
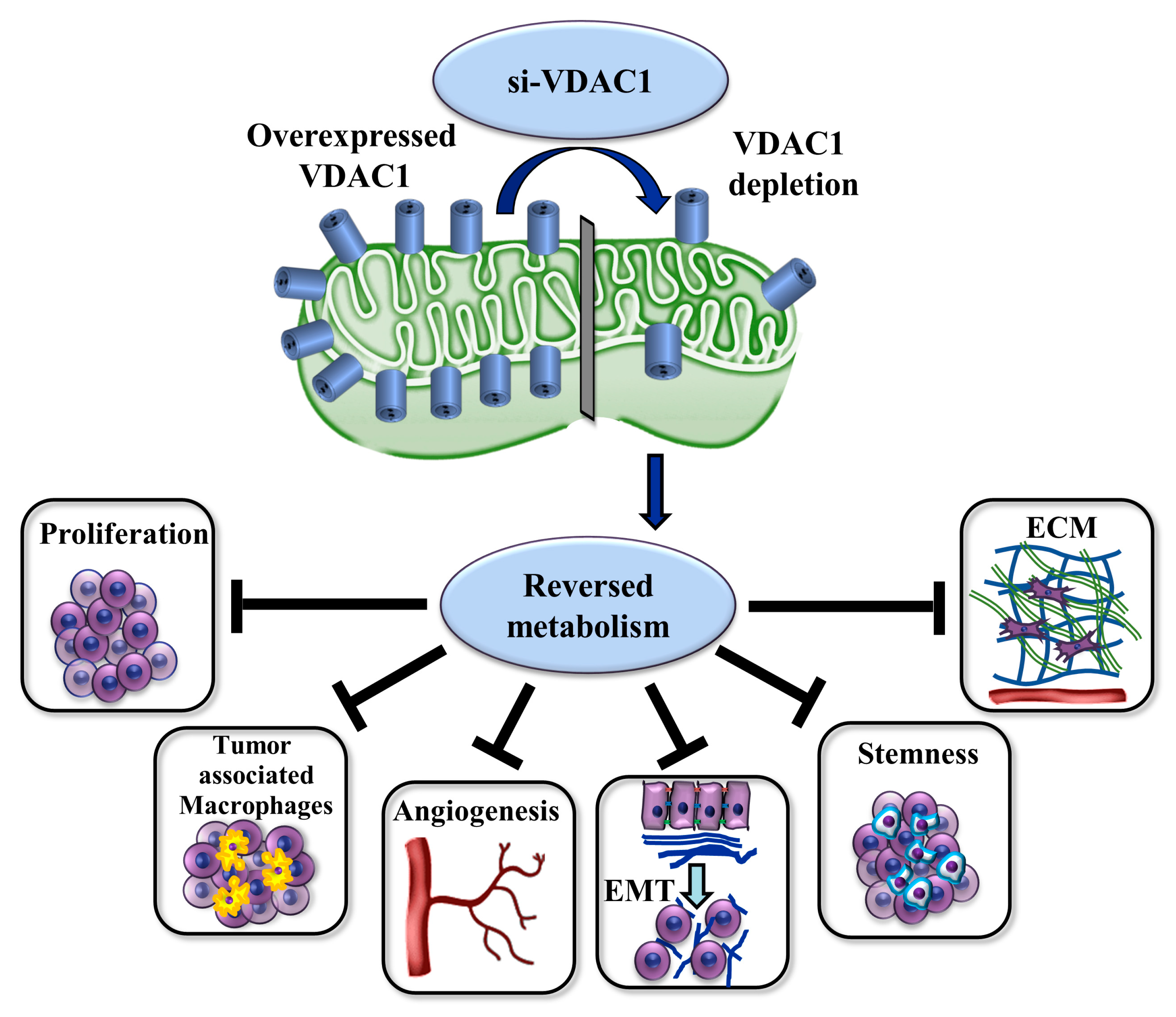
Silencing the Mitochondrial Gatekeeper VDAC1 as a Potential Treatment for Bladder Cancer


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. siRNA Transfection
2.4. Cell Viability Assay
2.5. Protein Extraction from Cells and Tumors, Gel Electrophoresis, and Immunoblot
2.6. Mitochondrial Membrane Potential Determination and Cellular ATP Levels
2.7. Immunocytochemistry (IF)
2.8. Preparation of siRNA Loaded–PLGA-PEI Nanoparticles
2.9. Bladder Cancer Mouse Models
2.10. Histological, Immunohistochemistry, and Immunofluorescence Analyses of Bladder Cancer
2.11. Sirius Red and Hematoxylin and Eosin Staining
2.12. Statistical Analysis
3. Results
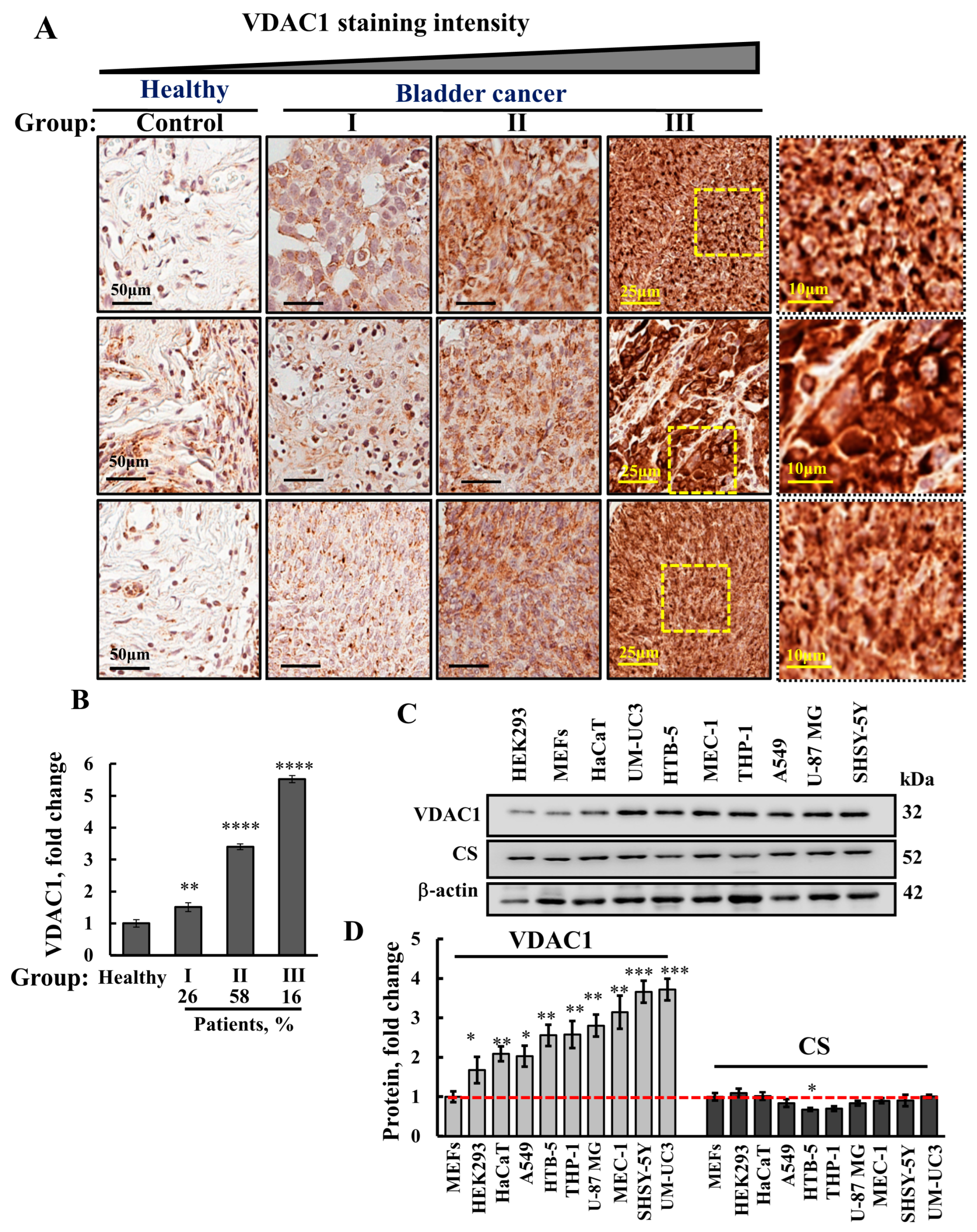
3.1. VDAC1 Is Highly Expressed in Human Bladder Cancer Tissue Compared to Healthy Tissues
3.2. VDAC1 Silencing Reduces Cell Viability, Proliferation, Mitochondrial Membrane Potential, and ATP Levels in Bladder Cancer Cell Lines
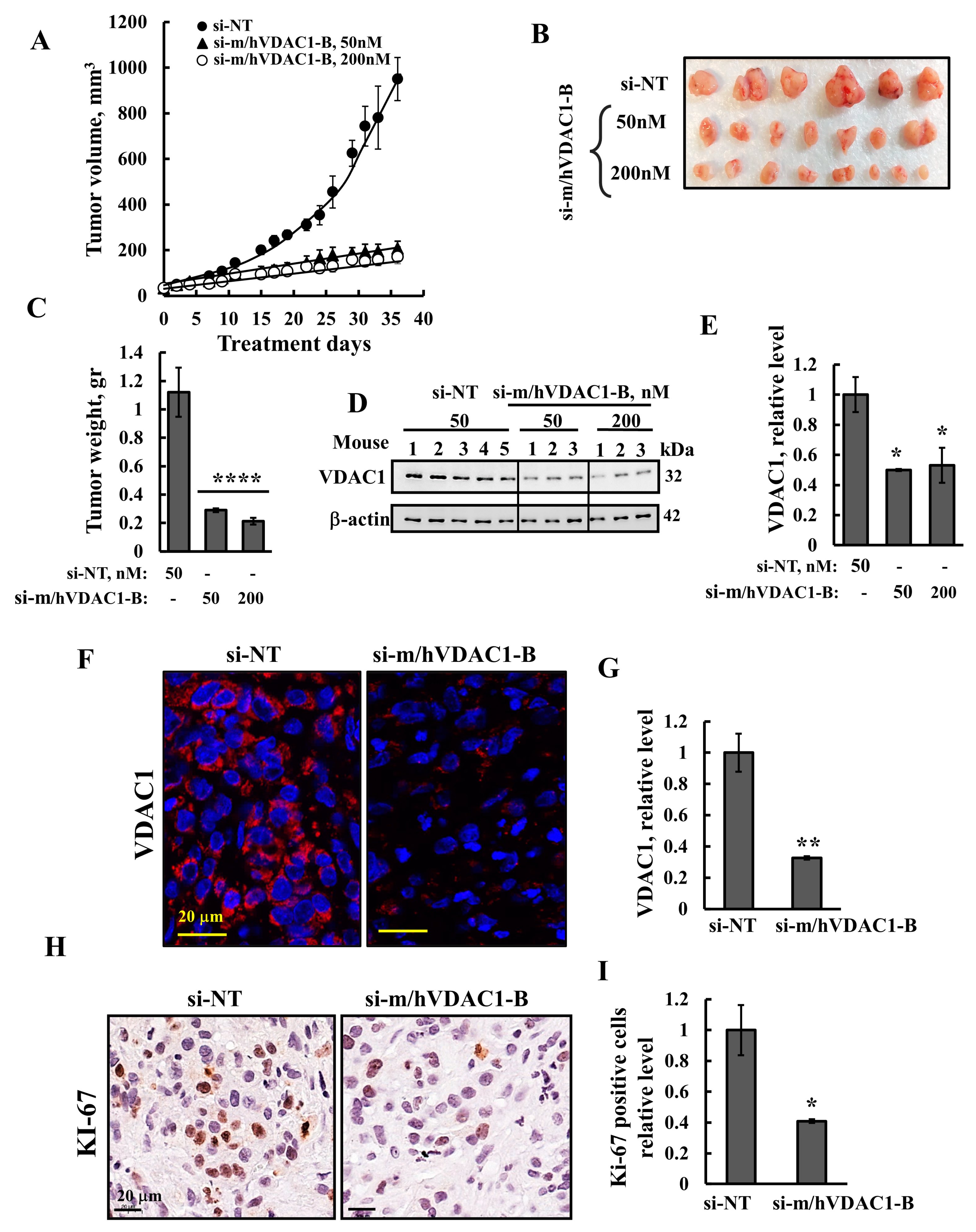
3.3. VDAC1 Silencing Reduced Tumor Growth of UM-UC3 Cell-Derived Tumor
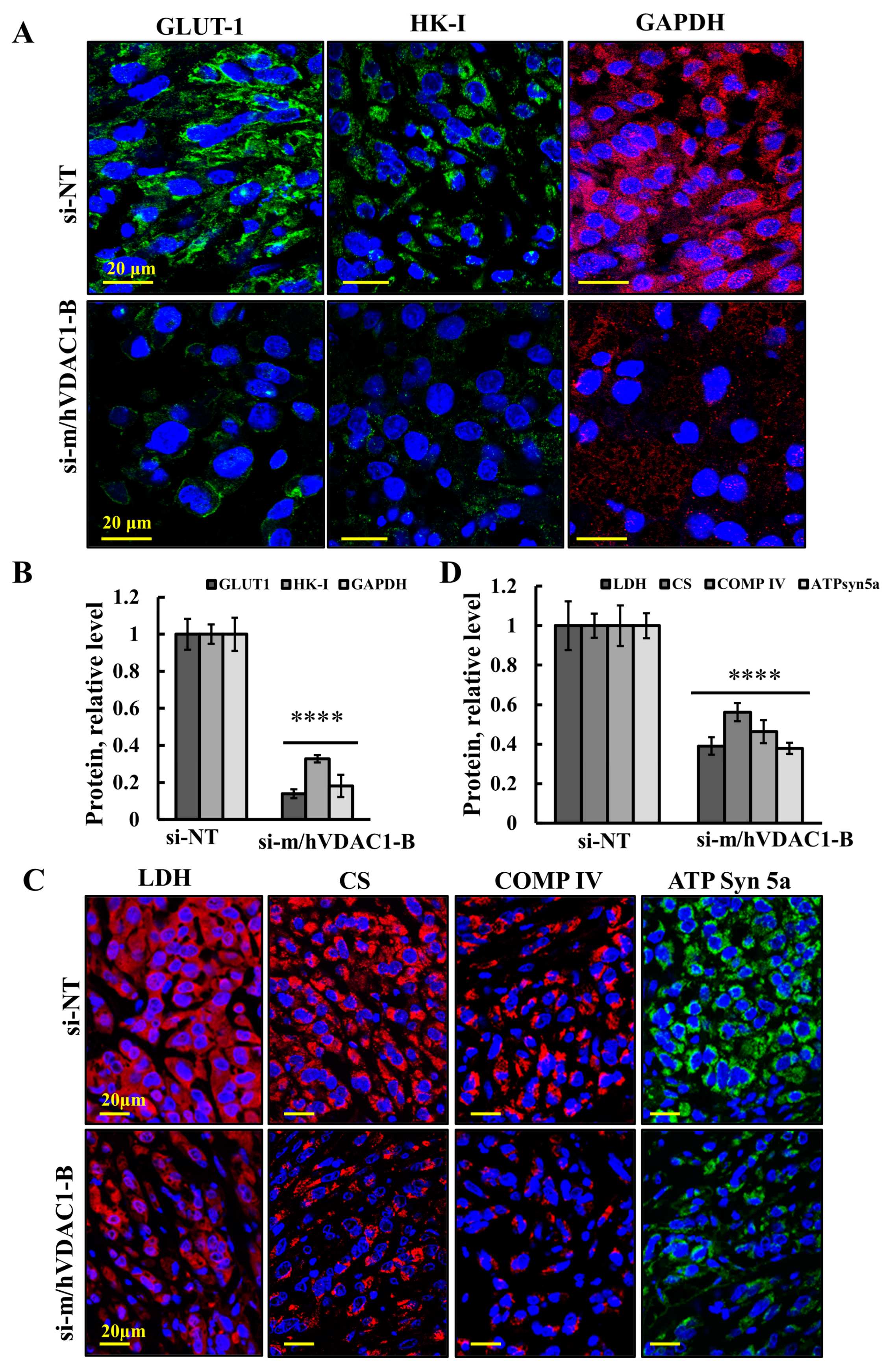
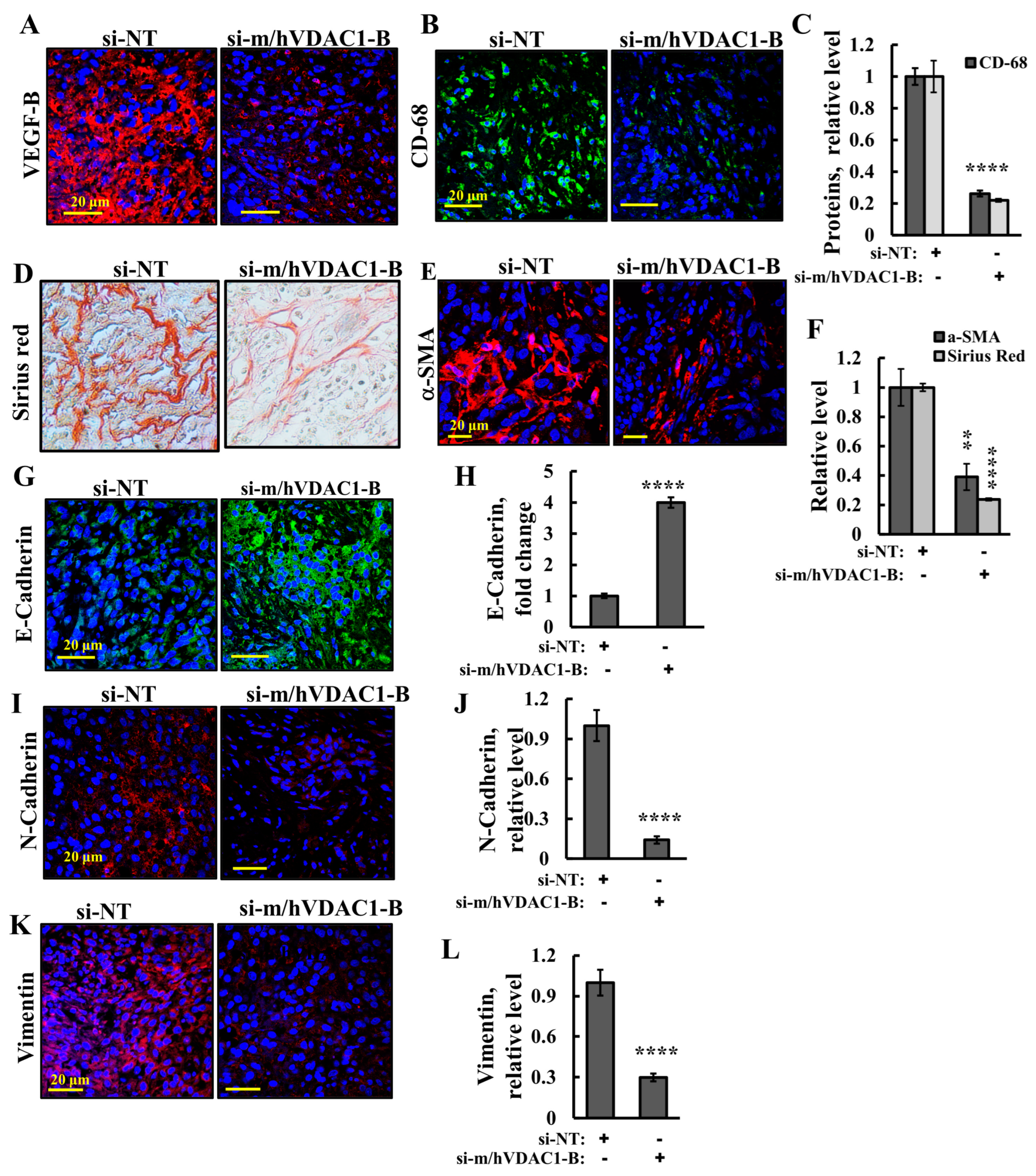
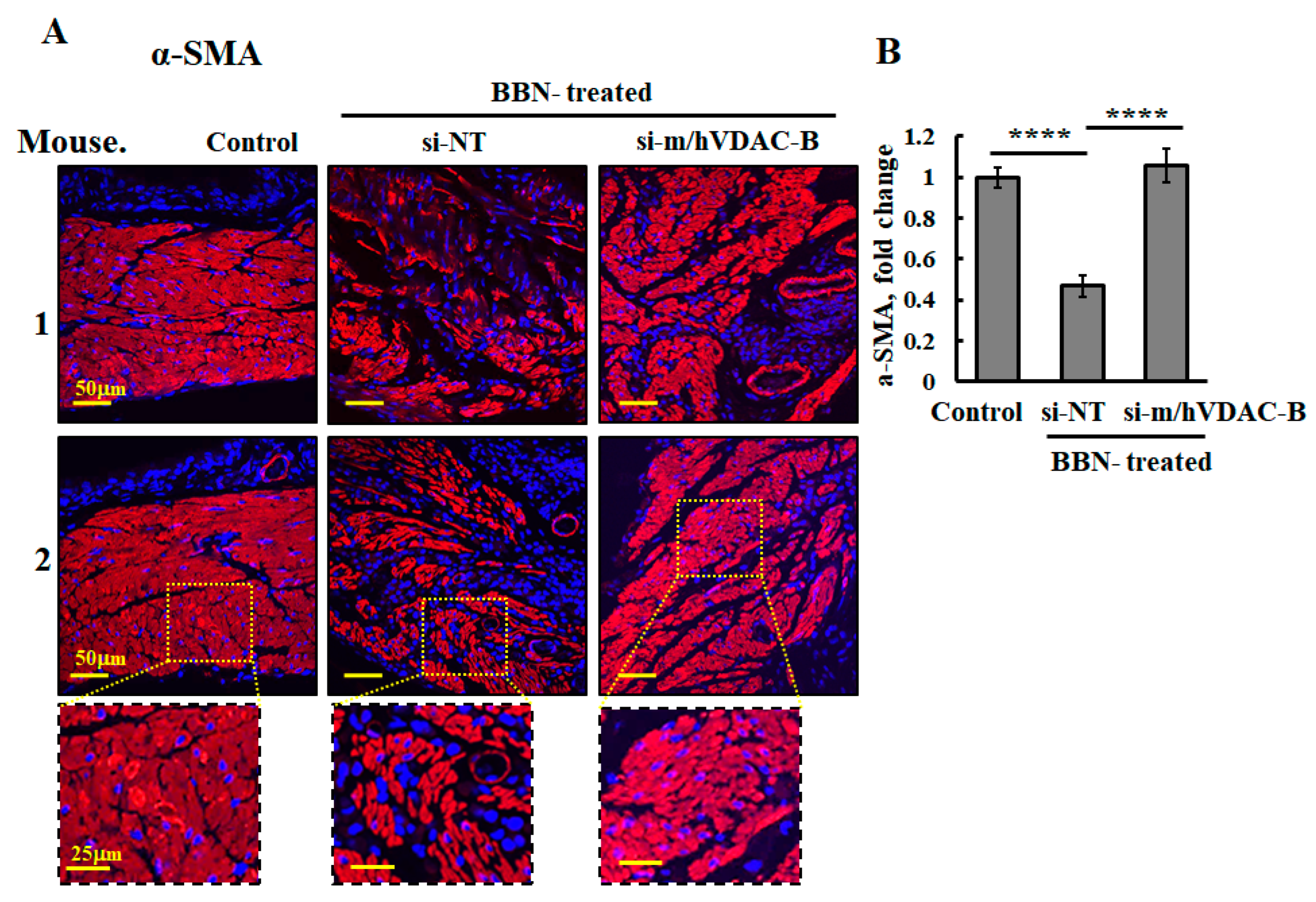
3.4. VDAC1 Reduced the Expression of Metabolism-Related Enzymes and Altered the Tumor Microenvironment
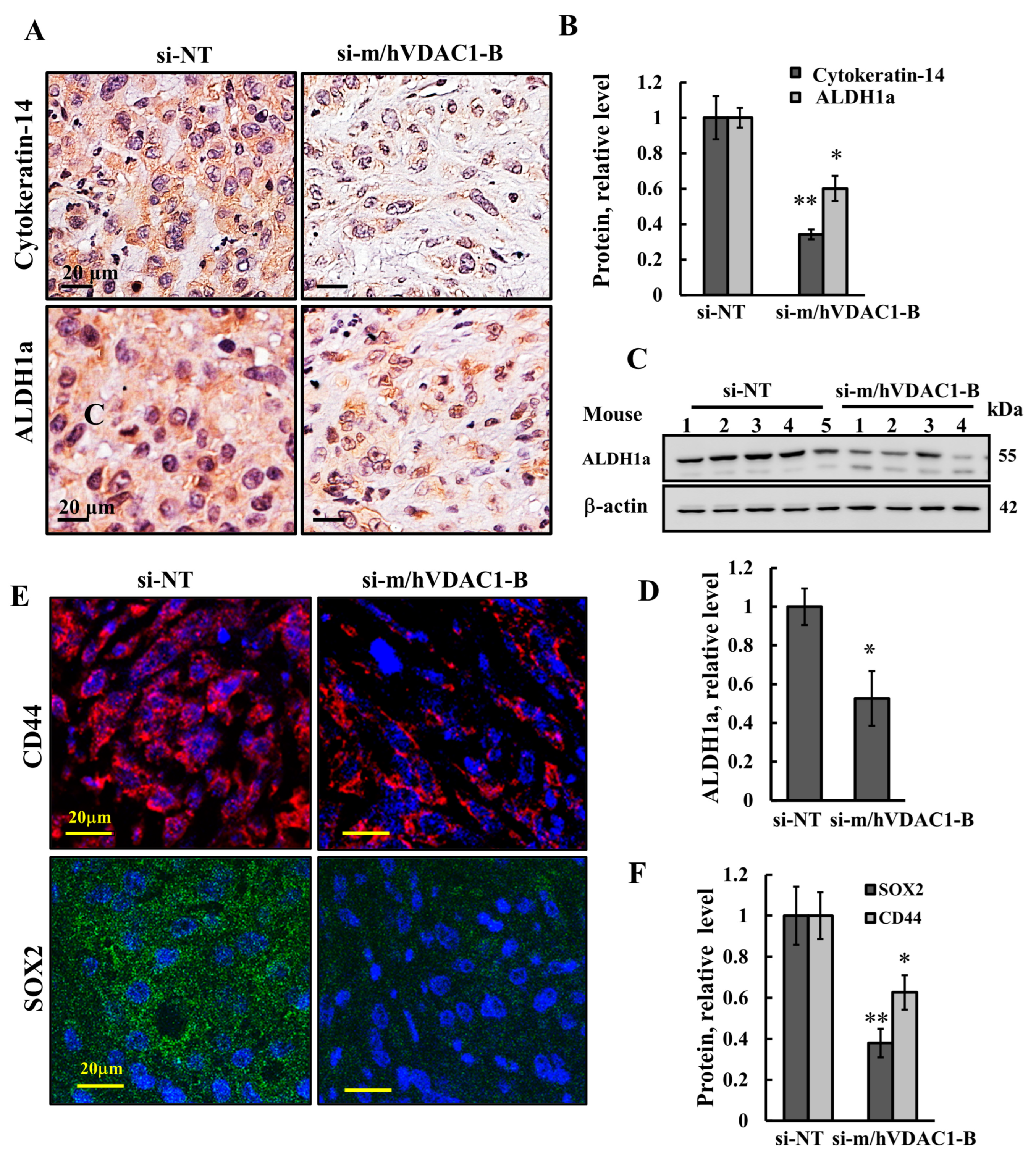
3.5. VDAC1 Depletion Reduced the Expression of Stem Cells
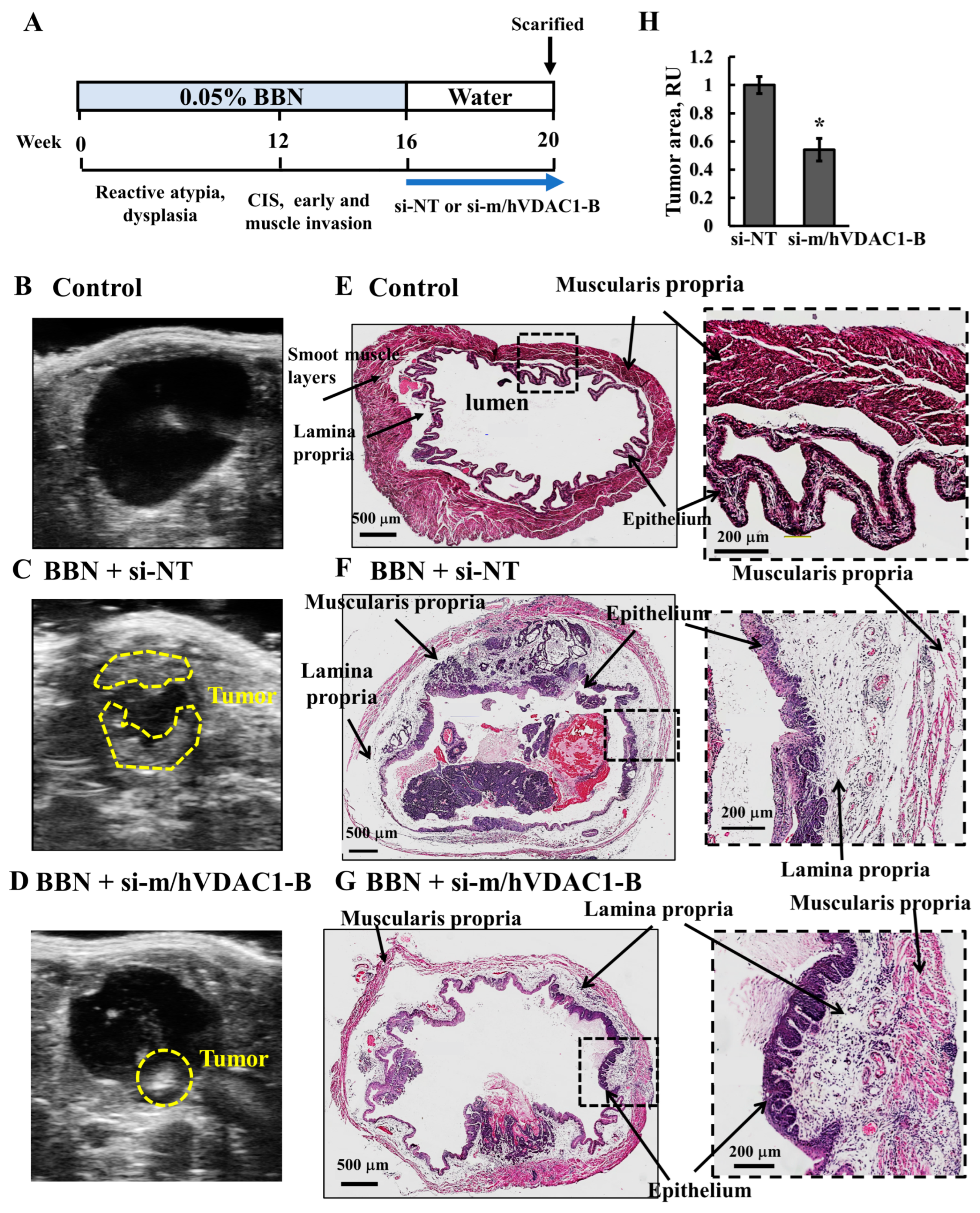
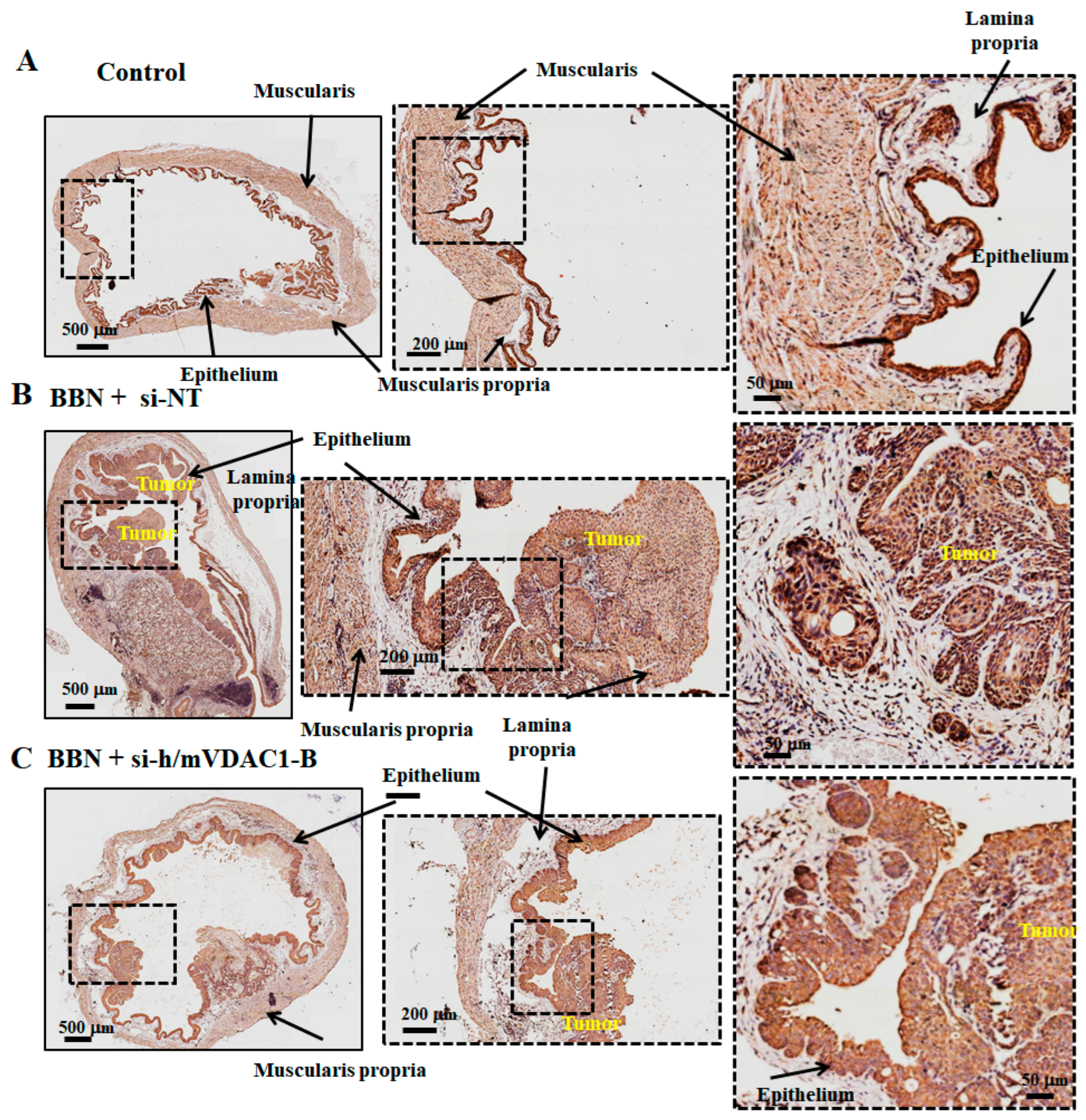
