
Lysosomes in Cancer—At the Crossroad of Good and Evil


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Lysosomal Characteristics
1.1.1. The Lysosomal Membrane
1.1.2. Calcium Signaling
1.2. Lysosomal Degradation
1.3. Endolysosomal Maturation and Lysosome Biogenesis
1.4. Nutrient Sensing and Transcriptional Regulation of Lysosomal Biogenesis
1.5. Lysosomal Involvement in Regulated Cell Death
2. Lysosomal Positioning
2.1. Regulation of Lysosomal Transport
2.2. Anterograde Transport
2.3. Retrograde Transport
3. Secretion from the Lysosomal Pathway
3.1. Release of Extracellular Vesicles
3.2. Lysosomal Exocytosis
3.3. Lysosome-Mediated Plasma Membrane Repair
4. Lysosomal Involvement in Cancer
4.1. Autophagic Rewiring
4.2. Tumor-Induced Regulation of pH
4.3. Lysosomes in Drug Resistance
4.4. Lysosomal Exocytosis and Release of Extracellular Vesicles in Cancer
4.4.1. Alterations of Lysosomal Function Regulate Release of Exosomes
4.4.2. Direct Shedding of EVs with Lysosomal Origin
4.4.3. Lysosomal Positioning Determines the Malignant Phenotype
4.4.4. Lysosomal Exocytosis Remodels the Extracellular Matrix and Activates EMT
4.4.5. The Lysosomal Membrane in Cancer Progression
4.4.6. Regulation of Cancer-Induced Lysosomal Repositioning
4.5. Induction of Lysosomal Damage as a Therapeutic Target
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lubke, T.; Lobel, P.; Sleat, D.E. Proteomics of the lysosome. Biochim. Biophys. Acta 2009, 1793, 625–635. [Google Scholar] [CrossRef]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Machado, E.R.; Annunziata, I.; van de Vlekkert, D.; Grosveld, G.C.; d’Azzo, A. Lysosomes and Cancer Progression: A Malignant Liaison. Front. Cell Dev. Biol. 2021, 9, 642494. [Google Scholar] [CrossRef]
- Eriksson, I. Dealing with Damaged Lysosomes: Impact of Lysosomal Membrane Stability in Health and Disease. Doctoral Thesis, Comprehensive Summary. Linköping University Electronic Press, Linköping, Sweden, 2022. [Google Scholar]
- Coffey, J.W.; De Duve, C. Digestive activity of lysosomes. I. The digestion of proteins by extracts of rat liver lysosomes. J. Biol. Chem. 1968, 243, 3255–3263. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Zhivotovsky, B. Proteases in autophagy. Biochim. Biophys. Acta 2012, 1824, 44–50. [Google Scholar] [CrossRef]
- Pungercar, J.R.; Caglic, D.; Sajid, M.; Dolinar, M.; Vasiljeva, O.; Pozgan, U.; Turk, D.; Bogyo, M.; Turk, V.; Turk, B. Autocatalytic processing of procathepsin B is triggered by proenzyme activity. FEBS J. 2009, 276, 660–668. [Google Scholar] [CrossRef]
- van der Stappen, J.W.; Williams, A.C.; Maciewicz, R.A.; Paraskeva, C. Activation of cathepsin B, secreted by a colorectal cancer cell line requires low pH and is mediated by cathepsin D. Int. J. Cancer 1996, 67, 547–554. [Google Scholar] [CrossRef]
- Laurent-Matha, V.; Derocq, D.; Prebois, C.; Katunuma, N.; Liaudet-Coopman, E. Processing of human cathepsin D is independent of its catalytic function and auto-activation: Involvement of cathepsins L and B. J. Biochem. 2006, 139, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization in cell death: New evidence and implications for health and disease. Ann. N. Y. Acad. Sci. 2016, 1371, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Dennemarker, J.; Lohmuller, T.; Muller, S.; Aguilar, S.V.; Tobin, D.J.; Peters, C.; Reinheckel, T. Impaired turnover of autophagolysosomes in cathepsin L deficiency. Biol. Chem. 2010, 391, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Felbor, U.; Kessler, B.; Mothes, W.; Goebel, H.H.; Ploegh, H.L.; Bronson, R.T.; Olsen, B.R. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc. Natl. Acad. Sci. USA 2002, 99, 7883–7888. [Google Scholar] [CrossRef]
- De Pasquale, V.; Moles, A.; Pavone, L.M. Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy. Cells 2020, 9, 979. [Google Scholar] [CrossRef]
- Chwieralski, C.E.; Welte, T.; Buhling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar] [CrossRef]
- Roberg, K.; Ollinger, K. Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am. J. Pathol. 1998, 152, 1151–1156. [Google Scholar] [PubMed]
- Soond, S.M.; Kozhevnikova, M.V.; Frolova, A.S.; Savvateeva, L.V.; Plotnikov, E.Y.; Townsend, P.A.; Han, Y.P.; Zamyatnin, A.A., Jr. Lost or Forgotten: The nuclear cathepsin protein isoforms in cancer. Cancer Lett. 2019, 462, 43–50. [Google Scholar] [CrossRef]
- Vidak, E.; Javorsek, U.; Vizovisek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Wilke, S.; Krausze, J.; Bussow, K. Crystal structure of the conserved domain of the DC lysosomal associated membrane protein: Implications for the lysosomal glycocalyx. BMC Biol. 2012, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Kundra, R.; Kornfeld, S. Asparagine-linked oligosaccharides protect Lamp-1 and Lamp-2 from intracellular proteolysis. J. Biol. Chem. 1999, 274, 31039–31046. [Google Scholar] [CrossRef]
- Schwake, M.; Schroder, B.; Saftig, P. Lysosomal membrane proteins and their central role in physiology. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef]
- Schroder, B.; Saftig, P. Intramembrane proteolysis within lysosomes. Ageing Res. Rev. 2016, 32, 51–64. [Google Scholar] [CrossRef]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagne, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell. Proteomics 2013, 12, 1572–1588. [Google Scholar] [CrossRef]
- Schroder, B.; Wrocklage, C.; Pan, C.; Jager, R.; Kosters, B.; Schafer, H.; Elsasser, H.P.; Mann, M.; Hasilik, A. Integral and associated lysosomal membrane proteins. Traffic 2007, 8, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.K.; Eskelinen, E.L.; Scott, C.C.; Malevanets, A.; Saftig, P.; Grinstein, S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007, 26, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Schmidt, C.K.; Neu, S.; Willenborg, M.; Fuertes, G.; Salvador, N.; Tanaka, Y.; Lullmann-Rauch, R.; Hartmann, D.; Heeren, J.; et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol. Biol. Cell 2004, 15, 3132–3145. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, K.; Tomabechi, Y.; Ikeda, M.; Ehara, H.; Kukimoto-Niino, M.; Wakiyama, M.; Podyma-Inoue, K.A.; Rajapakshe, A.R.; Watabe, T.; Shirouzu, M.; et al. Lysosome-associated membrane proteins-1 and -2 (LAMP-1 and LAMP-2) assemble via distinct modes. Biochem. Biophys. Res. Commun. 2016, 479, 489–495. [Google Scholar] [CrossRef]
- Yogalingam, G.; Bonten, E.J.; van de Vlekkert, D.; Hu, H.; Moshiach, S.; Connell, S.A.; d’Azzo, A. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev. Cell 2008, 15, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pfeffer, S.R. Lysosomal membrane glycoproteins bind cholesterol and contribute to lysosomal cholesterol export. eLife 2016, 5, e21635. [Google Scholar] [CrossRef]
- Heybrock, S.; Kanerva, K.; Meng, Y.; Ing, C.; Liang, A.; Xiong, Z.J.; Weng, X.; Ah Kim, Y.; Collins, R.; Trimble, W.; et al. Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export. Nat. Commun. 2019, 10, 3521. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schroder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Flannery, A.R.; Czibener, C.; Andrews, N.W. Palmitoylation-dependent association with CD63 targets the Ca2+ sensor synaptotagmin VII to lysosomes. J. Cell Biol. 2010, 191, 599–613. [Google Scholar] [CrossRef]
