
High-Resolution Microscopic Characterization of Tunneling Nanotubes in Living U87 MG and LN229 Glioblastoma Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Labeling, and Fixation
2.2. Cell Irradiation
2.3. TNT Identification
2.4. STED and Confocal Microscopy
2.5. Image Analysis
2.6. Deconvolution and SFP Rendering
2.7. Diameter Measuring Using the Autocorrelation Analysis Method of Van Steensel et al.
2.8. Statistical Analysis
3. Results
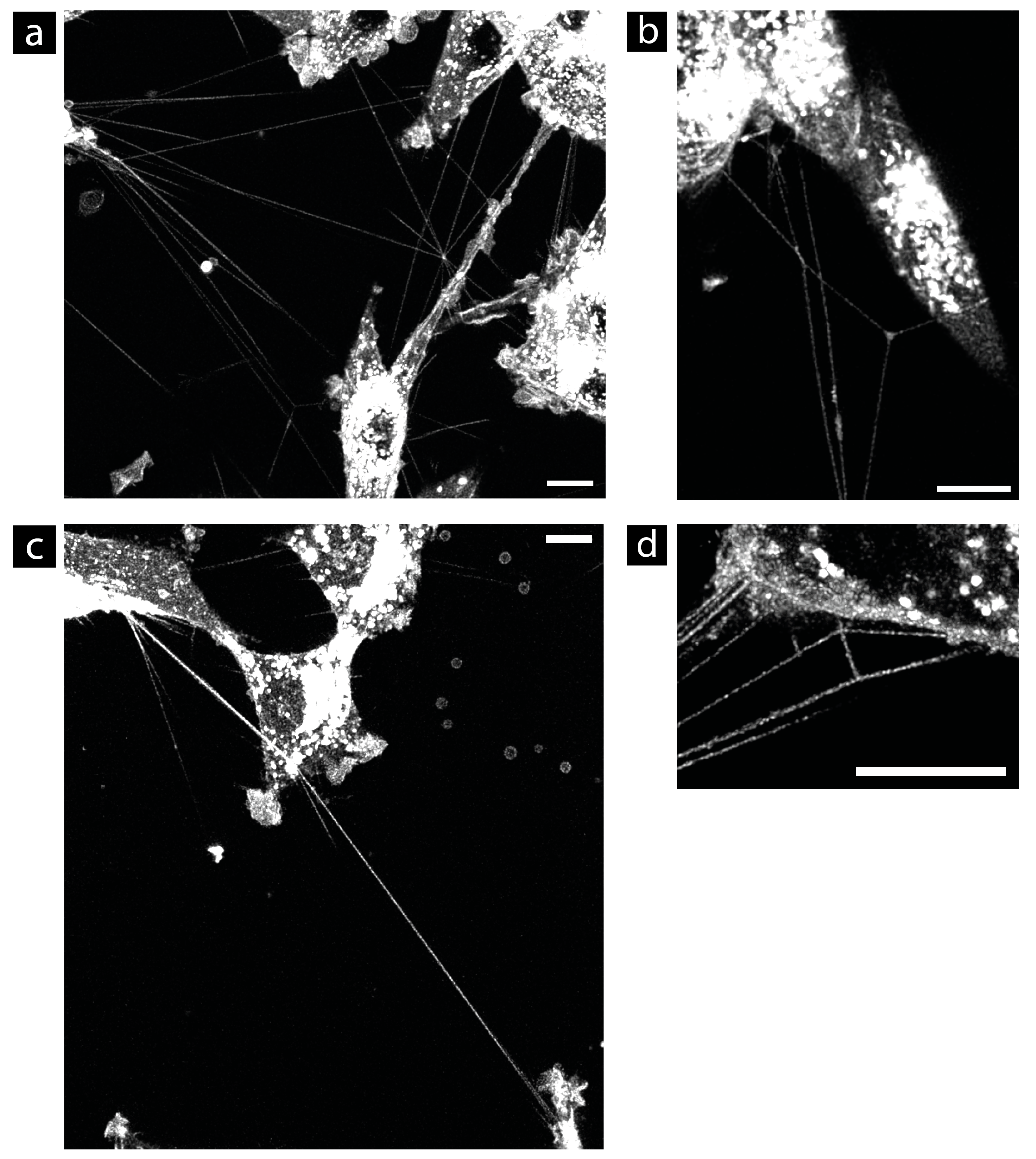
3.1. Suitable Fluorescent Membrane Marker for TNT Research
3.2. Size Measurement Using STED Microscopy
3.3. Analysis of Length, Lifetime, and Formation
3.4. Morphology
3.5. Stability during Fixation
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Diameter [nm] | Mean Length [µm] | Maximum Length [µm] | Lifetime | Reference |
|---|---|---|---|---|---|
| U87 MG | 195 ± 31 | 40 ± 27 | 189 | Mean (88 ± 60) min; max 360 min | Studied here |
| LN229 | 338 ± 91 | 20 ± 12 | 60 | Mean (41 ± 30) min; max 165 min | Studied here |
| PC12 | 50–200 | 15–60 | N.A. | <250 min | [7,62] |
| Jurkat T cells | 180–380 | 22 ± 3 | >100 | <1 h | [68] |
| ARPE-19 | 50–300 | 43.6 ± 18.1 | >120 | <1 h | [75] |
| HeLa | N.A. | 17.7 ± 8.3 | 40 | N.A. | [76] |
| Human monocyte-derived macrophages | Approx. 700 | N.A. | N.A. | Several hours | [77] |
| Normal rat kidney cells | N.A. | 10–70 | 70 | Several hours | [24] |
| Natural killer cells | N.A. | Approx. 30 | <140 | Observed for 40 min | [20,78] |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Cancer Report: Cancer Research for Cancer Development; IARC: Lyon, France, 2020; ISBN 978-92-832-0447-3. [Google Scholar]
- Brücher, B.L.D.M.; Jamall, I.S. Cell-cell communication in the tumor microenvironment, carcinogenesis, and anticancer treatment. Cell. Physiol. Biochem. 2014, 34, 213–243. [Google Scholar] [CrossRef]
- Calvo, F.; Sahai, E. Cell communication networks in cancer invasion. Curr. Opin. Cell Biol. 2011, 23, 621–629. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Thind, A.; Wilson, C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J. Extracell. Vesicles 2016, 5, 31292. [Google Scholar] [CrossRef]
- Fiorini, E.; Veghini, L.; Corbo, V. Modeling Cell Communication in Cancer With Organoids: Making the Complex Simple. Front. Cell Dev. Biol. 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Matejka, N.; Reindl, J. Perspectives of cellular communication through tunneling nanotubes in cancer cells and the connection to radiation effects. Radiat. Oncol. 2019, 14, 218. [Google Scholar] [CrossRef]
- Baker, M. How the Internet of cells has biologists buzzing. Nature 2017, 549, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, A.; Mehta, B.M.; Niu, X.; Kang, G.; Villafania, L.; Way, D.; Polycarpe, D.; Sadelain, M.; Larson, S.M. Intercellular transfer of P-glycoprotein mediates acquired multidrug resistance in tumor cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1933–1938. [Google Scholar] [CrossRef]
- Marzo, L.; Gousset, K.; Zurzolo, C. Multifaceted roles of tunneling nanotubes in intercellular communication. Front. Physiol. 2012, 3, 72. [Google Scholar] [CrossRef]