3.6. VDAC1 Reduction in Bladder Cancer in the Syngeneic Mouse Model as Induced by the Carcinogen BBN
4. Discussion
4.1. VDAC1 Is Highly Expressed in Bladder Cancer Cell Lines and Tissues
4.2. VDAC1 Silencing Reduced the Growth of BC Tumors and Induced Alterations in the Expression of Metabolism-, Microenvironment-, and Cancer-Stem-Cell-Related Proteins
4.3. si-VDAC1 Delivered Intravesical Attenuated BBN-Driven BC in a Mouse Model
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kaseb, H.; Aeddula, N.R. Bladder Cancer; StatPearls Publishing LLC: St. Petersburg, FL, USA, 2022. [Google Scholar]
- Pelucchi, C.; Bosetti, C.; Negri, E.; Malvezzi, M.; La Vecchia, C. Mechanisms of disease: The epidemiology of bladder cancer. Nat. Clin. Pract. Urol. 2006, 3, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Hinotsu, S.; Akaza, H.; Miki, T.; Fujimoto, H.; Shinohara, N.; Kikuchi, E.; Mizutani, Y.; Koga, H.; Okajima, E.; Okuyama, A.; et al. Bladder cancer develops 6 years earlier in current smokers: Analysis of bladder cancer registry data collected by the cancer registration committee of the Japanese Urological Association. Int. J. Urol. 2009, 16, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A.; Moch, H.; Cubilla, A.L.; Ulbright, T.M.; Reuter, V.E. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part B: Prostate and Bladder Tumours. Eur. Urol. 2016, 70, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Bochner, B.H.; Chou, R.; Dreicer, R.; Kamat, A.M.; Lerner, S.P.; Lotan, Y.; Meeks, J.J.; Michalski, J.M.; Morgan, T.M.; et al. Treatment of Non-Metastatic Muscle-Invasive Bladder Cancer: AUA/ASCO/ASTRO/SUO Guideline. J. Urol. 2017, 198, 552–559. [Google Scholar] [CrossRef]
