


Click Chemistry with Cell-Permeable Fluorophores Expands the Choice of Bioorthogonal Markers for Two-Color Live-Cell STED Nanoscopy


Abstract
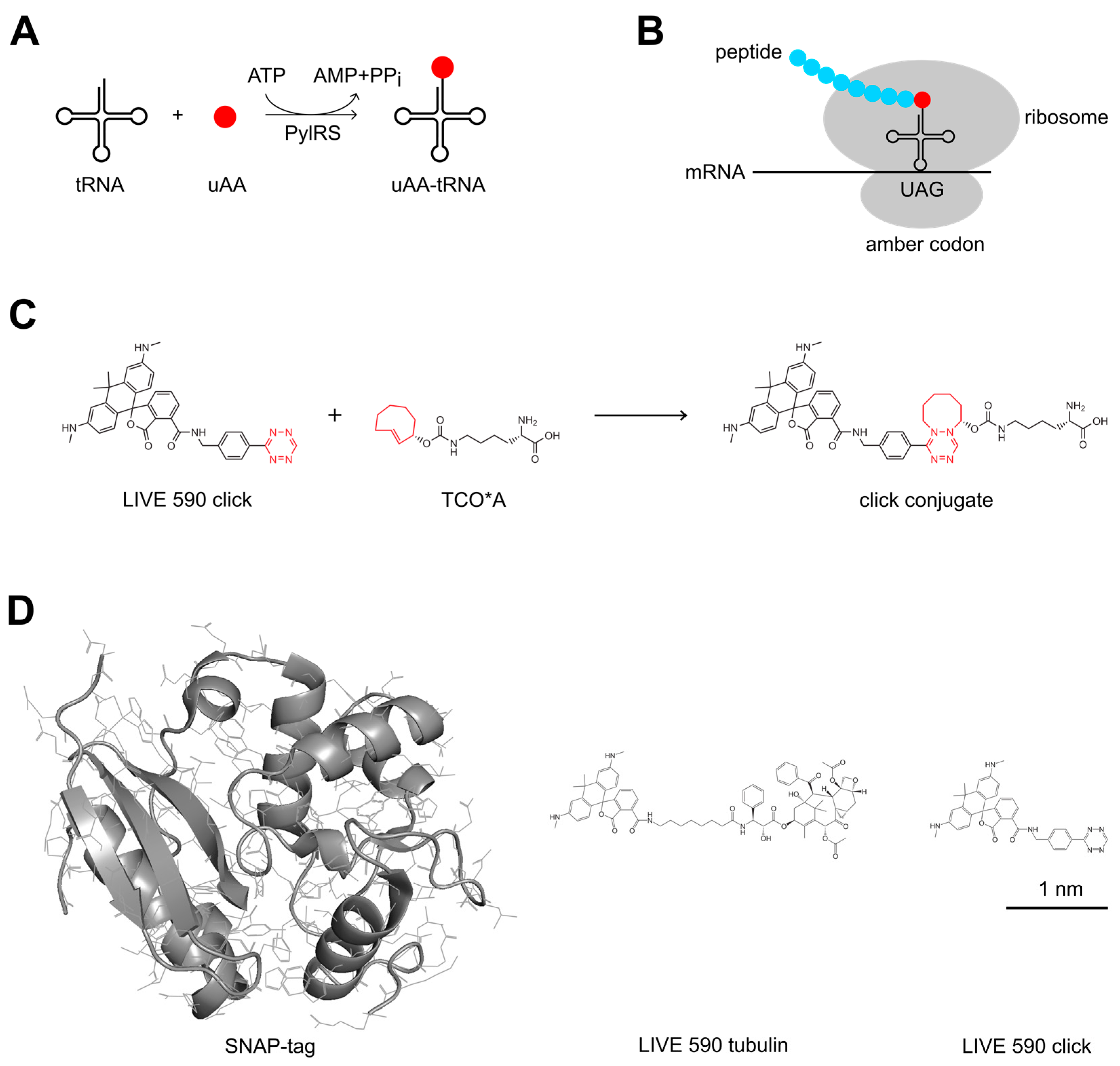
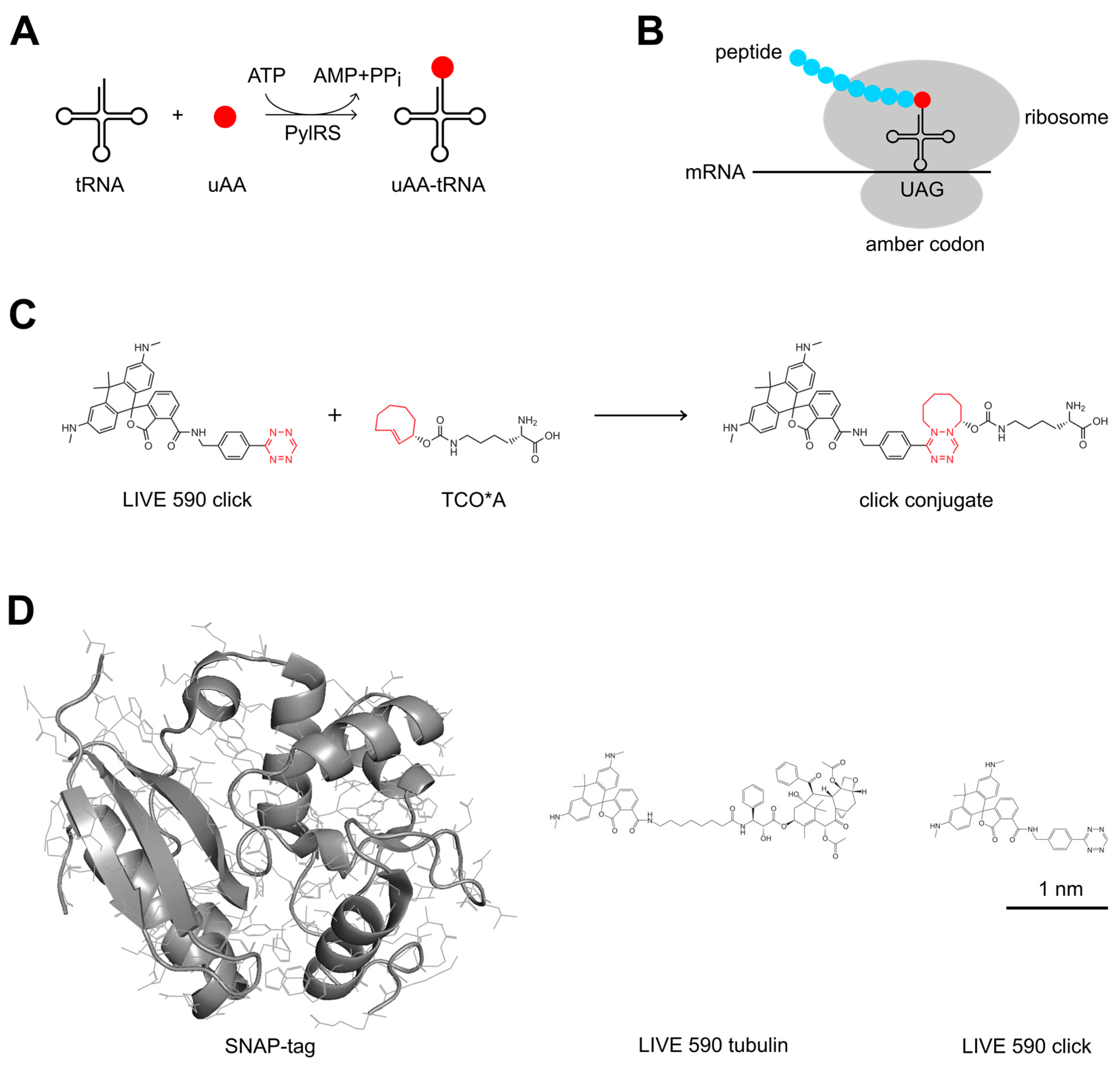
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Cell Culture
2.3. Live-Cell Labeling
2.4. Live-Cell Fluorescence Microscopy
3. Results
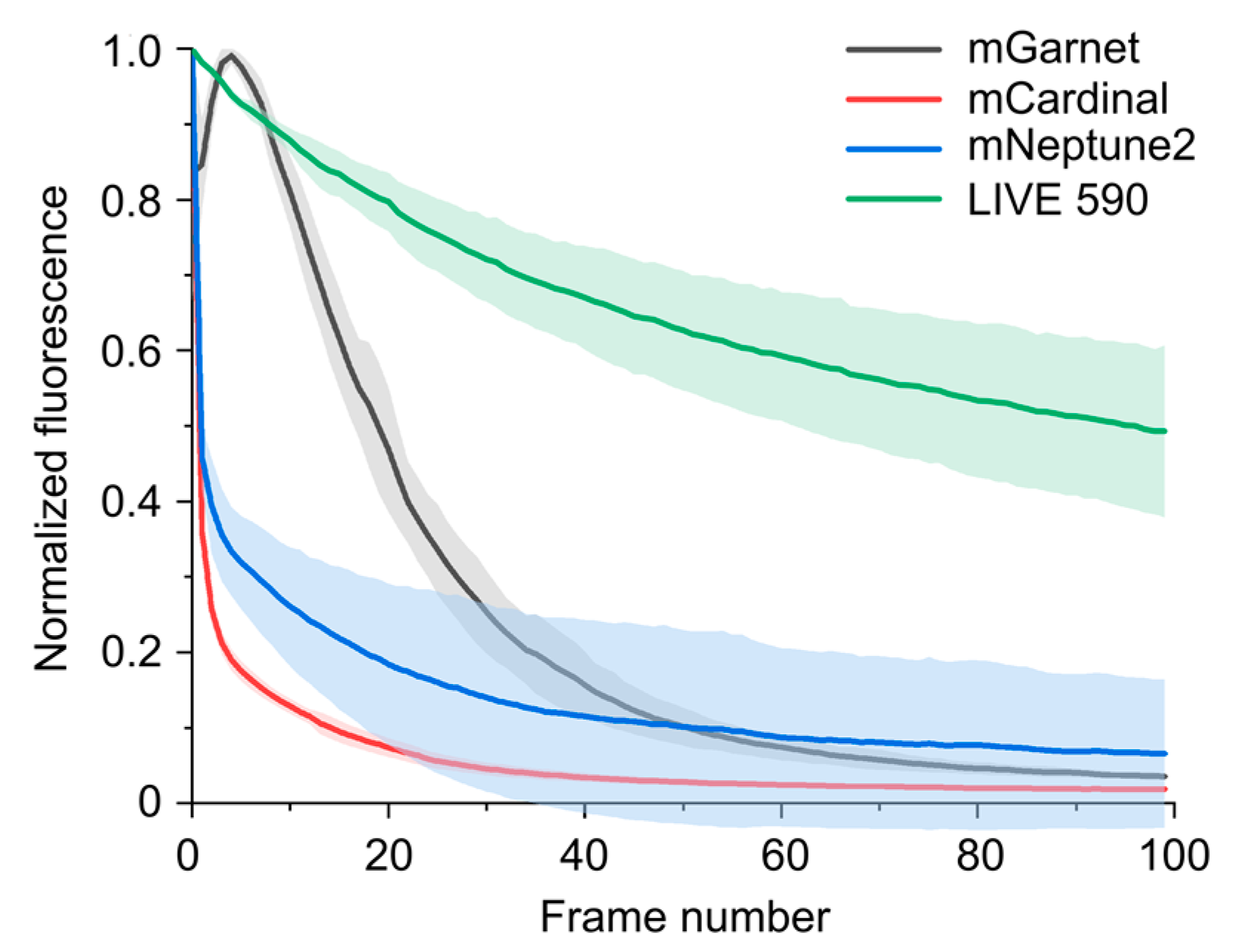
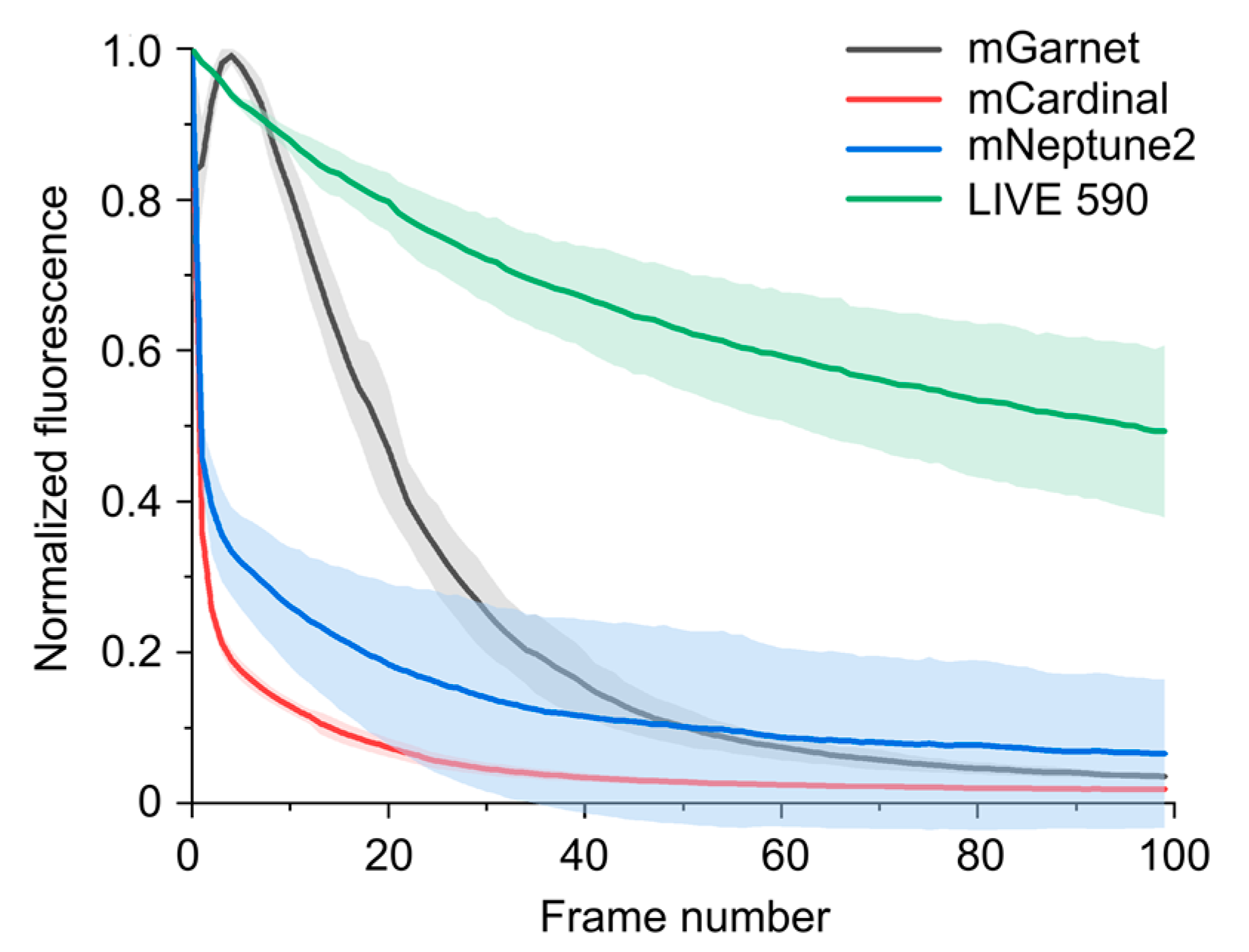
3.1. Photophysical Properties of the Dyes
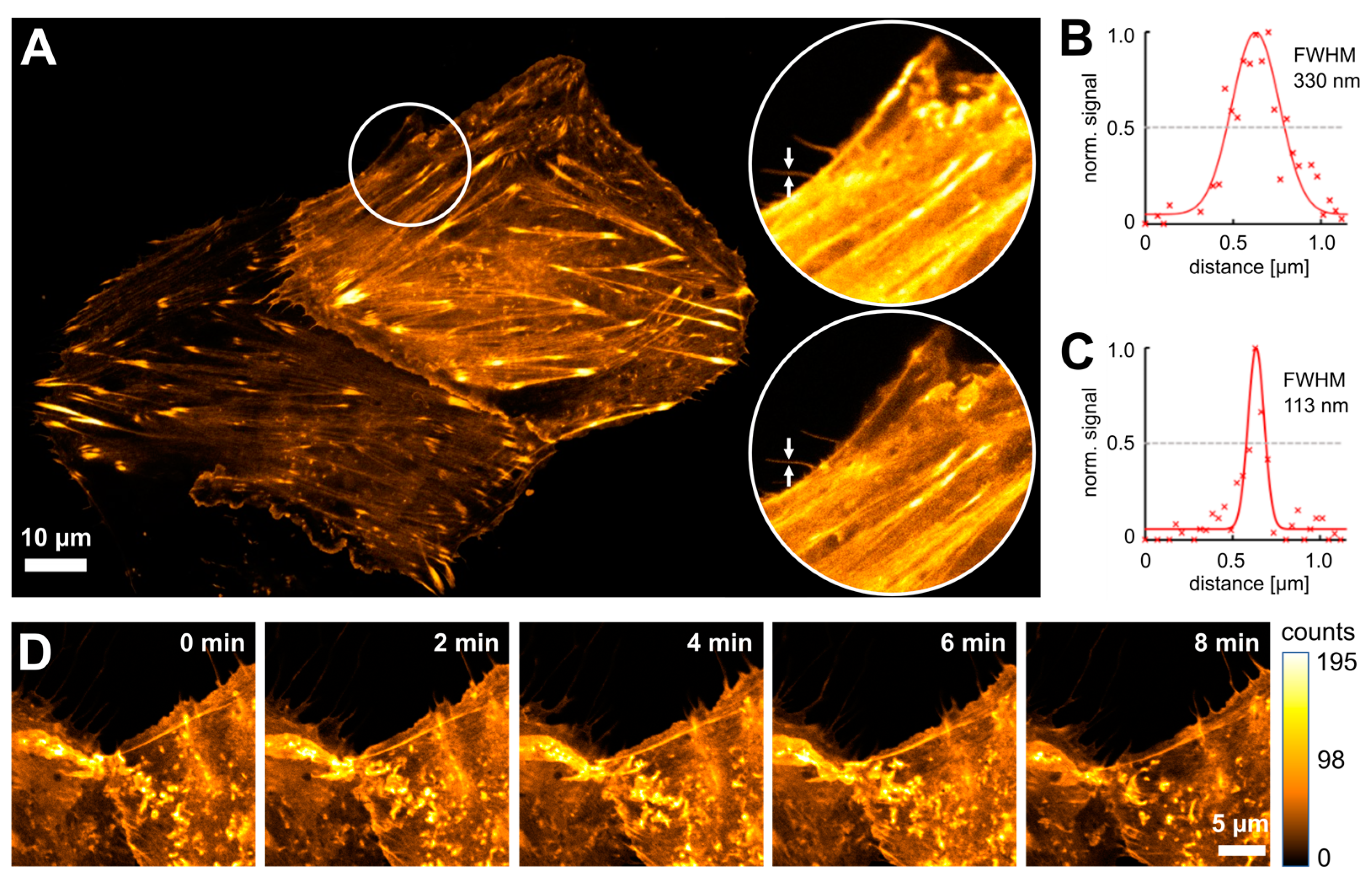
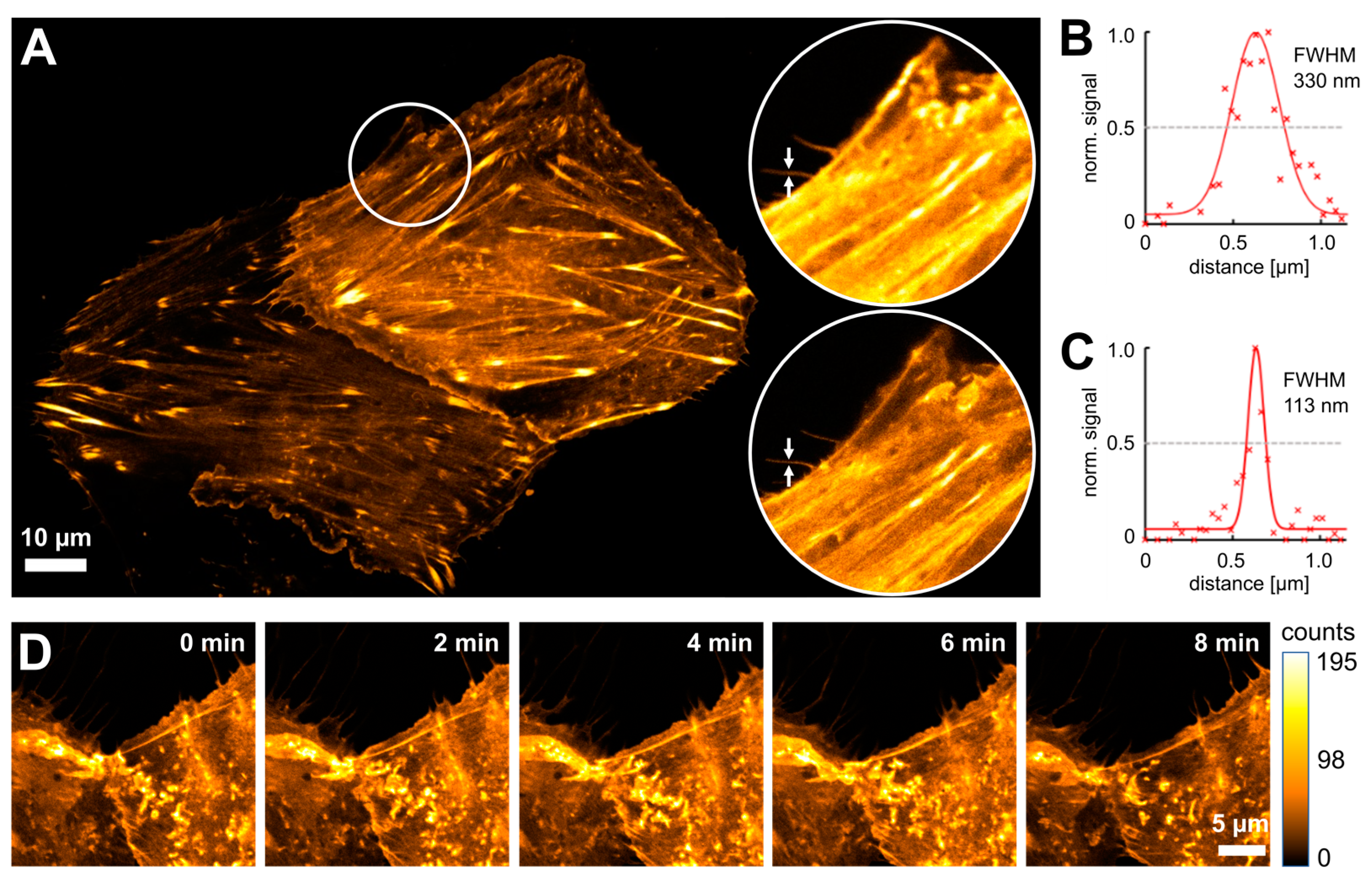
3.2. Live-Cell STED Nanoscopy of Click-Labeled Actin
3.3. Time-Lapse STED Imaging with Tetrazine Dyes and Fluorescent Proteins
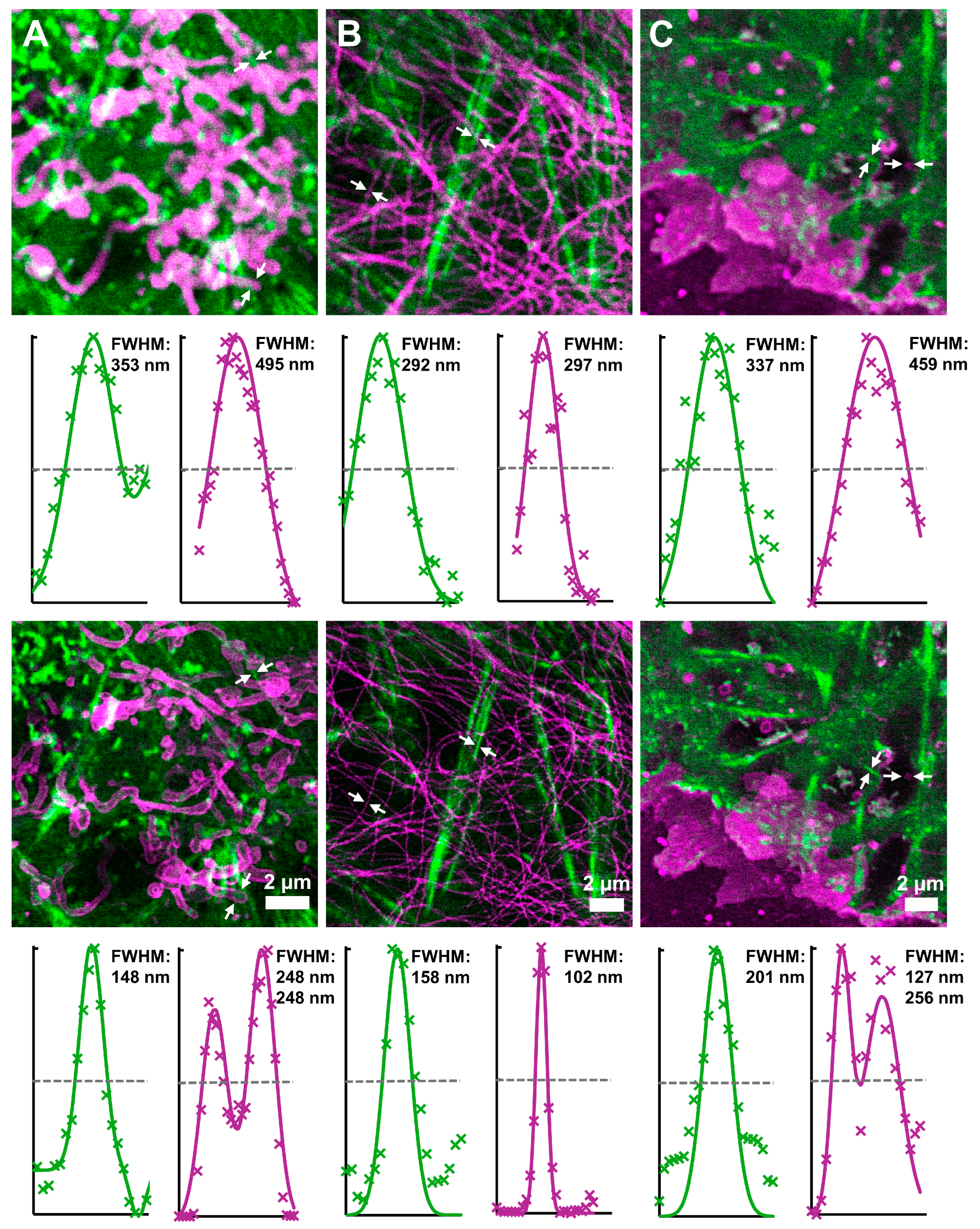
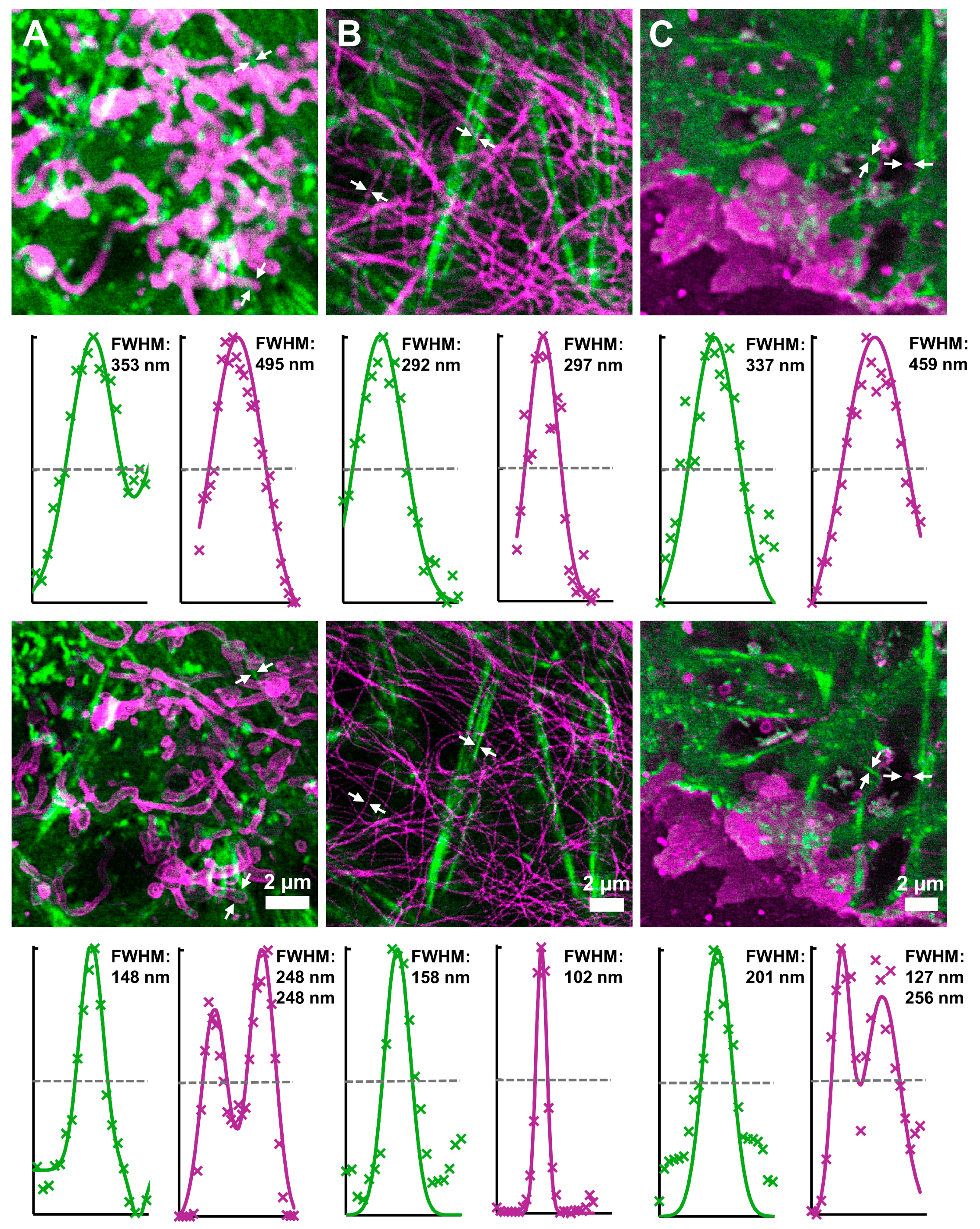
3.4. Two-Color Long-Term STED Nanoscopy in Living Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch. F Mikrosk. Anat. 1873, 9, 413–468. [Google Scholar] [CrossRef]
- Hein, B.; Willig, K.I.; Hell, S.W. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc. Natl. Acad. Sci. USA 2008, 105, 14271–14276. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.A.; Sun, X.; Gould, T.J.; Rothman, J.E.; Xu, M.Q.; Corrêa, I.R.; Bewersdorf, J. Two-color STED microscopy in living cells. Biomed. Opt. Express 2011, 2, 2364. [Google Scholar] [CrossRef]
- Berning, S.; Willig, K.I.; Steffens, H.; Dibaj, P.; Hell, S.W. Nanoscopy in a living mouse brain. Science 2012, 335, 551. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Hell, S.W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999, 24, 954–956. [Google Scholar] [CrossRef]