- Murray, L.M.A.; Krasnodembskaya, A.D. Concise Review: Intercellular Communication Via Organelle Transfer in the Biology and Therapeutic Applications of Stem Cells. Stem Cells 2019, 37, 14–25. [Google Scholar] [CrossRef]
- Thayanithy, V.; Dickson, E.L.; Steer, C.; Subramanian, S.; Lou, E. Tumor-stromal cross talk: Direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl. Res. 2014, 164, 359–365. [Google Scholar] [CrossRef]
- Vignais, M.-L.; Caicedo, A.; Brondello, J.-M.; Jorgensen, C. Cell Connections by Tunneling Nanotubes: Effects of Mitochondrial Trafficking on Target Cell Metabolism, Homeostasis, and Response to Therapy. Stem Cells Int. 2017, 2017, 6917941. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, H.R.; Pearlman, E.; McMenamin, P.G. Cutting edge: Membrane nanotubes in vivo: A feature of MHC class II+ cells in the mouse cornea. J. Immunol. 2008, 180, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Seyed-Razavi, Y.; Hickey, M.J.; Kuffová, L.; McMenamin, P.G.; Chinnery, H.R. Membrane nanotubes in myeloid cells in the adult mouse cornea represent a novel mode of immune cell interaction. Immunol. Cell Biol. 2013, 91, 89–95. [Google Scholar] [CrossRef]
- Rehberg, M.; Nekolla, K.; Sellner, S.; Praetner, M.; Mildner, K.; Zeuschner, D.; Krombach, F. Intercellular Transport of Nanomaterials is Mediated by Membrane Nanotubes In Vivo. Small 2016, 12, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Wick, W.; Winkler, F. Tunneling nanotube-like structures in brain tumors. Cancer Rep. 2019, 2, e118. [Google Scholar] [CrossRef]
- Pinto, G.; Brou, C.; Zurzolo, C. Tunneling Nanotubes: The Fuel of Tumor Progression? Trends Cancer 2020, 6, 874–888. [Google Scholar] [CrossRef]
- Davis, D.M.; Sowinski, S. Membrane nanotubes: Dynamic long-distance connections between animal cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 431–436. [Google Scholar] [CrossRef]
- Mittal, R.; Karhu, E.; Wang, J.-S.; Delgado, S.; Zukerman, R.; Mittal, J.; Jhaveri, V.M. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J. Cell. Physiol. 2019, 234, 1130–1146. [Google Scholar] [CrossRef]
- Austefjord, M.W.; Gerdes, H.-H.; Wang, X. Tunneling nanotubes: Diversity in morphology and structure. Commun. Integr. Biol. 2014, 7, e27934. [Google Scholar] [CrossRef]
- Cordero Cervantes, D.; Zurzolo, C. Peering into tunneling nanotubes-The path forward. EMBO J. 2021, 40, e105789. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.-H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef] [PubMed]
- Drab, M.; Stopar, D.; Kralj-Iglič, V.; Iglič, A. Inception Mechanisms of Tunneling Nanotubes. Cells 2019, 8, 626. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, X. Opportunities and Challenges in Tunneling Nanotubes Research: How Far from Clinical Application? Int. J. Mol. Sci. 2021, 22, 2306. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.; Jena, S.R.; Samanta, L. Tunneling Nanotubes: A Versatile Target for Cancer Therapy. Curr. Cancer Drug Targets 2018, 18, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Fujisawa, S.; Barlas, A.; Romin, Y.; Manova-Todorova, K.; Moore, M.A.S.; Subramanian, S. Tunneling Nanotubes: A new paradigm for studying intercellular communication and therapeutics in cancer. Commun. Integr. Biol. 2012, 5, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-G.; Liu, S.-L.; Tian, Z.-Q.; Zhang, Z.-L.; Tang, H.-W.; Pang, D.-W. Myosin-driven intercellular transportation of wheat germ agglutinin mediated by membrane nanotubes between human lung cancer cells. ACS Nano 2012, 6, 10033–10041. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Xiong, R.; Zhang, Y.; Yang, W.; Chen, J.-Y.; Wang, P.-N. Microscopic observation of the intercellular transport of CdTe quantum dot aggregates through tunneling-nanotubes. J. Biomater. Nanobiotechnol. 2011, 2, 172–179. [Google Scholar] [CrossRef]
- He, K.; Luo, W.; Zhang, Y.; Liu, F.; Liu, D.; Xu, L.; Qin, L.; Xiong, C.; Lu, Z.; Fang, X.; et al. Intercellular transportation of quantum dots mediated by membrane nanotubes. ACS Nano 2010, 4, 3015–3022. [Google Scholar] [CrossRef]
- Löscher, W.; Gericke, B. Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain? Pharmaceutics 2020, 12, 966. [Google Scholar] [CrossRef]
- Ottonelli, I.; Caraffi, R.; Tosi, G.; Vandelli, M.A.; Duskey, J.T.; Ruozi, B. Tunneling Nanotubes: A New Target for Nanomedicine? Int. J. Mol. Sci. 2022, 23, 2237. [Google Scholar] [CrossRef]
- Combs, S.E.; Kessel, K.; Habermehl, D.; Haberer, T.; Jäkel, O.; Debus, J. Proton and carbon ion radiotherapy for primary brain tumors and tumors of the skull base. Acta Oncol. 2013, 52, 1504–1509. [Google Scholar] [CrossRef]
- Babu, R.; Komisarow, J.M.; Agarwal, V.J.; Rahimpour, S.; Iyer, A.; Britt, D.; Karikari, I.O.; Grossi, P.M.; Thomas, S.; Friedman, A.H.; et al. Glioblastoma in the elderly: The effect of aggressive and modern therapies on survival. J. Neurosurg. 2016, 124, 998–1007. [Google Scholar] [CrossRef]
- Krex, D.; Klink, B.; Hartmann, C.; von Deimling, A.; Pietsch, T.; Simon, M.; Sabel, M.; Steinbach, J.P.; Heese, O.; Reifenberger, G.; et al. Long-term survival with glioblastoma multiforme. Brain 2007, 130, 2596–2606. [Google Scholar] [CrossRef]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef]
- Wank, M.; Schilling, D.; Reindl, J.; Meyer, B.; Gempt, J.; Motov, S.; Alexander, F.; Wilkens, J.J.; Schlegel, J.; Schmid, T.E.; et al. Evaluation of radiation-related invasion in primary patient-derived glioma cells and validation with established cell lines: Impact of different radiation qualities with differing LET. J. Neurooncol. 2018, 139, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Wank, M.; Schilling, D.; Schmid, T.E.; Meyer, B.; Gempt, J.; Barz, M.; Schlegel, J.; Liesche, F.; Kessel, K.A.; Wiestler, B.; et al. Human Glioma Migration and Infiltration Properties as a Target for Personalized Radiation Medicine. Cancers 2018, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Matejka, N.; Reindl, J. Influence of α-Particle Radiation on Intercellular Communication Networks of Tunneling Nanotubes in U87 Glioblastoma Cells. Front. Oncol. 2020, 10, 1691. [Google Scholar] [CrossRef] [PubMed]