- Kates, M.; Matoso, A.; Choi, W.; Baras, A.S.; Daniels, M.J.; Lombardo, K.; Brant, A.; Mikkilineni, N.; McConkey, D.J.; Kamat, A.M.; et al. Adaptive Immune Resistance to Intravesical BCG in Non-Muscle Invasive Bladder Cancer: Implications for Prospective BCG-Unresponsive Trials. Clin. Cancer Res. 2020, 26, 882–891. [Google Scholar] [CrossRef]
- Han, J.; Gu, X.; Li, Y.; Wu, Q. Mechanisms of BCG in the treatment of bladder cancer-current understanding and the prospect. Biomed. Pharmacother. 2020, 129, 110393. [Google Scholar] [CrossRef]
- Rhea, L.P.; Aragon-Ching, J.B. Advances and Controversies With Checkpoint Inhibitors in Bladder Cancer. Clin. Med. Insights Oncol. 2021, 15, 11795549211044963. [Google Scholar] [CrossRef]
- Nakano, I. Therapeutic potential of targeting glucose metabolism in glioma stem cells. Expert. Opin. Ther. Targets 2014, 18, 1233–1236. [Google Scholar] [CrossRef][Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, E.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. VDAC1 Silencing in Cancer Cells Leads to Metabolic Reprogramming That Modulates Tumor Microenvironment. Cancers 2021, 13, 2850. [Google Scholar] [CrossRef]