- Schmidt, R.; Wurm, C.A.; Jakobs, S.; Engelhardt, J.; Egner, A.; Hell, S.W. Spherical nanosized focal spot unravels the interior of cells. Nat. Methods 2008, 5, 539–544. [Google Scholar] [CrossRef]
- Göttfert, F.; Wurm, C.A.; Mueller, V.; Berning, S.; Cordes, V.C.; Honigmann, A.; Hell, S.W. Coaligned dual-channel STED nanoscopy and molecular diffusion analysis at 20 nm resolution. Biophys. J. 2013, 105, L01–L03. [Google Scholar] [CrossRef]
- Stockhammer, A.; Bottanelli, F. Appreciating the small things in life: STED microscopy in living cells. J. Phys. D Appl. Phys. 2021, 54, 033001. [Google Scholar] [CrossRef]
- Lukinavičius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Göttfert, F.; Ta, H.; Güther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef]
- Juillerat, A.; Gronemeyer, T.; Keppler, A.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003, 10, 313–317. [Google Scholar] [CrossRef]
- Mollwitz, B.; Brunk, E.; Schmitt, S.; Pojer, F.; Bannwarth, M.; Schiltz, M.; Rothlisberger, U.; Johnsson, K. Directed evolution of the suicide protein O6-alkylguanine-DNA alkyltransferase for increased reactivity results in an alkylated protein with exceptional stability. Biochemistry 2012, 51, 986–994. [Google Scholar] [CrossRef]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R.; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Kang, D.; Park, H.