- Enrich, C.; Rentero, C.; Grewal, T.; Futter, C.E.; Eden, E.R. Cholesterol Overload: Contact Sites to the Rescue! Contact 2019, 2, 2515256419893507. [Google Scholar] [CrossRef] [PubMed]
- Hoglinger, D.; Burgoyne, T.; Sanchez-Heras, E.; Hartwig, P.; Colaco, A.; Newton, J.; Futter, C.E.; Spiegel, S.; Platt, F.M.; Eden, E.R. NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 2019, 10, 4276. [Google Scholar] [CrossRef]
- Ohkuma, S.; Moriyama, Y.; Takano, T. Identification and characterization of a proton pump on lysosomes by fluorescein-isothiocyanate-dextran fluorescence. Proc. Natl. Acad. Sci. USA 1982, 79, 2758–2762. [Google Scholar] [CrossRef] [PubMed]
- Hinton, A.; Bond, S.; Forgac, M. V-ATPase functions in normal and disease processes. Pflugers Arch. 2009, 457, 589–598. [Google Scholar] [CrossRef]
- Peters, C.; Bayer, M.J.; Buhler, S.; Andersen, J.S.; Mann, M.; Mayer, A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature 2001, 409, 581–588. [Google Scholar] [CrossRef]
- Hiesinger, P.R.; Fayyazuddin, A.; Mehta, S.Q.; Rosenmund, T.; Schulze, K.L.; Zhai, R.G.; Verstreken, P.; Cao, Y.; Zhou, Y.; Kunz, J.; et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 2005, 121, 607–620. [Google Scholar] [CrossRef]
- Rudnik, S.; Damme, M. The lysosomal membrane-export of metabolites and beyond. FEBS J. 2021, 288, 4168–4182. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Neefjes, J.; Berlin, I. The journey of Ca(2+) through the cell—Pulsing through the network of ER membrane contact sites. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnoczky, G. Intracellular Ca(2+) Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Huang, P.; Dong, X.P. Lysosomal Calcium Channels in Autophagy and Cancer. Cancers 2021, 13, 1299. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Cai, X. Evolution of acidic Ca(2)(+) stores and their resident Ca(2)(+)-permeable channels. Cell Calcium 2015, 57, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, S.; Scotto-Rosato, A.; Medina, D.L. TRPML1: The Ca((2+))retaker of the lysosome. Cell Calcium 2018, 69, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Harnett, M.M.; Pineda, M.A.; Latre de Late, P.; Eason, R.J.; Besteiro, S.; Harnett, W.; Langsley, G. From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. Biomed. J. 2017, 40, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, P.; Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the switch in early-to-late endosome transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, E.; Tang, L.; Lei, Y.; Sun, X.; Hu, J.; Dong, H.; Yang, S.M.; Gao, M.; Tang, B. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun. Signal. 2018, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, L.; Lotfi, P.; Pal, R.; Ronza, A.D.; Sharma, J.; Sardiello, M. Lysosome biogenesis in health and disease. J. Neurochem. 2019, 148, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef]
- Shin, H.R.; Zoncu, R. The Lysosome at the Intersection of Cellular Growth and Destruction. Dev. Cell 2020, 54, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Franco-Juarez, B.; Coronel-Cruz, C.; Hernandez-Ochoa, B.; Gomez-Manzo, S.; Cardenas-Rodriguez, N.; Arreguin-Espinosa, R.; Bandala, C.; Canseco-Avila, L.M.; Ortega-Cuellar, D. TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator. Cells 2022, 11, 3153. [Google Scholar] [CrossRef]
- Martina, J.A.; Puertollano, R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem. 2018, 293, 12525–12534. [Google Scholar] [CrossRef]
- Annunziata, I.; van de Vlekkert, D.; Wolf, E.; Finkelstein, D.; Neale, G.; Machado, E.; Mosca, R.; Campos, Y.; Tillman, H.; Roussel, M.F.; et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat. Commun. 2019, 10, 3623. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.; Kravic, B.; Meyer, H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J. Mol. Biol. 2020, 432, 231–239. [Google Scholar] [CrossRef]
- Repnik, U.; Hafner Cesen, M.; Turk, B. Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion 2014, 19 Pt A, 49–57. [Google Scholar] [CrossRef]
- Oku, Y.; Murakami, K.; Irie, K.; Hoseki, J.; Sakai, Y. Synthesized Abeta42 Caused Intracellular Oxidative Damage, Leading to Cell Death, via Lysosome Rupture. Cell Struct. Funct. 2017, 42, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lonborg, A.; et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem. Soc. Trans. 2018, 46, 207–215. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T. Lysosomal iron, iron chelation, and cell death. Antioxid. Redox Signal. 2013, 18, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Villamil Giraldo, A.M.; Appelqvist, H.; Ederth, T.; Ollinger, K. Lysosomotropic agents: Impact on lysosomal membrane permeabilization and cell death. Biochem. Soc. Trans. 2014, 42, 1460–1464. [Google Scholar] [CrossRef]
- Bernheimer, A.W.; Schwartz, L.L. Lysosomal disruption by bacterial toxins. J. Bacteriol. 1964, 87, 1100–1104. [Google Scholar] [CrossRef]
- Helenius, A. Virus entry: What has pH got to do with it? Nat. Cell Biol. 2013, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Bivik, C.A.; Larsson, P.K.; Kagedal, K.M.; Rosdahl, I.K.; Ollinger, K.M. UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members. J. Investig. Dermatol. 2006, 126, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Andreau, K.; Poncet, D.; Zamzami, N.; Perfettini, J.L.; Metivier, D.; Ojcius, D.M.; Jaattela, M.; Kroemer, G. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J. Exp. Med. 2003, 197, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Steen, H.; Ollinger, K.; Roberg, K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003, 10, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Roberg, K. Relocalization of cathepsin D and cytochrome c early in apoptosis revealed by immunoelectron microscopy. Lab. Investig. 2001, 81, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.A.; Marriott, H.M.; Tulone, C.; Francis, S.E.; Mitchell, T.J.; Read, R.C.; Chain, B.; Kroemer, G.; Whyte, M.K.; Dockrell, D.H. A cardinal role for cathepsin d in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog. 2011, 7, e1001262. [Google Scholar] [CrossRef] [PubMed]
- Conus, S.; Perozzo, R.; Reinheckel, T.; Peters, C.; Scapozza, L.; Yousefi, S.; Simon, H.U. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J. Exp. Med. 2008, 205, 685–698. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef]
- Roberg, K.; Kagedal, K.; Ollinger, K. Microinjection of cathepsin d induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 2002, 161, 89–96. [Google Scholar] [CrossRef]
- Appelqvist, H.; Johansson, A.C.; Linderoth, E.; Johansson, U.; Antonsson, B.; Steinfeld, R.; Kagedal, K.; Ollinger, K. Lysosome-mediated apoptosis is associated with cathepsin D-specific processing of bid at Phe24, Trp48, and Phe183. Ann. Clin. Lab. Sci. 2012, 42, 231–242. [Google Scholar]
- Blomgran, R.; Zheng, L.; Stendahl, O. Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leukoc. Biol. 2007, 81, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Cirman, T.; Oresic, K.; Mazovec, G.D.; Turk, V.; Reed, J.C.; Myers, R.M.; Salvesen, G.S.; Turk, B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 2004, 279, 3578–3587. [Google Scholar] [CrossRef]
