
A Novel Approach for Glioblastoma Treatment by Combining Apoptosis Inducers (TMZ, MTX, and Cytarabine) with E.V.A. (Eltanexor, Venetoclax, and A1210477) Inhibiting XPO1, Bcl-2, and Mcl-1

 , , ,
, , ,  , ,
, ,  ,
,  and
and
Abstract
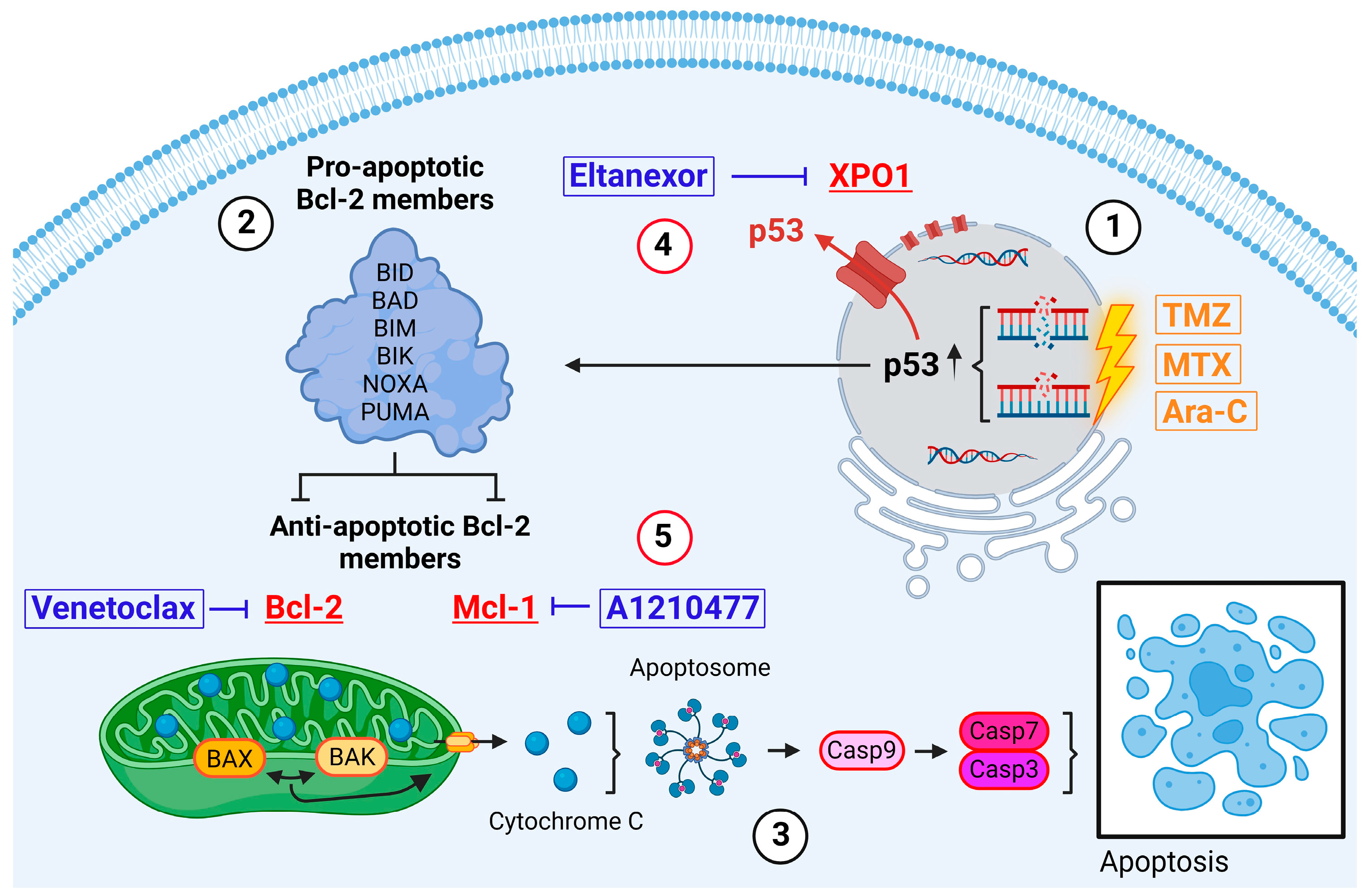