-S. Site-specific labeling of proteins using unnatural amino acids. Mol. Cells 2019, 42, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Chin, J.W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 2014, 114, 4764–4806. [Google Scholar] [CrossRef]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Kleiner, R.E. Cell- and polymerase-selective metabolic labeling of cellular RNA with 2′-azidocytidine. J. Am. Chem. Soc. 2020, 142, 14417–14421. [Google Scholar] [CrossRef] [PubMed]
- Neef, A.B.; Schultz, C. Selective fluorescence labeling of lipids in living cells. Angew. Chem. Int. Ed. Engl. 2009, 48, 1498–1500. [Google Scholar] [CrossRef]
- Chehrehasa, F.; Meedeniya, A.C.B.; Dwyer, P.; Abrahamsen, G.; Mackay-Sim, A. EdU, a new thymidine analogue for labelling proliferating cells in the nervous system. J. Neurosci. Methods 2009, 177, 122–130. [Google Scholar] [CrossRef]
- Mehl, R.A.; Anderson, J.C.; Santoro, S.W.; Wang, L.; Martin, A.B.; King, D.S.; Horn, D.M.; Schultz, P.G. Generation of a bacterium with a 21 amino acid. J. Am. Chem. Soc. 2003, 125, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Davis, L.; Wallace, S.; Mahesh, M.; Cox, D.J.; King, D.S.; Horn, D.M.; Schultz, P.G. Genetic encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels−Alder reactions. J. Am. Chem. Soc. 2012, 134, 10317–10320. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Paton, R.S.; Kim, S.; Liang, Y.; Houk, K.N. Diels-Alder reactivities of strained and unstrained cycloalkenes with normal and inverse-electron-demand dienes: Activation barriers and distortion/interaction analysis. J. Am. Chem. Soc. 2013, 135, 15642–15649. [Google Scholar] [CrossRef] [PubMed]