- Reindl, J.; Shevtsov, M.; Dollinger, G.; Stangl, S.; Multhoff, G. Membrane Hsp70-supported cell-to-cell connections via tunneling nanotubes revealed by live-cell STED nanoscopy. Cell Stress Chaperones 2019, 24, 213–221. [Google Scholar] [CrossRef]
- Valdebenito, S.; Audia, A.; Bhat, K.P.L.; Okafo, G.; Eugenin, E.A. Tunneling Nanotubes Mediate Adaptation of Glioblastoma Cells to Temozolomide and Ionizing Radiation Treatment. iScience 2020, 23, 101450. [Google Scholar] [CrossRef]
- Valdebenito, S.; Malik, S.; Luu, R.; Loudig, O.; Mitchell, M.; Okafo, G.; Bhat, K.; Prideaux, B.; Eugenin, E.A. Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci. Rep. 2021, 11, 14556. [Google Scholar] [CrossRef]
- Pinto, G.; Saenz-de-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y. Tunneling nanotubes between rat primary astrocytes and C6 glioma cells alter proliferation potential of glioma cells. Neurosci. Bull. 2015, 31, 371–378. [Google Scholar] [CrossRef]
- Formicola, B.; D’Aloia, A.; Dal Magro, R.; Stucchi, S.; Rigolio, R.; Ceriani, M.; Re, F. Differential Exchange of Multifunctional Liposomes Between Glioblastoma Cells and Healthy Astrocytes via Tunneling Nanotubes. Front. Bioeng. Biotechnol. 2019, 7, 403. [Google Scholar] [CrossRef] [PubMed]
- Carone, C.; Genedani, S.; Leo, G.; Filaferro, M.; Fuxe, K.; Agnati, L.F. In vitro effects of cocaine on tunneling nanotube formation and extracellular vesicle release in glioblastoma cell cultures. J. Mol. Neurosci. 2015, 55, 42–50. [Google Scholar] [CrossRef]
- Pontes, B.; Viana, N.B.; Campanati, L.; Farina, M.; Neto, V.M.; Nussenzveig, H.M. Structure and elastic properties of tunneling nanotubes. Eur. Biophys. J. 2008, 37, 121–129. [Google Scholar] [CrossRef]
- Khattar, K.E.; Safi, J.; Rodriguez, A.-M.; Vignais, M.-L. Intercellular Communication in the Brain through Tunneling Nanotubes. Cancers 2022, 14, 1207. [Google Scholar] [CrossRef]
- Winkler, F.; Wick, W. Harmful networks in the brain and beyond. Science 2018, 359, 1100–1101. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.S.; Lou, E. Tunneling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep. 2019, 2, e1185. [Google Scholar] [CrossRef]
- Taiarol, L.; Formicola, B.; Fagioli, S.; Sierri, G.; D’Aloia, A.; Kravicz, M.; Renda, A.; Viale, F.; Dal Magro, R.; Ceriani, M.; et al. The 3.0 Cell Communication: New Insights in the Usefulness of Tunneling Nanotubes for Glioblastoma Treatment. Cancers 2021, 13, 4001. [Google Scholar] [CrossRef]
- van Steensel, B.; van Binnendijk, E.P.; Hornsby, C.D.; van der Voort, H.T.; Krozowski, Z.S.; de Kloet, E.R.; van Driel, R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 1996, 109 Pt 4, 787–792. [Google Scholar] [CrossRef]
- Reindl, J.; Drexler, G.A.; Girst, S.; Greubel, C.; Siebenwirth, C.; Drexler, S.E.; Dollinger, G.; Friedl, A.A. Nanoscopic exclusion between Rad51 and 53BP1 after ion irradiation in human HeLa cells. Phys. Biol. 2015, 12, 66005. [Google Scholar] [CrossRef] [PubMed]