- Huai, J.; Vogtle, F.N.; Jockel, L.; Li, Y.; Kiefer, T.; Ricci, J.E.; Borner, C. TNFalpha-induced lysosomal membrane permeability is downstream of MOMP and triggered by caspase-mediated NDUFS1 cleavage and ROS formation. J. Cell Sci. 2013, 126, 4015–4025. [Google Scholar] [CrossRef]
- Oberle, C.; Huai, J.; Reinheckel, T.; Tacke, M.; Rassner, M.; Ekert, P.G.; Buellesbach, J.; Borner, C. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010, 17, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Yashin, D.V.; Romanova, E.A.; Ivanova, O.K.; Sashchenko, L.P. The Tag7-Hsp70 cytotoxic complex induces tumor cell necroptosis via permeabilisation of lysosomes and mitochondria. Biochimie 2016, 123, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Perez, P.; Sun, X.; Chen, K.; Fatirkhorani, R.; Mammadova, J.; Wang, Z. MLKL polymerization-induced lysosomal membrane permeabilization promotes necroptosis. Cell Death Differ. 2024, 31, 40–52. [Google Scholar] [CrossRef]
- Campden, R.I.; Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of cell death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H., Jr.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Henkel, L.; Linder, B.; Zielke, S.; Tascher, G.; Trautmann, S.; Geisslinger, G.; Munch, C.; Fulda, S.; Tegeder, I.; et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy 2021, 17, 3424–3443. [Google Scholar] [CrossRef] [PubMed]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, M.L.; Berlin, I.; Wijdeven, R.H.; Janssen, L.; Janssen, G.M.; Garstka, M.A.; Janssen, H.; Mensink, M.; van Veelen, P.A.; Spaapen, R.M.; et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell 2016, 166, 152–166. [Google Scholar] [CrossRef]
- Ba, Q.; Raghavan, G.; Kiselyov, K.; Yang, G. Whole-Cell Scale Dynamic Organization of Lysosomes Revealed by Spatial Statistical Analysis. Cell Rep. 2018, 23, 3591–3606. [Google Scholar] [CrossRef]
- Ferguson, S.M. Neuronal lysosomes. Neurosci. Lett. 2019, 697, 1–9. [Google Scholar] [CrossRef]
- Johnson, D.E.; Ostrowski, P.; Jaumouille, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef]
- Encarnacao, M.; Espada, L.; Escrevente, C.; Mateus, D.; Ramalho, J.; Michelet, X.; Santarino, I.; Hsu, V.W.; Brenner, M.B.; Barral, D.C.; et al. A Rab3a-dependent complex essential for lysosome positioning and plasma membrane repair. J. Cell Biol. 2016, 213, 631–640. [Google Scholar] [CrossRef]
- Bright, N.A.; Davis, L.J.; Luzio, J.P. Endolysosomes Are the Principal Intracellular Sites of Acid Hydrolase Activity. Curr. Biol. 2016, 26, 2233–2245. [Google Scholar] [CrossRef]
- Hong, Z.; Pedersen, N.M.; Wang, L.; Torgersen, M.L.; Stenmark, H.; Raiborg, C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J. Cell Biol. 2017, 216, 4217–4233. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef]
- Oyarzun, J.E.; Lagos, J.; Vazquez, M.C.; Valls, C.; De la Fuente, C.; Yuseff, M.I.; Alvarez, A.R.; Zanlungo, S. Lysosome motility and distribution: Relevance in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Takei, Y.; Kanai, Y.; Tanaka, Y.; Nonaka, S.; Hirokawa, N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 1998, 141, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J.; Swanson, J.A. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature 1990, 346, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Neefjes, J. Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 2017, 47, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Deitch, J.S.; Black, M.M.; Banker, G.A. Polarity orientation of microtubules in hippocampal neurons: Uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA 1988, 85, 8335–8339. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Schatzle, P.; Tortosa, E.; Pages, S.; Holtmaat, A.; Kapitein, L.C.; Hoogenraad, C.C. Dendrites In Vitro and In Vivo Contain Microtubules of Opposite Polarity and Axon Formation Correlates with Uniform Plus-End-Out Microtubule Orientation. J. Neurosci. 2016, 36, 1071–1085. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Marx, A.; Hoenger, A.; Mandelkow, E. Structures of kinesin motor proteins. Cell Motil. Cytoskelet. 2009, 66, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T.; Hirokawa, N. Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J. Cell Biol. 1995, 131, 1039–1053. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kanai, Y.; Okada, Y.; Nonaka, S.; Takeda, S.; Harada, A.; Hirokawa, N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 1998, 93, 1147–1158. [Google Scholar] [CrossRef]
- Rosa-Ferreira, C.; Munro, S. Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev. Cell 2011, 21, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.L.; Maier, K.C.; Stauber, T.; Ginkel, L.M.; Wordeman, L.; Vernos, I.; Schroer, T.A. Kinesin-2 is a motor for late endosomes and lysosomes. Traffic 2005, 6, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Tanaka, S.; Nakamura, N.; Inoue, H.; Kanazawa, H. A novel kinesin-like protein, KIF1Bbeta3 is involved in the movement of lysosomes to the cell periphery in non-neuronal cells. Traffic 2004, 5, 140–151. [Google Scholar] [CrossRef]
- Bentley, M.; Decker, H.; Luisi, J.; Banker, G. A novel assay reveals preferential binding between Rabs, kinesins, and specific endosomal subpopulations. J. Cell Biol. 2015, 208, 273–281. [Google Scholar] [CrossRef]
- Santama, N.; Krijnse-Locker, J.; Griffiths, G.; Noda, Y.; Hirokawa, N.; Dotti, C.G. KIF2beta, a new kinesin superfamily protein in non-neuronal cells, is associated with lysosomes and may be implicated in their centrifugal translocation. EMBO J. 1998, 17, 5855–5867. [Google Scholar] [CrossRef]
- Guardia, C.M.; Farias, G.G.; Jia, R.; Pu, J.; Bonifacino, J.S. BORC Functions Upstream of Kinesins 1 and 3 to Coordinate Regional Movement of Lysosomes along Different Microtubule Tracks. Cell Rep. 2016, 17, 1950–1961. [Google Scholar] [CrossRef]
- Huang, C.F.; Banker, G. The translocation selectivity of the kinesins that mediate neuronal organelle transport. Traffic 2012, 13, 549–564. [Google Scholar] [CrossRef]
- Lipka, J.; Kapitein, L.C.; Jaworski, J.; Hoogenraad, C.C. Microtubule-binding protein doublecortin-like kinase 1 (DCLK1) guides kinesin-3-mediated cargo transport to dendrites. EMBO J. 2016, 35, 302–318. [Google Scholar] [CrossRef]
- Nakata, T.; Hirokawa, N. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol. 2003, 162, 1045–1055. [Google Scholar] [CrossRef]
- Farias, G.G.; Guardia, C.M.; De Pace, R.; Britt, D.J.; Bonifacino, J.S. BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci. USA 2017, 114, E2955–E2964. [Google Scholar] [CrossRef]
- Pu, J.; Schindler, C.; Jia, R.; Jarnik, M.; Backlund, P.; Bonifacino, J.S. BORC, a multisubunit complex that regulates lysosome positioning. Dev. Cell 2015, 33, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Wenzel, E.M.; Pedersen, N.M.; Olsvik, H.; Schink, K.O.; Schultz, S.W.; Vietri, M.; Nisi, V.; Bucci, C.; Brech, A.; et al. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 2015, 520, 234–238. [Google Scholar] [CrossRef]
- Roberts, A.J. Emerging mechanisms of dynein transport in the cytoplasm versus the cilium. Biochem. Soc. Trans. 2018, 46, 967–982. [Google Scholar] [CrossRef]