- Arsić, A.; Hagemann, C.; Stajković, N.; Schubert, T.; Nikić-Spiegel, I. Minimal genetically encoded tags for fluorescent protein labeling in living neurons. Nat. Commun. 2022, 13, 314. [Google Scholar] [CrossRef] [PubMed]
- Lukinavičius, G.; Umezawa, K.; Olivier, N.; Honigmann, A.; Yang, G.; Plass, T.; Mueller, V.; Reymond, L.; Jr, I.R.C.; Luo, Z.-G. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Uttamapinant, C.; Howe, J.D.; Lang, K.; Beránek, V.; Davis, L.; Mahesh, M.; Barry, N.P.; Chin, J.W. Genetic code expansion enables live-cell and super-resolution imaging of site-specifically labeled cellular proteins. J. Am. Chem. Soc. 2015, 137, 4602–4605. [Google Scholar] [CrossRef]
- Beliu, G.; Kurz, A.J.; Kuhlemann, A.C.; Behringer-Pliess, L.; Meub, M.; Wolf, N.; Seibel, J.; Shi, Z.-D.; Schnermann, M.; Grimm, J.B. Bioorthogonal labeling with tetrazine-dyes for super-resolution microscopy. Commun. Biol. 2019, 2, 261. [Google Scholar] [CrossRef] [PubMed]
- Werther, P.; Yserentant, K.; Braun, F.; Kaltwasser, N.; Popp, C.; Baalmann, M.; Herten, D.; Wombacher, R. Live-cell localization microscopy with a fluorogenic and self-blinking tetrazine probe. Angew. Chem. Int. Ed. 2020, 59, 804–810. [Google Scholar] [CrossRef]
- Mitronova, G.Y.; Belov, V.; Bossi, M.; Wurm, C.; Meyer, L.; Medda, R.; Moneron, G.; Bretschneider, S.; Eggeling, C.; Jakobs, S. New fluorinated rhodamines for optical microscopy and nanoscopy. Chem. Eur. J. 2010, 16, 4477–4488. [Google Scholar] [CrossRef]