- Amsalem, Z.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers 2020, 12, 1031. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Lemasters, J.J. Warburg revisited: Regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J. Pharmacol. Exp. Ther. 2012, 342, 637–641. [Google Scholar] [CrossRef]
- Alshamsan, A.; Haddadi, A.; Hamdy, S.; Samuel, J.; El-Kadi, A.O.; Uludag, H.; Lavasanifar, A. STAT3 silencing in dendritic cells by siRNA polyplexes encapsulated in PLGA nanoparticles for the modulation of anticancer immune response. Mol. Pharm. 2010, 7, 1643–1654. [Google Scholar] [CrossRef]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Korac-Prlic, J.; Degoricija, M.; Vilovic, K.; Vujevic, S.; Terzic, J. BBN-driven urinary bladder cancer mouse model. Methods Cell Biol. 2021, 163, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.T.; Wolfe, D. Tissue Processing and Hematoxylin and Eosin Staining. In Histopathology: Methods in Molecular Biology; Day, C., Ed.; Humana Press: New York, NY, USA, 2014; Volume 1180. [Google Scholar]
- Pandey, S.K.; Machlof-Cohen, R.; Santhanam, M.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Silencing VDAC1 to Treat Mesothelioma Cancer: Tumor Reprograming and Altering Tumor Hallmarks. Biomolecules 2022, 12, 895. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef]
- Justus, C.R.; Sanderlin, E.J.; Yang, L.V. Molecular Connections between Cancer Cell Metabolism and the Tumor Microenvironment. Int. J. Mol. Sci. 2015, 16, 11055–11086. [Google Scholar] [CrossRef]
