21164–21169. [Google Scholar] [CrossRef]
- Cuitino, L.; Godoy, J.A.; Farias, G.G.; Couve, A.; Bonansco, C.; Fuenzalida, M.; Inestrosa, N.C. Wnt-5a modulates recycling of functional GABAA receptors on hippocampal neurons. J. Neurosci. 2010, 30, 8411–8420. [Google Scholar] [CrossRef]
- Pozzi, D.; Menna, E.; Canzi, A.; Desiato, G.; Mantovani, C.; Matteoli, M. The Communication Between the Immune and Nervous Systems: The Role of IL-1beta in Synaptopathies. Front. Mol. Neurosci. 2018, 11, 111. [Google Scholar] [CrossRef]
- Mishra, A.; Kim, H.J.; Shin, A.H.; Thayer, S.A. Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J. Neuroimmune Pharmacol. 2012, 7, 571–578. [Google Scholar] [CrossRef]
- Rutherford, L.C.; Nelson, S.B.; Turrigiano, G.G. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 1998, 21, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Kordek, R.; Nerurkar, V.R.; Liberski, P.P.; Isaacson, S.; Yanagihara, R.; Gajdusek, D.C. Heightened expression of tumor necrosis factor alpha, interleukin 1 alpha, and glial fibrillary acidic protein in experimental Creutzfeldt-Jakob disease in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9754–9758. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.W.; Basu, A.; Druckman, C.; Cicchese, M.; Krady, J.K.; Levison, S.W. Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J. Neuroinflammation 2006, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Hebert, J.M. Signaling pathways in reactive astrocytes, a genetic perspective. Mol. Neurobiol. 2011, 43, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sandkuhler, J.; Gruber-Schoffnegger, D. Hyperalgesia by synaptic long-term potentiation (LTP): An update. Curr. Opin. Pharmacol. 2012, 12, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Heinke, B.; Ruscheweyh, R.; Sandkuhler, J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science 2003, 299, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- Hjornevik, T.; Schoultz, B.W.; Marton, J.; Gjerstad, J.; Drzezga, A.; Henriksen, G.; Willoch, F. Spinal long-term potentiation is associated with reduced opioid neurotransmission in the rat brain. Clin. Physiol. Funct. Imaging 2010, 30, 285–293. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Tjia, M.; Thapliyal, S. Homeostatic plasticity and excitation-inhibition balance: The good, the bad, and the ugly. Curr. Opin. Neurobiol. 2022, 75, 102553. [Google Scholar] [CrossRef]
- Medrano, M.C.; Dhanasobhon, D.; Yalcin, I.; Schlichter, R.; Cordero-Erausquin, M. Loss of inhibitory tone on spinal cord dorsal horn spontaneously and nonspontaneously active neurons in a mouse model of neuropathic pain. Pain 2016, 157, 1432–1442. [Google Scholar] [CrossRef]
- Inoue, K.; Tsuda, M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19, 138–152. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Tang, S.-J. Neural Circuitry Polarization in the Spinal Dorsal Horn (SDH): A Novel Form of Dysregulated Circuitry Plasticity during Pain Pathogenesis. Cells 2024, 13, 398. https://doi.org/10.3390/cells13050398
Chen X, Tang S-J. Neural Circuitry Polarization in the Spinal Dorsal Horn (SDH): A Novel Form of Dysregulated Circuitry Plasticity during Pain Pathogenesis. Cells. 2024; 13(5):398. https://doi.org/10.3390/cells13050398
Chicago/Turabian StyleChen, Xufeng, and Shao-Jun Tang. 2024. "Neural Circuitry Polarization in the Spinal Dorsal Horn (SDH): A Novel Form of Dysregulated Circuitry Plasticity during Pain Pathogenesis" Cells 13, no. 5: 398. https://doi.org/10.3390/cells13050398
APA StyleChen, X., & Tang, S.-J. (2024). Neural Circuitry Polarization in the Spinal Dorsal Horn (SDH): A Novel Form of Dysregulated Circuitry Plasticity during Pain Pathogenesis. Cells, 13(5), 398. https://doi.org/10.3390/cells13050398