- Bucevičius, J.; Kostiuk, G.; Gerasimaitė, R.; Gilat, T.; Lukinavičius, G. Enhancing the biocompatibility of rhodamine fluorescent probes by a neighbouring group effect. Chem. Sci. 2020, 11, 7313–7323. [Google Scholar] [CrossRef]
- Chu, J.; Haynes, R.D.; Corbel, S.Y.; Li, P.; González-González, E.; Burg, J.S.; Ataie, N.J.; Lam, A.J.; Cranfill, P.J.; A Baird, M. Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat. Methods 2014, 11, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Hense, A.; Prunsche, B.; Gao, P.; Ishitsuka, Y.; Nienhaus, K.; Nienhaus, G.U. Monomeric Garnet, a far-red fluorescent protein for live-cell STED imaging. Sci. Rep. 2015, 5, 18006. [Google Scholar] [CrossRef] [PubMed]
- Serfling, R.; Lorenz, C.; Etzel, M.; Schicht, G.; Böttke, T.; Mörl, M.; Coin, I. Designer tRNAs for efficient incorporation of non-canonical amino acids by the pyrrolysine system in mammalian cells. Nucleic Acids Res. 2018, 46, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nikić, I.; Estrada Girona, G.; Kang, J.H.; Paci, G.; Mikhaleva, S.; Koehler, C.; Shymanska, N.V.; Santos, C.V.; Spitz, D.; Lemke, E.A. Debugging eukaryotic genetic code expansion for site-specific click-PAINT super-resolution microscopy. Angew. Chem. Int. Ed. 2016, 55, 16172–16176. [Google Scholar] [CrossRef] [PubMed]
- Grimm, F.; Nizamov, S.; Belov, V.N. Green-emitting rhodamine dyes for vital labeling of cell organelles using STED super-resolution microscopy. ChemBioChem 2019, 20, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Butkevich, A.N.; Lukinavičius, G.; D’Este, E.; Hell, S.W. Cell-permeant large Stokes shift dyes for transfection-free multicolor nanoscopy. J. Am. Chem. Soc. 2017, 139, 12378–12381. [Google Scholar] [CrossRef] [PubMed]