- Lin, S.X.; Collins, C.A. Immunolocalization of cytoplasmic dynein to lysosomes in cultured cells. J. Cell Sci. 1992, 101 Pt 1, 125–137. [Google Scholar] [CrossRef]
- Gill, S.R.; Schroer, T.A.; Szilak, I.; Steuer, E.R.; Sheetz, M.P.; Cleveland, D.W. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol. 1991, 115, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Schroer, T.A.; Sheetz, M.P. Two activators of microtubule-based vesicle transport. J. Cell Biol. 1991, 115, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef] [PubMed]
- Willett, R.; Martina, J.A.; Zewe, J.P.; Wills, R.; Hammond, G.R.V.; Puertollano, R. TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat. Commun. 2017, 8, 1580. [Google Scholar] [CrossRef] [PubMed]
- Takemasu, S.; Nigorikawa, K.; Yamada, M.; Tsurumi, G.; Kofuji, S.; Takasuga, S.; Hazeki, K. Phosphorylation of TMEM55B by Erk/MAPK regulates lysosomal positioning. J. Biochem. 2019, 166, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Murray, J.W.; Wang, X.; Pampliega, O.; Yin, D.; Patel, B.; Yuste, A.; Wolkoff, A.W.; Cuervo, A.M. Defective recruitment of motor proteins to autophagic compartments contributes to autophagic failure in aging. Aging Cell 2018, 17, e12777. [Google Scholar] [CrossRef]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef]
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321. [Google Scholar] [CrossRef]
- Griffiths, G.M. Secretory lysosomes—A special mechanism of regulated secretion in haemopoietic cells. Trends Cell Biol. 1996, 6, 329–332. [Google Scholar] [CrossRef]
- Andrews, N.W. Solving the secretory acid sphingomyelinase puzzle: Insights from lysosome-mediated parasite invasion and plasma membrane repair. Cell. Microbiol. 2019, 21, e13065. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Liu, J.; Ren, L.; Li, S.; Li, W.; Zheng, X.; Yang, Y.; Fu, W.; Yi, J.; Wang, J.; Du, G. The biology, function, and applications of exosomes in cancer. Acta Pharm. Sin. B 2021, 11, 2783–2797. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Gronborg, M.; Mobius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Hoshino, D.; Hong, N.H.; Kirkbride, K.C.; Grega-Larson, N.E.; Seiki, M.; Tyska, M.J.; Weaver, A.M. Cortactin promotes exosome secretion by controlling branched actin dynamics. J. Cell Biol. 2016, 214, 197–213. [Google Scholar] [CrossRef]
- Hyenne, V.; Apaydin, A.; Rodriguez, D.; Spiegelhalter, C.; Hoff-Yoessle, S.; Diem, M.; Tak, S.; Lefebvre, O.; Schwab, Y.; Goetz, J.G.; et al. RAL-1 controls multivesicular body biogenesis and exosome secretion. J. Cell Biol. 2015, 211, 27–37. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Pezzicoli, G.; Tucci, M.; Lovero, D.; Silvestris, F.; Porta, C.; Mannavola, F. Large Extracellular Vesicles-A New Frontier of Liquid Biopsy in Oncology. Int. J. Mol. Sci. 2020, 21, 6543. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015, 25, 24–38. [Google Scholar] [CrossRef]
- Piccin, A.; Murphy, W.G.; Smith, O.P. Circulating microparticles: Pathophysiology and clinical implications. Blood Rev. 2007, 21, 157–171. [Google Scholar] [CrossRef]
- Cadwell, K.; Debnath, J. Beyond self-eating: The control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol. 2018, 217, 813–822. [Google Scholar] [CrossRef]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef]
- Tancini, B.; Buratta, S.; Delo, F.; Sagini, K.; Chiaradia, E.; Pellegrino, R.M.; Emiliani, C.; Urbanelli, L. Lysosomal Exocytosis: The Extracellular Role of an Intracellular Organelle. Membranes 2020, 10, 406. [Google Scholar] [CrossRef]
- Miranda, A.M.; Lasiecka, Z.M.; Xu, Y.; Neufeld, J.; Shahriar, S.; Simoes, S.; Chan, R.B.; Oliveira, T.G.; Small, S.A.; Di Paolo, G. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat. Commun. 2018, 9, 291. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, G.; Zhou, W.; Song, A.; Xu, T.; Luo, Q.; Wang, W.; Gu, X.S.; Duan, S. Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol. 2007, 9, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gu, Y.; Wen, R.; Shen, F.; Tian, H.L.; Yang, G.Y.; Zhang, Z. Lysosome exocytosis is involved in astrocyte ATP release after oxidative stress induced by H(2)O(2). Neurosci. Lett. 2019, 705, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, X.; Gao, Q.; Ali Samie, M.; Azar, M.; Tsang, W.L.; Dong, L.; Sahoo, N.; Li, X.; Zhuo, Y.; et al. The intracellular Ca(2)(+) channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 2014, 20, 1187–1192. [Google Scholar] [CrossRef]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef]
- Sbano, L.; Bonora, M.; Marchi, S.; Baldassari, F.; Medina, D.L.; Ballabio, A.; Giorgi, C.; Pinton, P. TFEB-mediated increase in peripheral lysosomes regulates store-operated calcium entry. Sci. Rep. 2017, 7, 40797. [Google Scholar] [CrossRef]
- Martinez, I.; Chakrabarti, S.; Hellevik, T.; Morehead, J.; Fowler, K.; Andrews, N.W. Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J. Cell Biol. 2000, 148, 1141–1149. [Google Scholar] [CrossRef]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef]
- Andrews, N.W. Detection of Lysosomal Exocytosis by Surface Exposure of Lamp1 Luminal Epitopes. Methods Mol. Biol. 2017, 1594, 205–211. [Google Scholar] [CrossRef]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gomes, T.; Corrotte, M.; Tam, C.; Andrews, N.W. Plasma Membrane Repair Is Regulated Extracellularly by Proteases Released from Lysosomes. PLoS ONE 2016, 11, e0152583. [Google Scholar] [CrossRef] [PubMed]
- Lapaquette, P.; Ducreux, A.; Basmaciyan, L.; Paradis, T.; Bon, F.; Bataille, A.; Winckler, P.; Hube, B.; d’Enfert, C.; Esclatine, A.; et al. Membrane protective role of autophagic machinery during infection of epithelial cells by Candida albicans. Gut Microbes 2022, 14, 2004798. [Google Scholar] [CrossRef]
- Michelet, X.; Tuli, A.; Gan, H.; Geadas, C.; Sharma, M.; Remold, H.G.; Brenner, M.B. Lysosome-Mediated Plasma Membrane Repair Is Dependent on the Small GTPase Arl8b and Determines Cell Death Type in Mycobacterium tuberculosis Infection. J. Immunol. 2018, 200, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef]
- do Couto, N.F.; Pedersane, D.; Rezende, L.; Dias, P.P.; Corbani, T.L.; Bentini, L.C.; Oliveira, A.C.S.; Kelles, L.F.; Castro-Gomes, T.; Andrade, L.O. Correction: LAMP-2 absence interferes with plasma membrane repair and decreases T. cruzi host cell invasion. PLoS Negl. Trop. Dis. 2020, 14, e0008724. [Google Scholar] [CrossRef]
- Waster, P.; Eriksson, I.; Vainikka, L.; Rosdahl, I.; Ollinger, K. Extracellular vesicles are transferred from melanocytes to keratinocytes after UVA irradiation. Sci. Rep. 2016, 6, 27890. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, X.; Greene, K.S.; Si, H.; Antonyak, M.A.; Druso, J.E.; Wilson, K.F.; Cerione, R.A.; Feng, Q.; Wang, H. Cdc42 functions as a regulatory node for tumour-derived microvesicle biogenesis. J. Extracell. Vesicles 2021, 10, e12051. [Google Scholar] [CrossRef]
- Jin, Y.; Ma, L.; Zhang, W.; Yang, W.; Feng, Q.; Wang, H. Extracellular signals regulate the biogenesis of extracellular vesicles. Biol. Res. 2022, 55, 35. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Hamalisto, S.; Jaattela, M. Lysosomes in cancer-living on the edge (of the cell). Curr. Opin. Cell Biol. 2016, 39, 69–76. [Google Scholar] [CrossRef]