- Reindl, J.; Girst, S.; Walsh, D.W.M.; Greubel, C.; Schwarz, B.; Siebenwirth, C.; Drexler, G.A.; Friedl, A.A.; Dollinger, G. Chromatin organization revealed by nanostructure of irradiation induced γH2AX, 53BP1 and Rad51 foci. Sci. Rep. 2017, 7, 40616. [Google Scholar] [CrossRef] [PubMed]
- Veranic, P.; Lokar, M.; Schütz, G.J.; Weghuber, J.; Wieser, S.; Hägerstrand, H.; Kralj-Iglic, V.; Iglic, A. Different types of cell-to-cell connections mediated by nanotubular structures. Biophys. J. 2008, 95, 4416–4425. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Delage, E.; Zurzolo, C. Identification and Characterization of Tunneling Nanotubes for Intercellular Trafficking. Curr. Protoc. Cell Biol. 2015, 67, 12.10.1–12.10.21. [Google Scholar] [CrossRef] [PubMed]
- Cotte, Y.; Toy, F.; Jourdain, P.; Pavillon, N.; Boss, D.; Magistretti, P.; Marquet, P.; Depeursinge, C. Marker-free phase nanoscopy. Nat. Photon 2013, 7, 113–117. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A.S. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed]
- Bukoreshtliev, N.V.; Wang, X.; Hodneland, E.; Gurke, S.; Barroso, J.F.V.; Gerdes, H.-H. Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett. 2009, 583, 1481–1488. [Google Scholar] [CrossRef]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef]
- MacLean-Fletcher, S.; Pollard, T.D. Mechanism of action of cytochalasin B on actin. Cell 1980, 20, 329–341. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, H.-H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef]
- Pasquier, J.; Galas, L.; Boulangé-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Le Foll, F. Different modalities of intercellular membrane exchanges mediate cell-to-cell p-glycoprotein transfers in MCF-7 breast cancer cells. J. Biol. Chem. 2012, 287, 7374–7387. [Google Scholar] [CrossRef]
- Sowinski, S.; Alakoskela, J.-M.; Jolly, C.; Davis, D.M. Optimized methods for imaging membrane nanotubes between T cells and trafficking of HIV-1. Methods 2011, 53, 27–33. [Google Scholar] [CrossRef]
- Kiernan, J.A. Formaldehyde, Formalin, Paraformaldehyde And Glutaraldehyde: What They Are And What They Do. Microsc. Today 2000, 8, 8–13. [Google Scholar] [CrossRef]
- Tagliaferro, P.; Tandler, C.J.; Ramos, A.J.; Pecci Saavedra, J.; Brusco, A. Immunofluorescence and glutaraldehyde fixation. A new procedure based on the Schiff-quenching method. J. Neurosci. Methods 1997, 77, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Jiang, W.; Li, A.; Gao, M.; Liu, H.; Gao, Y.; Tian, X.; Gong, G. The Combination of Paraformaldehyde and Glutaraldehyde Is a Potential Fixative for Mitochondria. Biomolecules 2021, 11, 711. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Bénard, M.; Jean-Jacques, B.; Schapman, D.; Roberge, H.; Lebon, A.; Goux, D.; Monterroso, B.; Elie, N.; Komuro, H.; et al. Investigating Tunneling Nanotubes in Cancer Cells: Guidelines for Structural and Functional Studies through Cell Imaging. Biomed. Res. Int. 2020, 2020, 2701345. [Google Scholar] [CrossRef]
- Bénard, M.; Schapman, D.; Lebon, A.; Monterroso, B.; Bellenger, M.; Le Foll, F.; Pasquier, J.; Vaudry, H.; Vaudry, D.; Galas, L. Structural and functional analysis of tunneling nanotubes (TnTs) using gCW STED and gconfocal approaches. Biol. Cell 2015, 107, 419–425. [Google Scholar] [CrossRef]