- Soliman, K.; Grimm, F.; Wurm, C.A.; Egner, A. Predicting the membrane permeability of organic fluorescent probes by the deep neural network based lipophilicity descriptor DeepFl-LogP. Sci. Rep. 2021, 11, 6991. [Google Scholar] [CrossRef] [PubMed]
- Butkevich, A.N.; Mitronova, G.Y.; Sidenstein, S.C.; Klocke, J.L.; Kamin, D.; Meineke, D.N.H.; D’Este, E.; Kraemer, P.; Danzl, J.G.; Belov, V.N. Fluorescent rhodamines and fluorogenic carbopyronines for super-resolution STED microscopy in living cells. Angew. Chem. Int. Ed. 2016, 55, 3290–3294. [Google Scholar] [CrossRef]
- Bucevičius, J.; Gerasimaitė, R.; Kiszka, K.A.; Pradhan, S.; Kostiuk, G.; Koenen, T.; Lukinavičius, G. A general highly efficient synthesis of biocompatible rhodamine dyes and probes for live-cell multicolor nanoscopy. Nat. Commun. 2023, 14, 1306. [Google Scholar] [CrossRef]
- Nikić, I.; Plass, T.; Schraidt, O.; Szymánski, J.; Briggs, J.A.G.; Schultz, C.; Lemke, E.A. Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew. Chem. Int. Ed. 2014, 53, 2245–2249. [Google Scholar] [CrossRef]
- Hoffmann, J.E.; Plass, T.; Nikić, I.; Aramburu, I.V.; Koehler, C.; Gillandt, H.; Lemke, E.A.; Schultz, C. Highly stable trans-cyclooctene amino acids for live-cell labeling. Chem. Eur. J. 2015, 21, 12266–12270. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode Nε-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef]