- Eriksson, I.; Vainikka, L.; Waster, P.; Ollinger, K. Lysosomal function and intracellular position determine the malignant phenotype in malignant melanoma. J. Investig. Dermatol. 2023, 143, 1769–1778.e12. [Google Scholar] [CrossRef] [PubMed]
- Assi, M.; Kimmelman, A.C. Impact of context-dependent autophagy states on tumor progression. Nat. Cancer 2023, 4, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Gavilan, E.; Sanchez-Aguayo, I.; Daza, P.; Ruano, D. GSK-3beta signaling determines autophagy activation in the breast tumor cell line MCF7 and inclusion formation in the non-tumor cell line MCF10A in response to proteasome inhibition. Cell Death Dis. 2013, 4, e572. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Perera, R.M.; Di Malta, C.; Ballabio, A. MiT/TFE Family of Transcription Factors, Lysosomes, and Cancer. Annu. Rev. Cancer Biol. 2019, 3, 203–222. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Kauffman, E.C.; Ricketts, C.J.; Rais-Bahrami, S.; Yang, Y.; Merino, M.J.; Bottaro, D.P.; Srinivasan, R.; Linehan, W.M. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol. 2014, 11, 465–475. [Google Scholar] [CrossRef]
- Malouf, G.G.; Su, X.; Yao, H.; Gao, J.; Xiong, L.; He, Q.; Comperat, E.; Couturier, J.; Molinie, V.; Escudier, B.; et al. Next-generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin-remodeling genes. Clin. Cancer Res. 2014, 20, 4129–4140. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Schepens, M.; Thijssen, J.; van Asseldonk, M.; van den Berg, E.; Bridge, J.; Schuuring, E.; Schoenmakers, E.F.; van Kessel, A.G. Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum. Mol. Genet. 2003, 12, 1661–1669. [Google Scholar] [CrossRef]
- Moller, K.; Sigurbjornsdottir, S.; Arnthorsson, A.O.; Pogenberg, V.; Dilshat, R.; Fock, V.; Brynjolfsdottir, S.H.; Bindesboll, C.; Bessadottir, M.; Ogmundsdottir, H.M.; et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci. Rep. 2019, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Hong, S.B.; Oh, H.; Valera, V.A.; Baba, M.; Schmidt, L.S.; Linehan, W.M. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS ONE 2010, 5, e15793. [Google Scholar] [CrossRef] [PubMed]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef]
- Singla, M.; Bhattacharyya, S. Autophagy as a potential therapeutic target during epithelial to mesenchymal transition in renal cell carcinoma: An in vitro study. Biomed. Pharmacother. 2017, 94, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ma, Y.; He, H.W.; Zhao, W.L.; Shao, R.G. SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy 2017, 13, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Janda, E.; Nevolo, M.; Lehmann, K.; Downward, J.; Beug, H.; Grieco, M. Raf plus TGFbeta-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene 2006, 25, 7117–7130. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front. Oncol. 2020, 10, 317. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; White, K.A.; Barber, D.L. Intracellular pH Regulates Cancer and Stem Cell Behaviors: A Protein Dynamics Perspective. Front. Oncol. 2020, 10, 1401. [Google Scholar] [CrossRef]
- Chen, R.; Jaattela, M.; Liu, B. Lysosome as a Central Hub for Rewiring PH Homeostasis in Tumors. Cancers 2020, 12, 2437. [Google Scholar] [CrossRef]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef]
- Heuser, J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J. Cell Biol. 1989, 108, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Guggino, S.E.; Solaiyappan, M.; Pathak, A.P.; Ichikawa, Y.; Bhujwalla, Z.M. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia 2003, 5, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updat. 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; Gomez-Casal, R.; Cruz-Rangel, S.; Villanueva, H.; Sikora, A.G.; Rajagopalan, P.; Basu, D.; Pacheco, J.; Hammond, G.R.V.; Kiselyov, K.; et al. Lysosomal inhibition sensitizes TMEM16A-expressing cancer cells to chemotherapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2100670119. [Google Scholar] [CrossRef]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef]
- Herlevsen, M.; Oxford, G.; Owens, C.R.; Conaway, M.; Theodorescu, D. Depletion of major vault protein increases doxorubicin sensitivity and nuclear accumulation and disrupts its sequestration in lysosomes. Mol. Cancer Ther. 2007, 6, 1804–1813. [Google Scholar] [CrossRef]
- Groth-Pedersen, L.; Ostenfeld, M.S.; Hoyer-Hansen, M.; Nylandsted, J.; Jaattela, M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 2007, 67, 2217–2225. [Google Scholar] [CrossRef]
- Geisslinger, F.; Muller, M.; Vollmar, A.M.; Bartel, K. Targeting Lysosomes in Cancer as Promising Strategy to Overcome Chemoresistance-A Mini Review. Front. Oncol. 2020, 10, 1156. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Koch, R.; Radunski, U.; Corsham, S.; Cheong, N.; Inagaki, N.; Ban, N.; Wenzel, D.; Reinhardt, D.; Zapf, A.; et al. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia 2008, 22, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Rovithi, M.; de Haas, R.R.; Honeywell, R.J.; Dekker, H.; Poel, D.; Azijli, K.; Peters, G.J.; Broxterman, H.J.; Verheul, H.M. Cross-resistance to clinically used tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib. Cell. Oncol. 2015, 38, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Su, S.H.; Su, S.J.; Huang, L.Y.; Chiang, Y.C. Leukemic cells resist lysosomal inhibition through the mitochondria-dependent reduction of intracellular pH and oxidants. Free Radic. Biol. Med. 2023, 198, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, G.; Kim, H.; Song, Y.S.; Jung, J. Modulation of Cisplatin Sensitivity through TRPML1-Mediated Lysosomal Exocytosis in Ovarian Cancer Cells: A Comprehensive Metabolomic Approach. Cells 2024, 13, 115. [Google Scholar] [CrossRef]
- Machado, E.; White-Gilbertson, S.; van de Vlekkert, D.; Janke, L.; Moshiach, S.; Campos, Y.; Finkelstein, D.; Gomero, E.; Mosca, R.; Qiu, X.; et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 2015, 1, e1500603. [Google Scholar] [CrossRef]
- Hrabeta, J.; Belhajova, M.; Subrtova, H.; Merlos Rodrigo, M.A.; Heger, Z.; Eckschlager, T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. Int. J. Mol. Sci. 2020, 21, 4392. [Google Scholar] [CrossRef]
- Mlejnek, P. Lysosomal-mediated drug resistance—Fact or illusion? Pharmacol. Res. 2024, 199, 107025. [Google Scholar] [CrossRef]
- Mlejnek, P.; Havlasek, J.; Pastvova, N.; Dolezel, P.; Dostalova, K. Lysosomal sequestration of weak base drugs, lysosomal biogenesis, and cell cycle alteration. Biomed. Pharmacother. 2022, 153, 113328. [Google Scholar] [CrossRef] [PubMed]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.Y.; Shi, Y.Y.; Cui, X.W.; Pan, Y.F.; Lin, Y.K.; Feng, X.F.; Ding, Z.W.; Yang, C.; Tan, Y.X.; Dong, L.W.; et al. PTEN Deficiency Facilitates Exosome Secretion and Metastasis in Cholangiocarcinoma by Impairing TFEB-mediated Lysosome Biogenesis. Gastroenterology 2023, 164, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, R.; Zhai, P.; Liu, Z.; Xia, R.; Zhang, Z.; Qin, X.; Li, C.; Chen, W.; Li, J.; et al. Hypoxia promotes EV secretion by impairing lysosomal homeostasis in HNSCC through negative regulation of ATP6V1A by HIF-1alpha. J. Extracell. Vesicles 2023, 12, e12310. [Google Scholar] [CrossRef]