- Leblond, M.M.; Zdimerova, H.; Desponds, E.; Verdeil, G. Tumor-Associated Macrophages in Bladder Cancer: Biological Role, Impact on Therapeutic Response and Perspectives for Immunotherapy. Cancers 2021, 13, 4712. [Google Scholar] [CrossRef]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Investig. 2009, 119, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Res 2018, 7, 1796. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C.; et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 2009, 385, 307–313. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55. [Google Scholar] [CrossRef]
- Zhu, F.; Qian, W.; Zhang, H.; Liang, Y.; Wu, M.; Zhang, Y.; Zhang, X.; Gao, Q.; Li, Y. SOX2 Is a Marker for Stem-like Tumor Cells in Bladder Cancer. Stem Cell Rep. 2017, 9, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Papafotiou, G.; Paraskevopoulou, V.; Vasilaki, E.; Kanaki, Z.; Paschalidis, N.; Klinakis, A. KRT14 marks a subpopulation of bladder basal cells with pivotal role in regeneration and tumorigenesis. Nat. Commun. 2016, 7, 11914. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smith, C.C.; Utsumi, T.; Bixby, L.M.; Kardos, J.; Wobker, S.E.; Stewart, K.G.; Chai, S.; Manocha, U.; Byrd, K.M.; et al. Molecular Subtype-Specific Immunocompetent Models of High-Grade Urothelial Carcinoma Reveal Differential Neoantigen Expression and Response to Immunotherapy. Cancer Res. 2018, 78, 3954–3968. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.O.; Sopena, J.M.; Favaro, W.J.; Martin, M.C.; Simao, A.F.; Reis, R.B.; Andrade, M.F.; Domenech, J.D.; Cardo, C.C. Anatomical features of the urethra and urinary bladder catheterization in female mice and rats. An essential translational tool. Acta Cir. Bras. 2011, 26 (Suppl. S2), 106–110. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Marin-Hernandez, A.; Saavedra, E.; Pardo, J.P.; Ralph, S.J.; Rodriguez-Enriquez, S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 2014, 50, 10–23. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef]
- Alexa, A.; Baderca, F.; Lighezan, R.; Izvernariu, D. Myofibroblasts reaction in urothelial carcinomas. Rom. J. Morphol. Embryol. 2009, 50, 639–643. [Google Scholar] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Aghaalikhani, N.; Rashtchizadeh, N.; Shadpour, P.; Allameh, A.; Mahmoodi, M. Cancer stem cells as a therapeutic target in bladder cancer. J. Cell. Physiol. 2019, 234, 3197–3206. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Bertucci, F.; Cabaud, O.; Wicinski, J.; Finetti, P.; Josselin, E.; Adelaide, J.; Nguyen, T.T.; Monville, F.; et al. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013, 73, 7290–7300. [Google Scholar] [CrossRef]
- Wang, L.; Park, P.; Zhang, H.; La Marca, F.; Lin, C.Y. Prospective identification of tumorigenic osteosarcoma cancer stem cells in OS99-1 cells based on high aldehyde dehydrogenase activity. Int. J. Cancer 2011, 128, 294–303. [Google Scholar] [CrossRef]