- Nagasaki, A.; Kijima, S.T.; Yumoto, T.; Imaizumi, M.; Yamagishi, A.; Kim, H.; Nakamura, C.; Uyeda, T.Q. The position of the GFP tag on actin affects the filament formation in mammalian cells. Cell Struct. Funct. 2017, 42, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Wegner, W.; Ilgen, P.; Gregor, C.; van Dort, J.; Mott, A.C.; Steffens, H.; Willig, K.I. In vivo mouse and live cell STED microscopy of neuronal actin plasticity using far-red emitting fluorescent proteins. Sci. Rep. 2017, 7, 11781. [Google Scholar] [CrossRef]
- Mobarak, E.; Javanainen, M.; Kulig, W.; Honigmann, A.; Sezgin, E.; Aho, N.; Eggeling, C.; Rog, T.; Vattulainen, I. How to minimize dye-induced perturbations while studying biomembrane structure and dynamics: PEG linkers as a rational alternative. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef]
- Weber, M.; Leutenegger, M.; Stoldt, S.; Jakobs, S.; Mihaila, T.S.; Butkevich, A.N.; Hell, S.W. MINSTED fluorescence localization and nanoscopy. Nat. Photonics 2021, 15, 361–366. [Google Scholar] [CrossRef]
- Mihaila, T.S.; Bäte, C.; Ostersehlt, L.M.; Pape, J.K.; Keller-Findeisen, J.; Sahl, S.J.; Hell, S.W. Enhanced incorporation of subnanometer tags into cellular proteins for fluorescence nanoscopy via optimized genetic code expansion. Proc. Natl. Acad. Sci. USA 2022, 119, e2201861119. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | λabs.max [nm] | ε [M−1∙cm−1] | λem.max [nm] | τ [ns] | QY | ε∙QY [M−1∙cm−1] |
|---|---|---|---|---|---|---|
| LIVE 510 [29] | 498 | 53,000 | 529 | 3.8 | 0.94 | 49,820 |