- Hikita, T.; Uehara, R.; Itoh, R.E.; Mitani, F.; Miyata, M.; Yoshida, T.; Yamaguchi, R.; Oneyama, C. MEK/ERK-mediated oncogenic signals promote secretion of extracellular vesicles by controlling lysosome function. Cancer Sci. 2022, 113, 1264–1276. [Google Scholar] [CrossRef]
- Latifkar, A.; Ling, L.; Hingorani, A.; Johansen, E.; Clement, A.; Zhang, X.; Hartman, J.; Fischbach, C.; Lin, H.; Cerione, R.A.; et al. Loss of Sirtuin 1 Alters the Secretome of Breast Cancer Cells by Impairing Lysosomal Integrity. Dev. Cell 2019, 49, 393–408.e7. [Google Scholar] [CrossRef]
- Latifkar, A.; Wang, F.; Mullmann, J.J.; Panizza, E.; Fernandez, I.R.; Ling, L.; Miller, A.D.; Fischbach, C.; Weiss, R.S.; Lin, H.; et al. IGF2BP2 promotes cancer progression by degrading the RNA transcript encoding a v-ATPase subunit. Proc. Natl. Acad. Sci. USA 2022, 119, e2200477119. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Waster, P.; Eriksson, I.; Vainikka, L.; Ollinger, K. Extracellular vesicles released by melanocytes after UVA irradiation promote intercellular signaling via miR21. Pigment Cell Melanoma Res. 2020, 33, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.V.; Narendradev, N.D.; Nambiar, R.P.; Kumar, R.; Srinivasula, S.M. Naturally occurring and tumor-associated variants of RNF167 promote lysosomal exocytosis and plasma membrane resealing. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; McCann, A.P.; Sereesongsaeng, N.; Burden, J.M.; Alsa’d, A.A.; Burden, R.E.; Micu, I.; Williams, R.; Van Schaeybroeck, S.; Evergren, E.; et al. USP17 is required for peripheral trafficking of lysosomes. EMBO Rep. 2022, 23, e51932. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Almasi, S.; Yang, Y.; Yan, C.; Sterea, A.M.; Rizvi Syeda, A.K.; Shen, B.; Richard Derek, C.; Huang, P.; Gujar, S.; et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium 2019, 79, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.D.; Yan, J.; Cao, K.Y.; Yin, Z.Q.; Xin, W.W.; Zhang, M.F. MCOLN1 Promotes Proliferation and Predicts Poor Survival of Patients with Pancreatic Ductal Adenocarcinoma. Dis. Markers 2019, 2019, 9436047. [Google Scholar] [CrossRef]
- Nguyen, O.N.; Grimm, C.; Schneider, L.S.; Chao, Y.K.; Atzberger, C.; Bartel, K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; et al. Two-Pore Channel Function Is Crucial for the Migration of Invasive Cancer Cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakayama, J.; Yamamoto, Y.; Kuroda, M.; Hattori, Y.; Ochiya, T. SORT1/LAMP2-mediated extracellular vesicle secretion and cell adhesion are linked to lenalidomide resistance in multiple myeloma. Blood Adv. 2022, 6, 2480–2495. [Google Scholar] [CrossRef]
- Nishimura, Y.; Sameni, M.; Sloane, B.F. Malignant transformation alters intracellular trafficking of lysosomal cathepsin D in human breast epithelial cells. Pathol. Oncol. Res. 1998, 4, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Sameni, M.; Elliott, E.; Ziegler, G.; Fortgens, P.H.; Dennison, C.; Sloane, B.F. Cathepsin B and D are Localized at the Surface of Human Breast Cancer Cells. Pathol. Oncol. Res. 1995, 1, 43–53. [Google Scholar] [CrossRef]
- Brix, D.M.; Rafn, B.; Bundgaard Clemmensen, K.; Andersen, S.H.; Ambartsumian, N.; Jaattela, M.; Kallunki, T. Screening and identification of small molecule inhibitors of ErbB2-induced invasion. Mol. Oncol. 2014, 8, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Rafn, B.; Nielsen, C.F.; Andersen, S.H.; Szyniarowski, P.; Corcelle-Termeau, E.; Valo, E.; Fehrenbacher, N.; Olsen, C.J.; Daugaard, M.; Egebjerg, C.; et al. ErbB2-driven breast cancer cell invasion depends on a complex signaling network activating myeloid zinc finger-1-dependent cathepsin B expression. Mol. Cell 2012, 45, 764–776. [Google Scholar] [CrossRef]
- Cavallo-Medved, D.; Dosescu, J.; Linebaugh, B.E.; Sameni, M.; Rudy, D.; Sloane, B.F. Mutant K-ras regulates cathepsin B localization on the surface of human colorectal carcinoma cells. Neoplasia 2003, 5, 507–519. [Google Scholar] [CrossRef]
- Saitoh, O.; Wang, W.C.; Lotan, R.; Fukuda, M. Differential glycosylation and cell surface expression of lysosomal membrane glycoproteins in sublines of a human colon cancer exhibiting distinct metastatic potentials. J. Biol. Chem. 1992, 267, 5700–5711. [Google Scholar] [CrossRef]
- Damaghi, M.; Tafreshi, N.K.; Lloyd, M.C.; Sprung, R.; Estrella, V.; Wojtkowiak, J.W.; Morse, D.L.; Koomen, J.M.; Bui, M.M.; Gatenby, R.A.; et al. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nat. Commun. 2015, 6, 8752. [Google Scholar] [CrossRef]
- Levicar, N.; Strojnik, T.; Kos, J.; Dewey, R.A.; Pilkington, G.J.; Lah, T.T. Lysosomal enzymes, cathepsins in brain tumour invasion. J. Neurooncol. 2002, 58, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Berquin, I.M.; Sloane, B.F. Cathepsin B expression in human tumors. Adv. Exp. Med. Biol. 1996, 389, 281–294. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Gavert, N.; Brabletz, T.; Haase, G.; Ben-Ze’ev, A. Increased expression of cathepsin D is required for L1-mediated colon cancer progression. Oncotarget 2019, 10, 5217–5228. [Google Scholar] [CrossRef] [PubMed]
- Eding, C.B.; Domert, J.; Waster, P.; Jerhammar, F.; Rosdahl, I.; Ollinger, K. Melanoma growth and progression after ultraviolet a irradiation: Impact of lysosomal exocytosis and cathepsin proteases. Acta Derm. Venereol. 2015, 95, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, K.; Lu, S.L.; Zhang, Z.; Kato, Y.; Chen, S.; Noda, K.; Hirose, K.; Usami, Y.; Uzawa, N.; Murakami, S.; et al. Rab32 and Rab38 maintain bone homeostasis by regulating intracellular traffic in osteoclasts. Cell Struct. Funct. 2023, 48, 223–239. [Google Scholar] [CrossRef]
- Cotter, K.; Capecci, J.; Sennoune, S.; Huss, M.; Maier, M.; Martinez-Zaguilan, R.; Forgac, M. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J. Biol. Chem. 2015, 290, 3680–3692. [Google Scholar] [CrossRef]
- Cai, M.; Liu, P.; Wei, L.; Wang, J.; Qi, J.; Feng, S.; Deng, L. Atp6v1c1 may regulate filament actin arrangement in breast cancer cells. PLoS ONE 2014, 9, e84833. [Google Scholar] [CrossRef]
- Kobayashi, H.; Schmitt, M.; Goretzki, L.; Chucholowski, N.; Calvete, J.; Kramer, M.; Gunzler, W.A.; Janicke, F.; Graeff, H. Cathepsin B efficiently activates the soluble and the tumor cell receptor-bound form of the proenzyme urokinase-type plasminogen activator (Pro-uPA). J. Biol. Chem. 1991, 266, 5147–5152. [Google Scholar] [CrossRef]
- Sameni, M.; Dosescu, J.; Moin, K.; Sloane, B.F. Functional imaging of proteolysis: Stromal and inflammatory cells increase tumor proteolysis. Mol. Imaging 2003, 2, 159–175. [Google Scholar] [CrossRef]