- Kitamura, H.; Torigoe, T.; Hirohashi, Y.; Asanuma, H.; Inoue, R.; Nishida, S.; Tanaka, T.; Fukuta, F.; Masumori, N.; Sato, N.; et al. Prognostic impact of the expression of ALDH1 and SOX2 in urothelial cancer of the upper urinary tract. Mod. Pathol. 2013, 26, 117–124. [Google Scholar] [CrossRef]
- Li, Y.P.; Jia, X.P.; Jiang, Y.Q.; Wang, W.; Wang, Y.L.; Wang, X.L.; Guo, Y.X. Differential expression of cytokeratin 14 and 18 in bladder cancer tumorigenesis. Exp. Biol. Med. 2018, 243, 344–349. [Google Scholar] [CrossRef]
- Wu, C.T.; Lin, W.Y.; Chang, Y.H.; Chen, W.C.; Chen, M.F. Impact of CD44 expression on radiation response for bladder cancer. J. Cancer 2017, 8, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Lin, W.Y.; Chen, W.C.; Chen, M.F. Predictive Value of CD44 in Muscle-Invasive Bladder Cancer and Its Relationship with IL-6 Signaling. Ann. Surg. Oncol. 2018, 25, 3518–3526. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J., Jr.; Chang, H.Y.; van de Rijn, M.; et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.V.; Edin, S.; Eklöf, V.; Öberg, Å.; Palmqvist, R.; Wikberg, M.L. SOX2 expression is associated with a cancer stem cell state and down-regulation of CDX2 in colorectal cancer. BMC Cancer 2016, 16, 471. [Google Scholar] [CrossRef]
- Ruan, J.; Wei, B.; Xu, Z.; Yang, S.; Zhou, Y.; Yu, M.; Liang, J.; Jin, K.; Huang, X.; Lu, P.; et al. Predictive value of Sox2 expression in transurethral resection specimens in patients with T1 bladder cancer. Med. Oncol. 2013, 30, 445. [Google Scholar] [CrossRef]