| LIVE 460L [30] | 455 | 28,000 | 617 | 2.0 | 0.17 | 4760 |
| LIVE 550 [30] | 551 | 75,000 | 573 | 2.0 | 0.41 | 30,750 |
| LIVE 590 [30] | 585 | 105,000 | 609 | 3.5 | 0.60 | 63,000 |
| LIVE 610 [30] | 611 | 100,000 | 636 | 3.1 | 0.50 | 50,000 |
| mNeptune2 [31] | 599 | 89,000 | 651 | ND | 0.24 | 21,360 |
| mCardinal [31] | 604 | 87,000 | 659 | ND | 0.19 | 16,530 |
| mGarnet [32] | 598 | 95,000 | 670 | 0.8 | 0.09 | 8550 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregor, C.; Grimm, F.; Rehman, J.; Wurm, C.A.; Egner, A. Click Chemistry with Cell-Permeable Fluorophores Expands the Choice of Bioorthogonal Markers for Two-Color Live-Cell STED Nanoscopy. Cells 2024, 13, 683. https://doi.org/10.3390/cells13080683
Gregor C, Grimm F, Rehman J, Wurm CA, Egner A. Click Chemistry with Cell-Permeable Fluorophores Expands the Choice of Bioorthogonal Markers for Two-Color Live-Cell STED Nanoscopy. Cells. 2024; 13(8):683. https://doi.org/10.3390/cells13080683
Chicago/Turabian StyleGregor, Carola, Florian Grimm, Jasmin Rehman, Christian A. Wurm, and Alexander Egner. 2024. "Click Chemistry with Cell-Permeable Fluorophores Expands the Choice of Bioorthogonal Markers for Two-Color Live-Cell STED Nanoscopy" Cells 13, no. 8: 683. https://doi.org/10.3390/cells13080683
APA StyleGregor, C., Grimm, F., Rehman, J., Wurm, C. A., & Egner, A. (2024). Click Chemistry with Cell-Permeable Fluorophores Expands the Choice of Bioorthogonal Markers for Two-Color Live-Cell STED Nanoscopy. Cells, 13(8), 683. https://doi.org/10.3390/cells13080683