- Small, D.M.; Burden, R.E.; Jaworski, J.; Hegarty, S.M.; Spence, S.; Burrows, J.F.; McFarlane, C.; Kissenpfennig, A.; McCarthy, H.O.; Johnston, J.A.; et al. Cathepsin S from both tumor and tumor-associated cells promote cancer growth and neovascularization. Int. J. Cancer 2013, 133, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Chen, X.; Peters, C.; Reinheckel, T.; Joyce, J.A. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol. Chem. 2010, 391, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Waster, P.; Orfanidis, K.; Eriksson, I.; Rosdahl, I.; Seifert, O.; Ollinger, K. UV radiation promotes melanoma dissemination mediated by the sequential reaction axis of cathepsins-TGF-beta1-FAP-alpha. Br. J. Cancer 2017, 117, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Soikkeli, J.; Jahkola, T.; Virolainen, S.; Saksela, O.; Holtta, E. TGF-beta signaling, activated stromal fibroblasts, and cysteine cathepsins B and L drive the invasive growth of human melanoma cells. Am. J. Pathol. 2012, 181, 2202–2216. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Mathieu, P.A.; Linebaugh, B.; Sloane, B.F.; Reiners, J.J., Jr. Phorbol ester activation of a proteolytic cascade capable of activating latent transforming growth factor-betaL a process initiated by the exocytosis of cathepsin B. J. Biol. Chem. 2002, 277, 14829–14837. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Jin, X.; Hu, T.; Chi, F. LAPTM5 regulated by FOXP3 promotes the malignant phenotypes of breast cancer through activating the Wnt/beta-catenin pathway. Oncol. Rep. 2023, 49, 60. [Google Scholar] [CrossRef]
- Umeda, S.; Kanda, M.; Shimizu, D.; Nakamura, S.; Sawaki, K.; Inokawa, Y.; Hattori, N.; Hayashi, M.; Tanaka, C.; Nakayama, G.; et al. Lysosomal-associated membrane protein family member 5 promotes the metastatic potential of gastric cancer cells. Gastric Cancer 2022, 25, 558–572. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Xiong, Y.; Wang, W.; Fei, Y.; Tan, C.; Liang, Z. K-ras mutation promotes ionizing radiation-induced invasion and migration of lung cancer in part via the Cathepsin L/CUX1 pathway. Exp. Cell Res. 2018, 362, 424–435. [Google Scholar] [CrossRef]
- Mitrovic, A.; Pecar Fonovic, U.; Kos, J. Cysteine cathepsins B and X promote epithelial-mesenchymal transition of tumor cells. Eur. J. Cell Biol. 2017, 96, 622–631. [Google Scholar] [CrossRef]
- Han, M.L.; Zhao, Y.F.; Tan, C.H.; Xiong, Y.J.; Wang, W.J.; Wu, F.; Fei, Y.; Wang, L.; Liang, Z.Q. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol. Sin. 2016, 37, 1606–1622. [Google Scholar] [CrossRef]
- Wei, L.; Shao, N.; Peng, Y.; Zhou, P. Inhibition of Cathepsin S Restores TGF-beta-induced Epithelial-to-mesenchymal Transition and Tight Junction Turnover in Glioblastoma Cells. J. Cancer 2021, 12, 1592–1603. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, M.; Wang, W.; Song, Y.; Chen, G.; Wang, Z.; Liang, Z. Downregulation of cathepsin L suppresses cancer invasion and migration by inhibiting transforming growth factor-beta-mediated epithelial-mesenchymal transition. Oncol. Rep. 2015, 33, 1851–1859. [Google Scholar] [CrossRef]
- Dykes, S.S.; Gao, C.; Songock, W.K.; Bigelow, R.L.; Woude, G.V.; Bodily, J.M.; Cardelli, J.A. Zinc finger E-box binding homeobox-1 (Zeb1) drives anterograde lysosome trafficking and tumor cell invasion via upregulation of Na+/H+ Exchanger-1 (NHE1). Mol. Carcinog. 2017, 56, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Lohmer, L.L.; Kelley, L.C.; Hagedorn, E.J.; Sherwood, D.R. Invadopodia and basement membrane invasion in vivo. Cell Adhes. Migr. 2014, 8, 246–255. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef]
- Naegeli, K.M.; Hastie, E.; Garde, A.; Wang, Z.; Keeley, D.P.; Gordon, K.L.; Pani, A.M.; Kelley, L.C.; Morrissey, M.A.; Chi, Q.; et al. Cell Invasion In Vivo via Rapid Exocytosis of a Transient Lysosome-Derived Membrane Domain. Dev. Cell 2017, 43, 403–417.e10. [Google Scholar] [CrossRef]
- Dange, M.C.; Agarwal, A.K.; Kalraiya, R.D. Extracellular galectin-3 induces MMP9 expression by activating p38 MAPK pathway via lysosome-associated membrane protein-1 (LAMP1). Mol. Cell. Biochem. 2015, 404, 79–86. [Google Scholar] [CrossRef]
- Steffan, J.J.; Williams, B.C.; Welbourne, T.; Cardelli, J.A. HGF-induced invasion by prostate tumor cells requires anterograde lysosome trafficking and activity of Na+-H+ exchangers. J. Cell Sci. 2010, 123, 1151–1159. [Google Scholar] [CrossRef]
- Dykes, S.S.; Gray, A.L.; Coleman, D.T.; Saxena, M.; Stephens, C.A.; Carroll, J.L.; Pruitt, K.; Cardelli, J.A. The Arf-like GTPase Arl8b is essential for three-dimensional invasive growth of prostate cancer in vitro and xenograft formation and growth in vivo. Oncotarget 2016, 7, 31037–31052. [Google Scholar] [CrossRef]
- Jung, J.; Cho, K.J.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Verma, R.; Aggarwal, P.; Bischoff, M.E.; Reigle, J.; Secic, D.; Wetzel, C.; VandenHeuvel, K.; Biesiada, J.; Ehmer, B.; Landero Figueroa, J.A.; et al. Microtubule-associated protein MAP1LC3C regulates lysosomal exocytosis and induces zinc reprogramming in renal cancer cells. J. Biol. Chem. 2023, 299, 104663. [Google Scholar] [CrossRef]
- Cardoso, C.M.; Groth-Pedersen, L.; Hoyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jaattela, M.; Nylandsted, J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.H.; Onodera, Y.; Giaccia, A.J.; Le, Q.T.; Shimizu, S.; Shirato, H.; Nam, J.M. Lysosomal trafficking mediated by Arl8b and BORC promotes invasion of cancer cells that survive radiation. Commun. Biol. 2020, 3, 620. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.J.; Dykes, S.S.; Coleman, D.T.; Adams, L.K.; Rogers, D.; Carroll, J.L.; Williams, B.J.; Cardelli, J.A. Supporting a role for the GTPase Rab7 in prostate cancer progression. PLoS ONE 2014, 9, e87882. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Curbelo, D.; Riveiro-Falkenbach, E.; Perez-Guijarro, E.; Cifdaloz, M.; Karras, P.; Osterloh, L.; Megias, D.; Canon, E.; Calvo, T.G.; Olmeda, D.; et al. RAB7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 2014, 26, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Xia, K.; Zheng, D.; Gong, C.; Guo, W. RILP inhibits tumor progression in osteosarcoma via Grb10-mediated inhibition of the PI3K/AKT/mTOR pathway. Mol. Med. 2023, 29, 133. [Google Scholar] [CrossRef]
- Tzeng, H.T.; Wang, Y.C. Rab-mediated vesicle trafficking in cancer. J. Biomed. Sci. 2016, 23, 70. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sorensen, T.; Rafn, B.; Bottzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jaattela, M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [PubMed]
- Kima, P.E.; Burleigh, B.; Andrews, N.W. Surface-targeted lysosomal membrane glycoprotein-1 (Lamp-1) enhances lysosome exocytosis and cell invasion by Trypanosoma cruzi. Cell. Microbiol. 2000, 2, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Pezze, L.; Ciribilli, Y. LAMPs: Shedding light on cancer biology. Semin. Oncol. 2017, 44, 239–253. [Google Scholar] [CrossRef]