- Papadaki, S.; Magklara, A. Regulation of Metabolic Plasticity in Cancer Stem Cells and Implications in Cancer Therapy. Cancers 2022, 14, 5912. [Google Scholar] [CrossRef]
- Nogawa, M.; Yuasa, T.; Kimura, S.; Tanaka, M.; Kuroda, J.; Sato, K.; Yokota, A.; Segawa, H.; Toda, Y.; Kageyama, S.; et al. Intravesical administration of small interfering RNA targeting PLK-1 successfully prevents the growth of bladder cancer. J. Clin. Investig. 2005, 115, 978–985. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhozeel, B.; Pandey, S.K.; Shteinfer-Kuzmine, A.; Santhanam, M.; Shoshan-Barmatz, V. Silencing the Mitochondrial Gatekeeper VDAC1 as a Potential Treatment for Bladder Cancer. Cells 2024, 13, 627. https://doi.org/10.3390/cells13070627
Alhozeel B, Pandey SK, Shteinfer-Kuzmine A, Santhanam M, Shoshan-Barmatz V. Silencing the Mitochondrial Gatekeeper VDAC1 as a Potential Treatment for Bladder Cancer. Cells. 2024; 13(7):627. https://doi.org/10.3390/cells13070627
Chicago/Turabian StyleAlhozeel, Belal, Swaroop Kumar Pandey, Anna Shteinfer-Kuzmine, Manikandan Santhanam, and Varda Shoshan-Barmatz. 2024. "Silencing the Mitochondrial Gatekeeper VDAC1 as a Potential Treatment for Bladder Cancer" Cells 13, no. 7: 627. https://doi.org/10.3390/cells13070627
APA StyleAlhozeel, B., Pandey, S. K., Shteinfer-Kuzmine, A., Santhanam, M., & Shoshan-Barmatz, V. (2024). Silencing the Mitochondrial Gatekeeper VDAC1 as a Potential Treatment for Bladder Cancer. Cells, 13(7), 627. https://doi.org/10.3390/cells13070627