- Sartori-Rupp, A.; Cordero Cervantes, D.; Pepe, A.; Gousset, K.; Delage, E.; Corroyer-Dulmont, S.; Schmitt, C.; Krijnse-Locker, J.; Zurzolo, C. Correlative cryo-electron microscopy reveals the structure of TNTs in neuronal cells. Nat. Commun. 2019, 10, 342. [Google Scholar] [CrossRef]
- Wittig, D.; Wang, X.; Walter, C.; Gerdes, H.-H.; Funk, R.H.W.; Roehlecke, C. Multi-level communication of human retinal pigment epithelial cells via tunneling nanotubes. PLoS ONE 2012, 7, e33195. [Google Scholar] [CrossRef] [PubMed]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef]
- Onfelt, B.; Nedvetzki, S.; Benninger, R.K.P.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.A.; French, P.M.W.; Davis, D.M. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [PubMed]
- Onfelt, B.; Nedvetzki, S.; Yanagi, K.; Davis, D.M. Cutting edge: Membrane nanotubes connect immune cells. J. Immunol. 2004, 173, 1511–1513. [Google Scholar] [CrossRef] [PubMed]







| Dye | Excitation Wavelength [nm] | Detector Range [nm] | Gating [ns] |
|---|---|---|---|
| CellMask™ Orange Plasma Membrane Stain | 554 | 567–635 | 0.3–6 |
| CellMask™ Green Plasma Membrane Stain | 522 | 530–583 | 0.3–6 |
| Vybrant™ DiO | 484 | 501–583 | 0.3–7 |
| PKH | 551 | 559–630 | 0.3–12 |
| WGA Alexa Fluor 633 | 625 | 634–751 | 0.3–6 |
| CellLight™ Plasma Membrane-GFP | 485 | 500–580 | 0.3–6 |
| MemGlow™ | 595 | 613–750 | 0.3–6 |
| Dye | Cell-Off Background Noise | Quantum Yield | Homogeneity | Internalization Speed | Loading Buffer |
|---|---|---|---|---|---|
| CellMask™ Plasma Membrane | + | + | + | + | Medium |
| DiO | −/+ | −/+ | − | − | Medium |
| PKH | −/+ | −/+ | − | − | Diluent C (iso-osmotic, aqueous solution) |
| WGA | − | − | + | N.A. | HBSS |
| CellLight™ | −/+ | − | − | Not relevant | Medium |
| MemGlow™ | − | −/+ | − | N.A. | Serum-free Medium |
| Cell Line | Mean Length ± SD [µm] | Max. Measured Length [µm] | Mean Lifetime ± SD [min] | Max. Recorded Lifetime [min] | Formation Type |
|---|---|---|---|---|---|
| U87 MG | 40 ± 27 n = 426 | 189 | 88 ± 60 n = 234 | 360 | Cell movement |
| LN229 | 20 ± 12 n = 197 | 60 | 41 ± 30 n = 85 | 165 | Cell movement |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matejka, N.; Amarlou, A.; Neubauer, J.; Rudigkeit, S.; Reindl, J. High-Resolution Microscopic Characterization of Tunneling Nanotubes in Living U87 MG and LN229 Glioblastoma Cells. Cells 2024, 13, 464. https://doi.org/10.3390/cells13050464
Matejka N, Amarlou A, Neubauer J, Rudigkeit S, Reindl J. High-Resolution Microscopic Characterization of Tunneling Nanotubes in Living U87 MG and LN229 Glioblastoma Cells. Cells. 2024; 13(5):464. https://doi.org/10.3390/cells13050464
Chicago/Turabian StyleMatejka, Nicole, Asieh Amarlou, Jessica Neubauer, Sarah Rudigkeit, and Judith Reindl. 2024. "High-Resolution Microscopic Characterization of Tunneling Nanotubes in Living U87 MG and LN229 Glioblastoma Cells" Cells 13, no. 5: 464. https://doi.org/10.3390/cells13050464