- Tian, Y.; Liang, L.; Chen, J.; Liu, J.; Su, Y.; Shi, M.; Li, W.; Zhang, J.; Feng, Y.; He, L.; et al. Knockdown LIMP2 inhibits colorectal cancer cells migration, invasion, and metastasis. Exp. Cell Res. 2023, 431, 113757. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, S.; Wang, S.; Yang, Q.; Wu, Z.; Zhang, M.; Chen, L.; Sun, Z. LIMP-2 enhances cancer stem-like cell properties by promoting autophagy-induced GSK3beta degradation in head and neck squamous cell carcinoma. Int. J. Oral Sci. 2023, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Maldonado, G.; Clark, J.; Burwinkel, M.; Greenslade, B.; Wunderlich, M.; Salomonis, N.; Leone, D.; Gatti, E.; Pierre, P.; Kumar, A.R.; et al. LAMP-5 is an essential inflammatory-signaling regulator and novel immunotherapy target for mixed lineage leukemia-rearranged acute leukemia. Haematologica 2022, 107, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.T.; Grzeskowiak, C.L.; Fradette, J.J.; Gibson, L.A.; Rodriguez, L.B.; Creighton, C.J.; Scott, K.L.; Gibbons, D.L. TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat. Commun. 2018, 9, 2731. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Gyrd-Hansen, M.; Poulsen, B.; Felbor, U.; Kallunki, T.; Boes, M.; Weber, E.; Leist, M.; Jaattela, M. Sensitization to the lysosomal cell death pathway upon immortalization and transformation. Cancer Res. 2004, 64, 5301–5310. [Google Scholar] [CrossRef]
- Groth-Pedersen, L.; Aits, S.; Corcelle-Termeau, E.; Petersen, N.H.; Nylandsted, J.; Jaattela, M. Identification of cytoskeleton-associated proteins essential for lysosomal stability and survival of human cancer cells. PLoS ONE 2012, 7, e45381. [Google Scholar] [CrossRef]
- Cao, Y.; Li, Y.; Liu, R.; Zhou, J.; Wang, K. Preclinical and Basic Research Strategies for Overcoming Resistance to Targeted Therapies in HER2-Positive Breast Cancer. Cancers 2023, 15, 2568. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.B.; Postol, M.; Tvingsholm, S.; Nielsen, I.O.; Dietrich, T.N.; Puustinen, P.; Maeda, K.; Dinant, C.; Strauss, R.; Egan, D.; et al. Identification of lysosome-targeting drugs with anti-inflammatory activity as potential invasion inhibitors of treatment resistant HER2 positive cancers. Cell. Oncol. 2021, 44, 805–820. [Google Scholar] [CrossRef] [PubMed]
- de Duve, C.; de Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; Van Hoof, F. Commentary. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- Stark, M.; Silva, T.F.D.; Levin, G.; Machuqueiro, M.; Assaraf, Y.G. The Lysosomotropic Activity of Hydrophobic Weak Base Drugs is Mediated via Their Intercalation into the Lysosomal Membrane. Cells 2020, 9, 1082. [Google Scholar] [CrossRef]
- Gallala, H.D.; Sandhoff, K. Biological function of the cellular lipid BMP-BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem. Res. 2011, 36, 1594–1600. [Google Scholar] [CrossRef]
- Petersen, N.H.; Kirkegaard, T.; Olsen, O.D.; Jaattela, M. Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle 2010, 9, 2305–2309. [Google Scholar] [CrossRef]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef]
- Teres, S.; Llado, V.; Higuera, M.; Barcelo-Coblijn, G.; Martin, M.L.; Noguera-Salva, M.A.; Marcilla-Etxenike, A.; Garcia-Verdugo, J.M.; Soriano-Navarro, M.; Saus, C.; et al. 2-Hydroxyoleate, a nontoxic membrane binding anticancer drug, induces glioma cell differentiation and autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, 8489–8494. [Google Scholar] [CrossRef]
- Barcelo-Coblijn, G.; Martin, M.L.; de Almeida, R.F.; Noguera-Salva, M.A.; Marcilla-Etxenike, A.; Guardiola-Serrano, F.; Luth, A.; Kleuser, B.; Halver, J.E.; Escriba, P.V. Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574. [Google Scholar] [CrossRef]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef]
- Granato, M.; Lacconi, V.; Peddis, M.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Santarelli, R.; Trivedi, P.; Frati, L.; D’Orazi, G.; et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013, 4, e730. [Google Scholar] [CrossRef]
- Leu, J.I.; Pimkina, J.; Pandey, P.; Murphy, M.E.; George, D.L. HSP70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol. Cancer Res. 2011, 9, 936–947. [Google Scholar] [CrossRef]
- Hu, M.; Carraway, K.L., 3rd. Repurposing Cationic Amphiphilic Drugs and Derivatives to Engage Lysosomal Cell Death in Cancer Treatment. Front. Oncol. 2020, 10, 605361. [Google Scholar] [CrossRef]
- Ellegaard, A.M.; Bach, P.; Jaattela, M. Targeting Cancer Lysosomes with Good Old Cationic Amphiphilic Drugs. Rev. Physiol. Biochem. Pharmacol. 2021, 185, 107–152. [Google Scholar] [CrossRef]
- Halliwell, W.H. Cationic amphiphilic drug-induced phospholipidosis. Toxicol. Pathol. 1997, 25, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Muhle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.; Cardelli, J.; Barr, M.P.; O’Byrne, K.; Mills, G.; El-Osta, H. Modulating lysosomal function through lysosome membrane permeabilization or autophagy suppression restores sensitivity to cisplatin in refractory non-small-cell lung cancer cells. PLoS ONE 2017, 12, e0184922. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.R.; Kim, J.H.; Woo, Y.H.; Kim, H.S.; Yoon, S. Anti-malarial Drugs Primaquine and Chloroquine Have Different Sensitization Effects with Anti-mitotic Drugs in Resistant Cancer Cells. Anticancer Res. 2016, 36, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Groth-Pedersen, L.; Jaattela, M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013, 332, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, A.M.; Dehlendorff, C.; Vind, A.C.; Anand, A.; Cederkvist, L.; Petersen, N.H.T.; Nylandsted, J.; Stenvang, J.; Mellemgaard, A.; Osterlind, K.; et al. Repurposing Cationic Amphiphilic Antihistamines for Cancer Treatment. EBioMedicine 2016, 9, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Allemailem, K.S.; Almatroudi, A.; Alrumaihi, F.; Almatroodi, S.A.; Alkurbi, M.O.; Basfar, G.T.; Rahmani, A.H.; Khan, A.A. Novel Approaches of Dysregulating Lysosome Functions in Cancer Cells by Specific Drugs and Its Nanoformulations: A Smart Approach of Modern Therapeutics. Int. J. Nanomed. 2021, 16, 5065–5098. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Xu, Y.D.; Ma, Y.Y.; Qiu, L.L.; Wang, Y.; Kong, J.L.; Xiong, H.M. Biodegradable ZnO@polymer core-shell nanocarriers: pH-triggered release of doxorubicin in vitro. Angew. Chem. Int. Ed. Engl. 2013, 52, 4127–4131. [Google Scholar] [CrossRef]
- Domenech, M.; Marrero-Berrios, I.; Torres-Lugo, M.; Rinaldi, C. Lysosomal membrane permeabilization by targeted magnetic nanoparticles in alternating magnetic fields. ACS Nano 2013, 7, 5091–5101. [Google Scholar] [CrossRef] [PubMed]
- Joris, F.; De Backer, L.; Van de Vyver, T.; Bastiancich, C.; De Smedt, S.C.; Raemdonck, K. Repurposing cationic amphiphilic drugs as adjuvants to induce lysosomal siRNA escape in nanogel transfected cells. J. Control. Release 2018, 269, 266–276. [Google Scholar] [CrossRef] [PubMed]
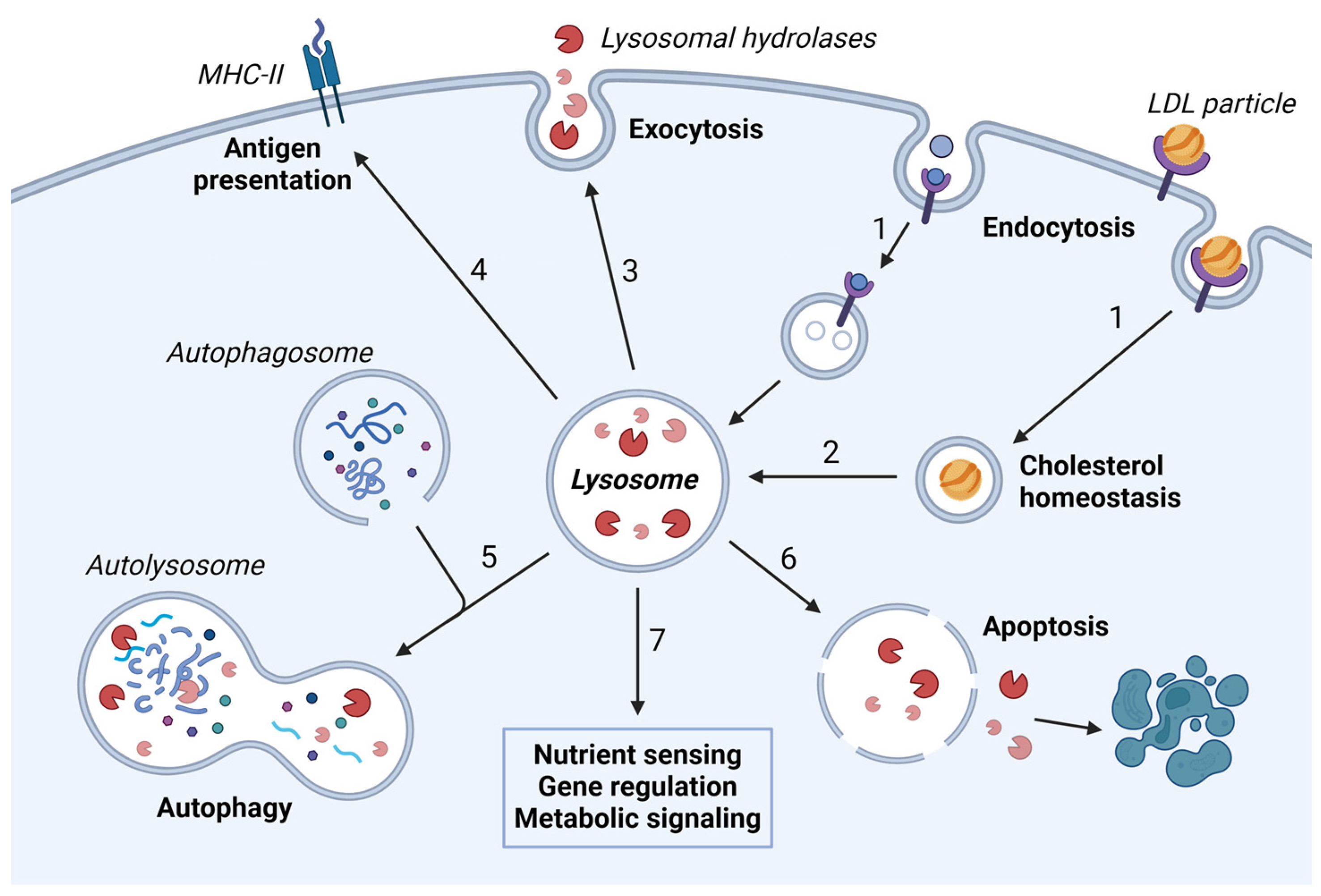
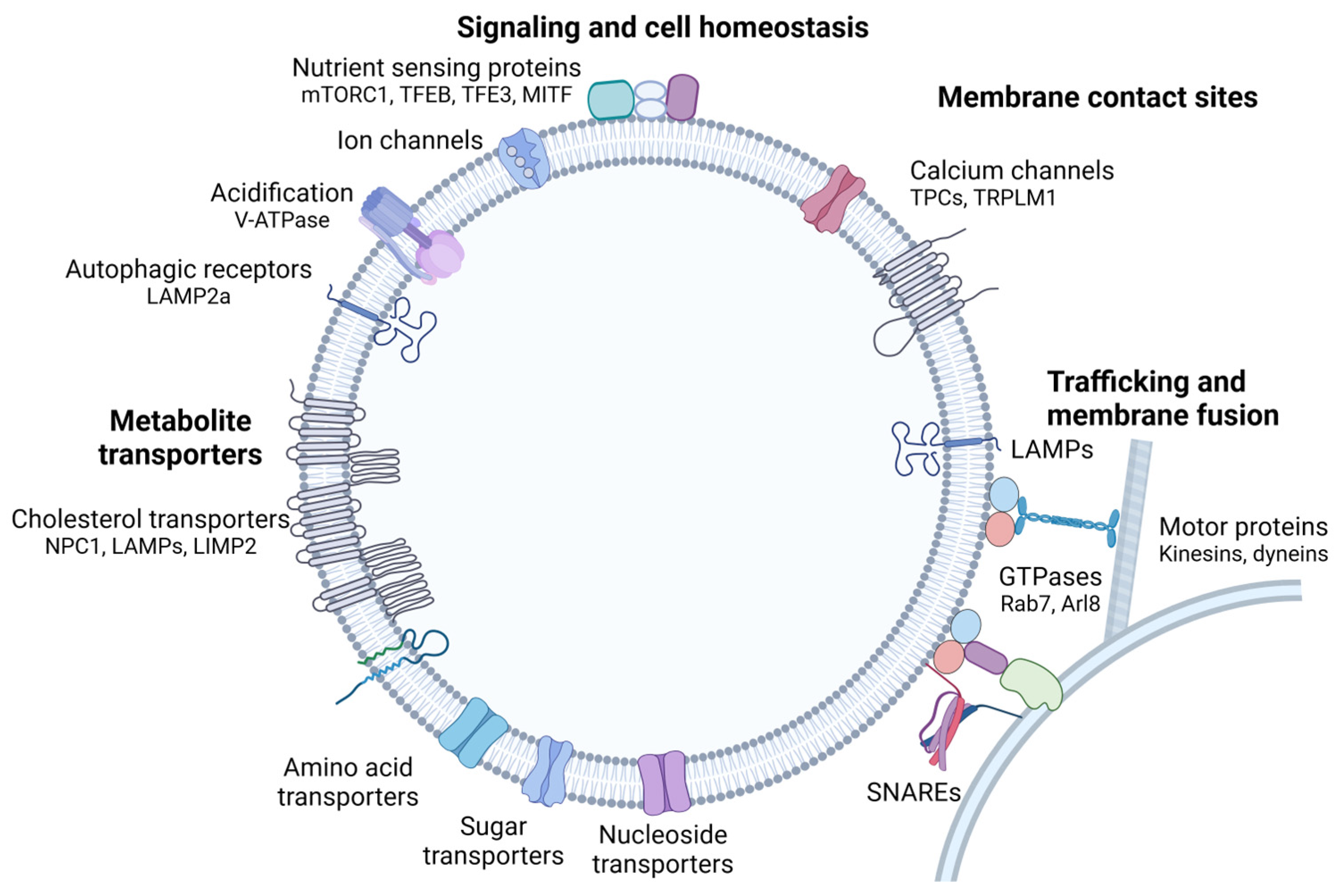


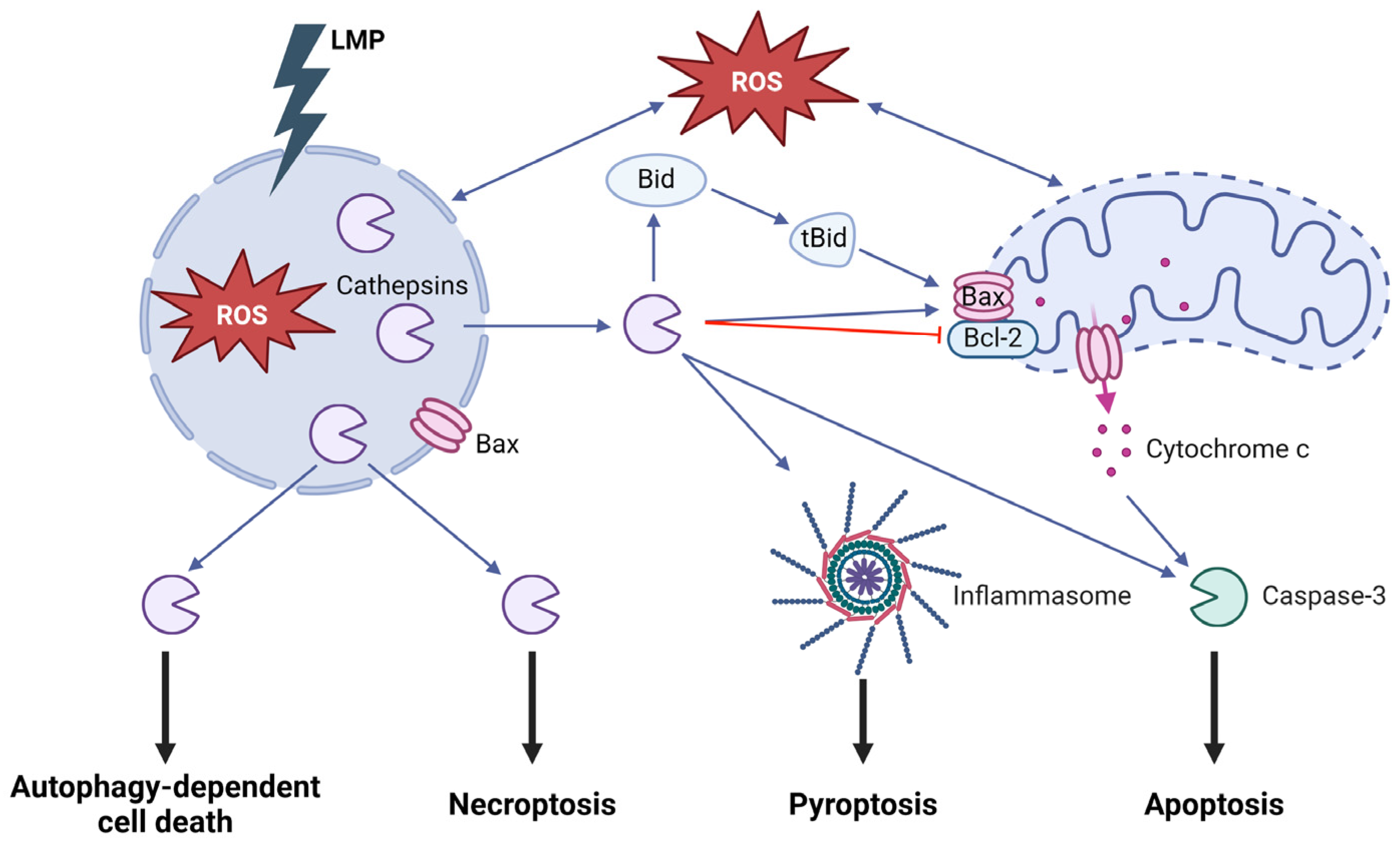
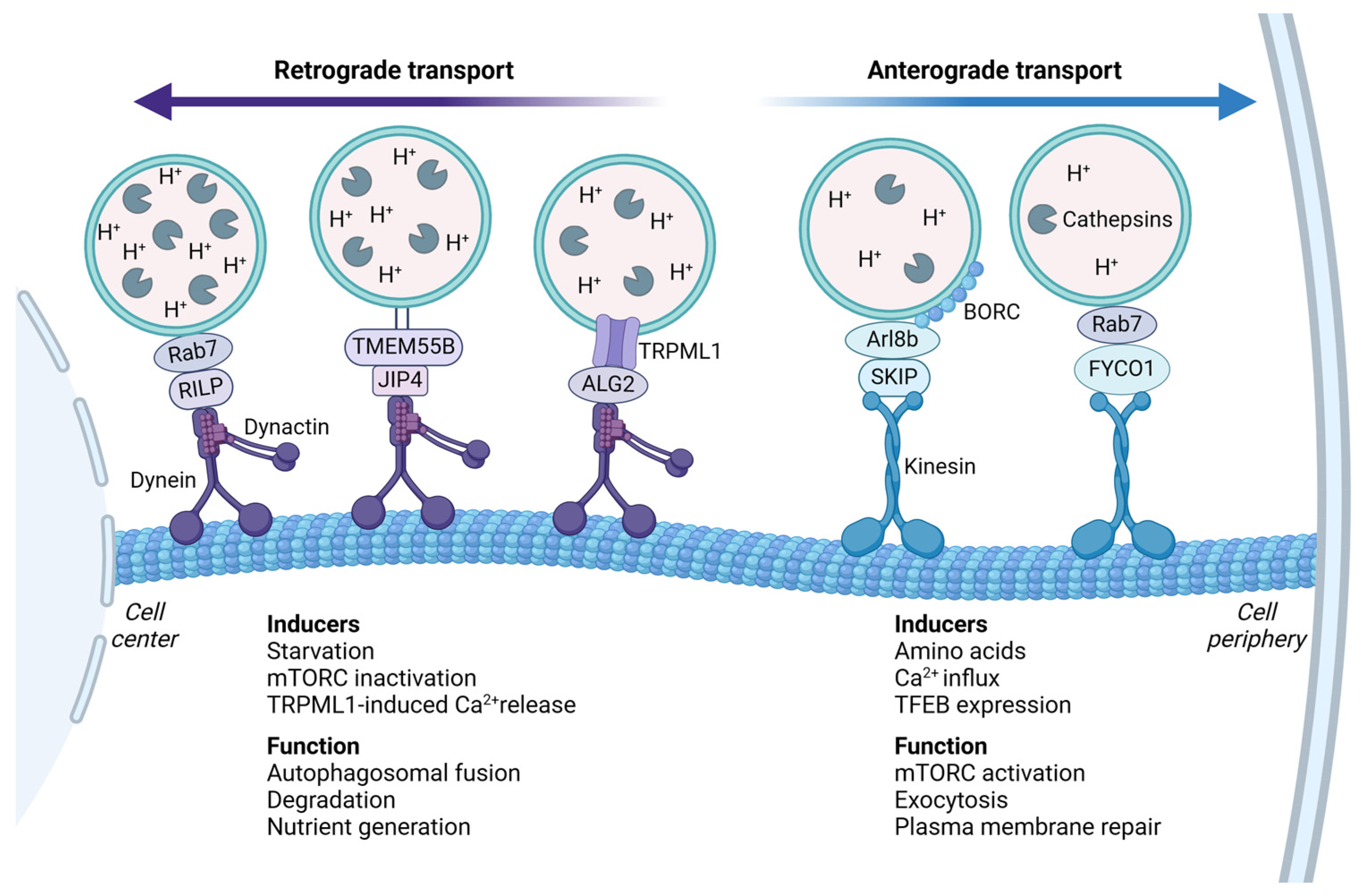
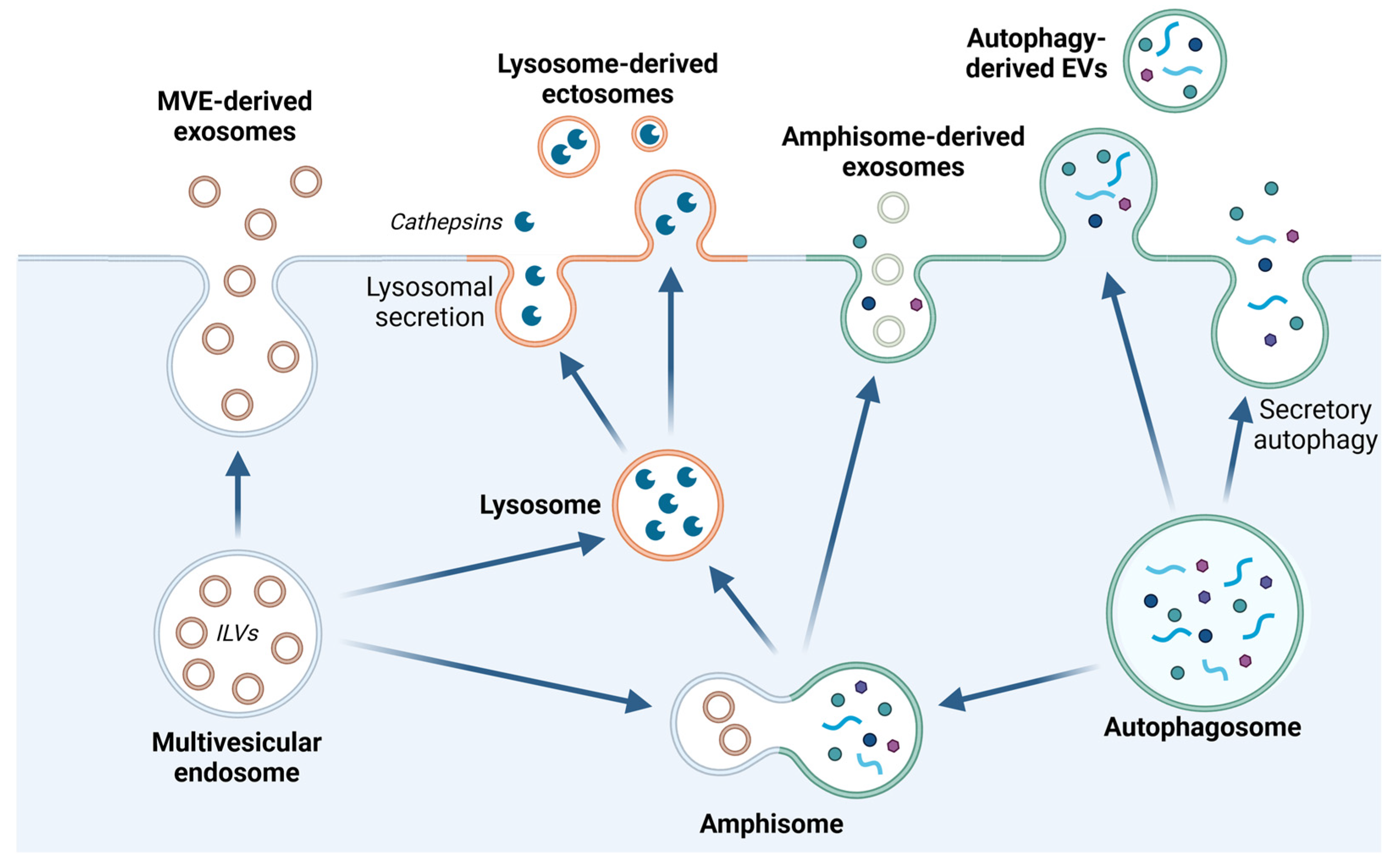
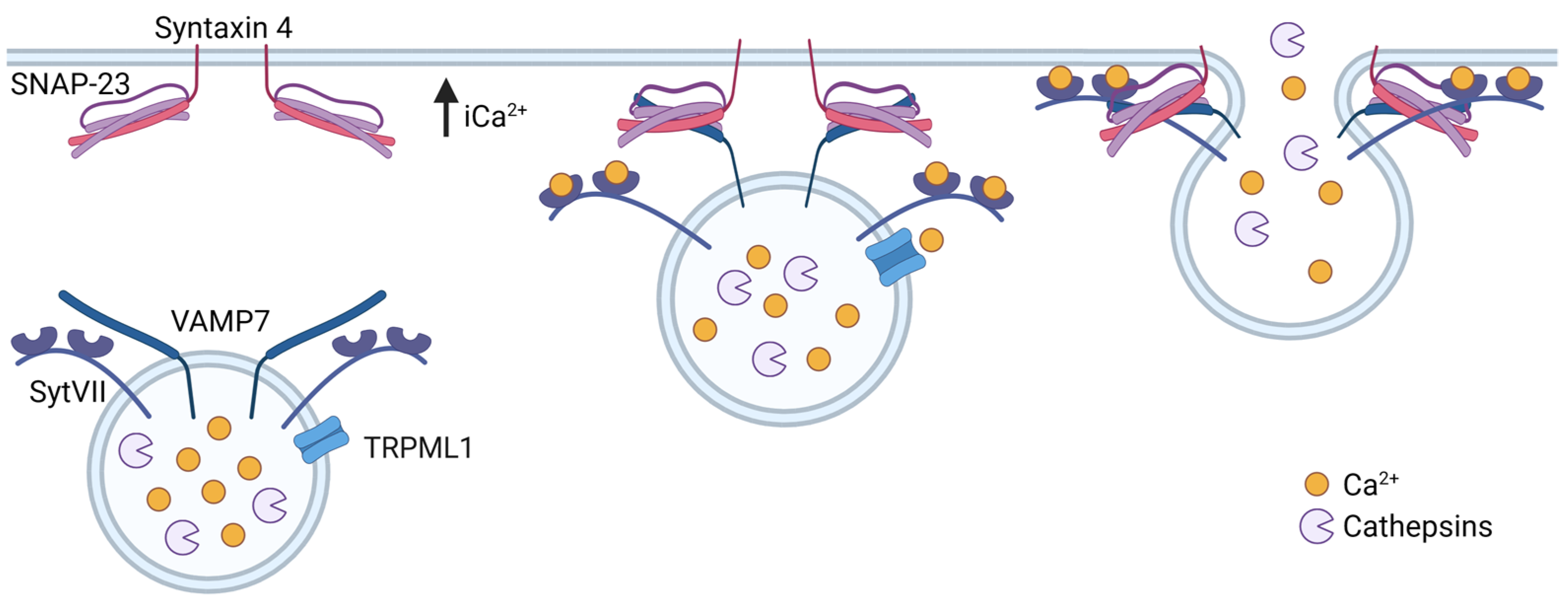


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eriksson, I.; Öllinger, K. Lysosomes in Cancer—At the Crossroad of Good and Evil. Cells 2024, 13, 459. https://doi.org/10.3390/cells13050459
Eriksson I, Öllinger K. Lysosomes in Cancer—At the Crossroad of Good and Evil. Cells. 2024; 13(5):459. https://doi.org/10.3390/cells13050459
Chicago/Turabian StyleEriksson, Ida, and Karin Öllinger. 2024. "Lysosomes in Cancer—At the Crossroad of Good and Evil" Cells 13, no. 5: 459. https://doi.org/10.3390/cells13050459
APA StyleEriksson, I., & Öllinger, K. (2024). Lysosomes in Cancer—At the Crossroad of Good and Evil. Cells, 13(5), 459. https://doi.org/10.3390/cells13050459