1. Introduction
1.1. Resistance to Apoptosis
1.2. Accessibility
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs
2.3. Cell Viability Assay
2.4. RNA Isolation and Real-Time RT-PCR
2.5. Gene Expression Analysis and Survival Curve Analysis
2.6. Protein Isolation and Western Blot Analysis
2.7. Apoptosis Assay
2.8. Flow Cytometry Analysis
2.9. Brain Slice Culture
2.10. Immunofluorescence Staining for Brain Slice Culture
2.11. Quantification of Neuronal Apoptosis in Mouse Brain Slice Cultures
2.12. Sampling of Liquor and Blood Samples from Patients
2.13. Quantification of Venetoclax in Patient-Derived Material
2.14. Statistical Analyses
3. Results
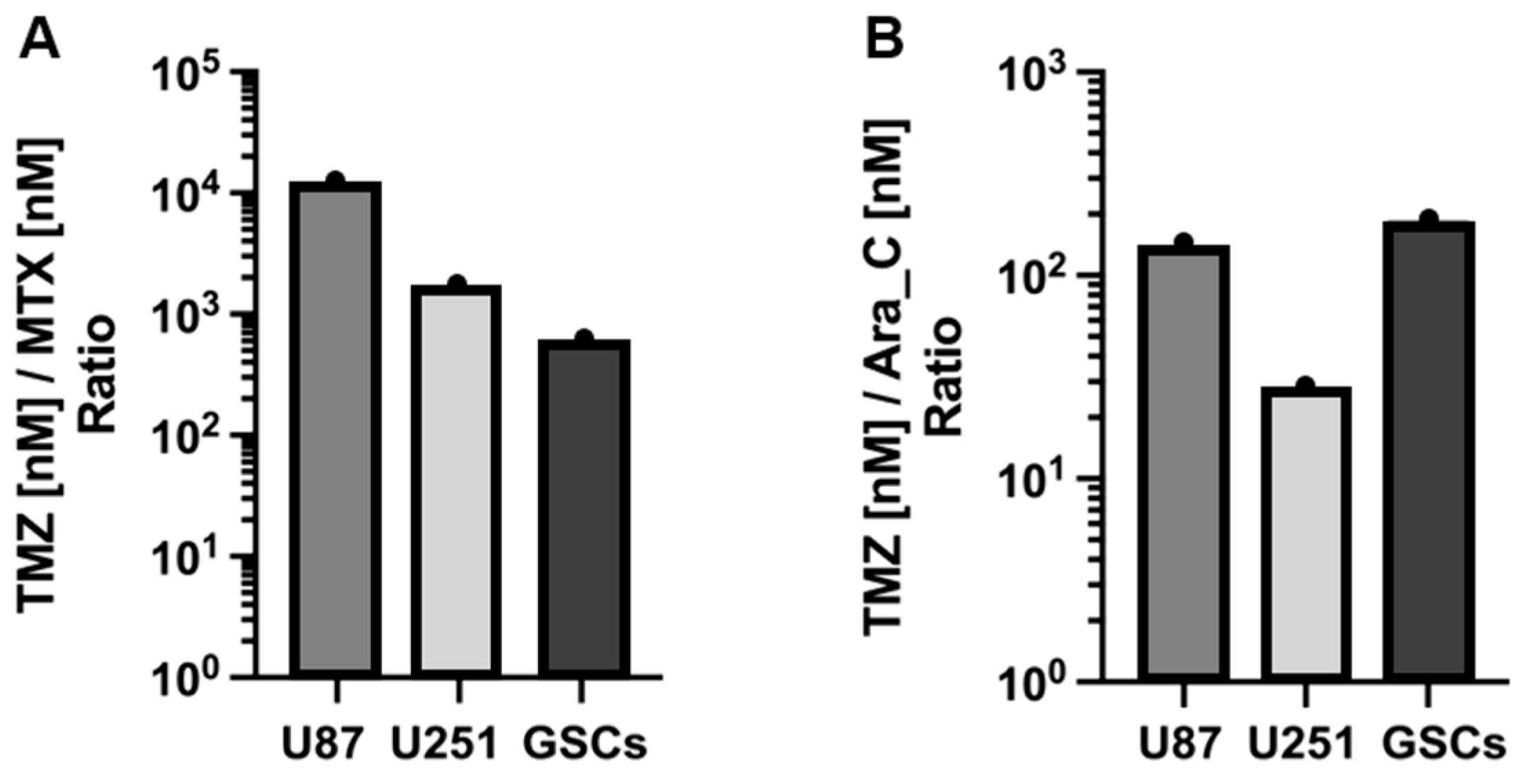
3.1. Cytotoxicity of TMZ, MTX, and Ara-C in GB Cell Lines and GSCs
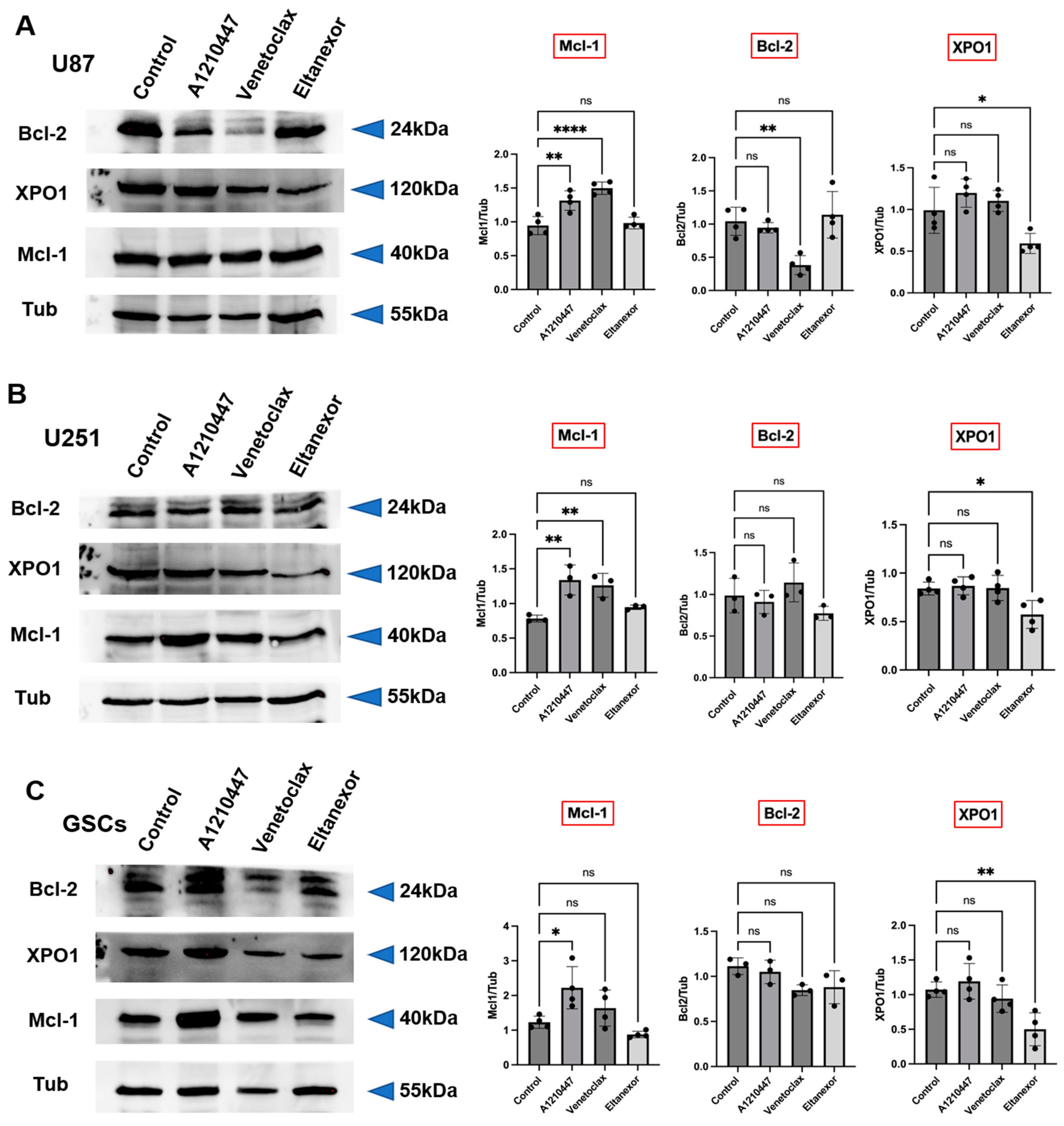
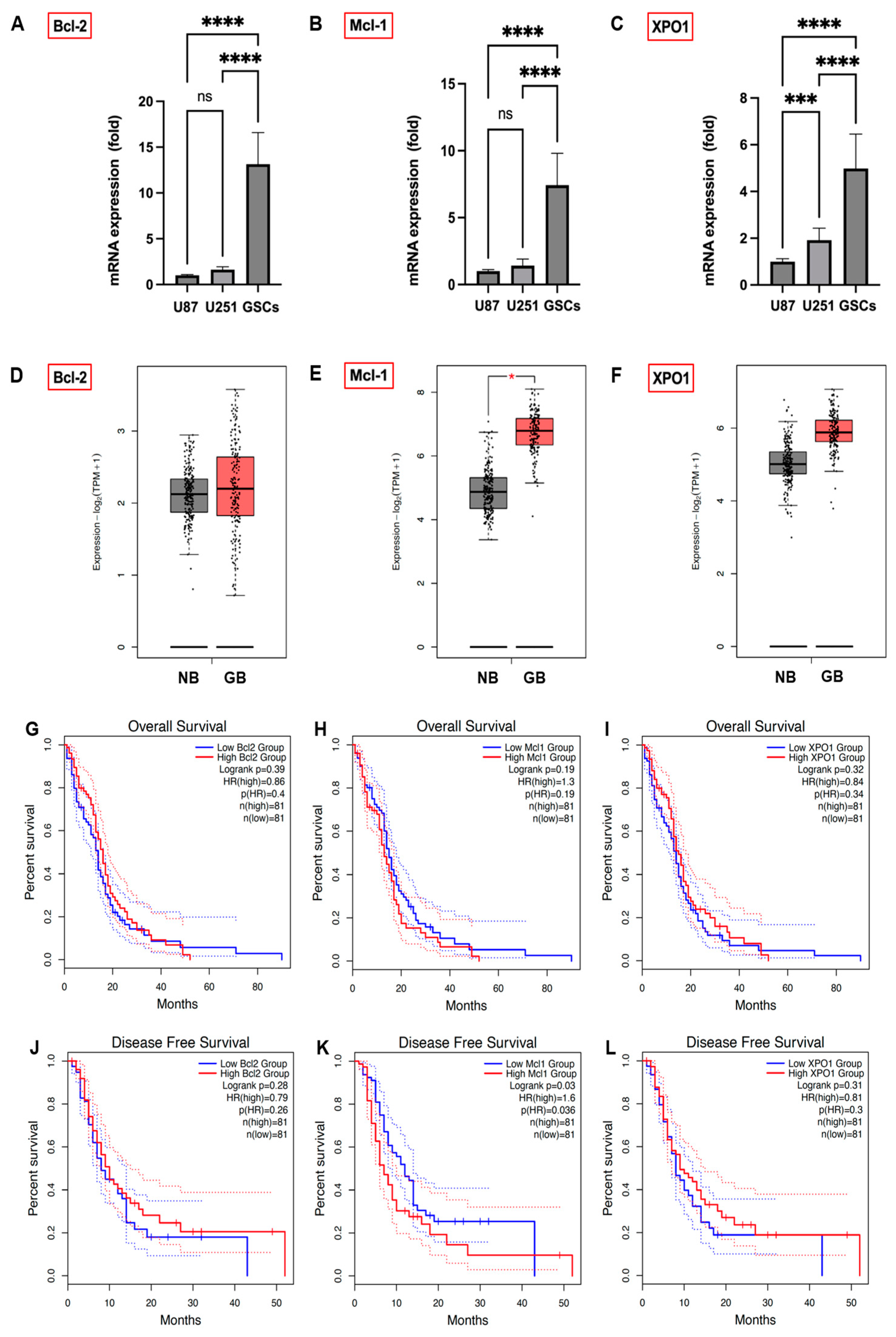
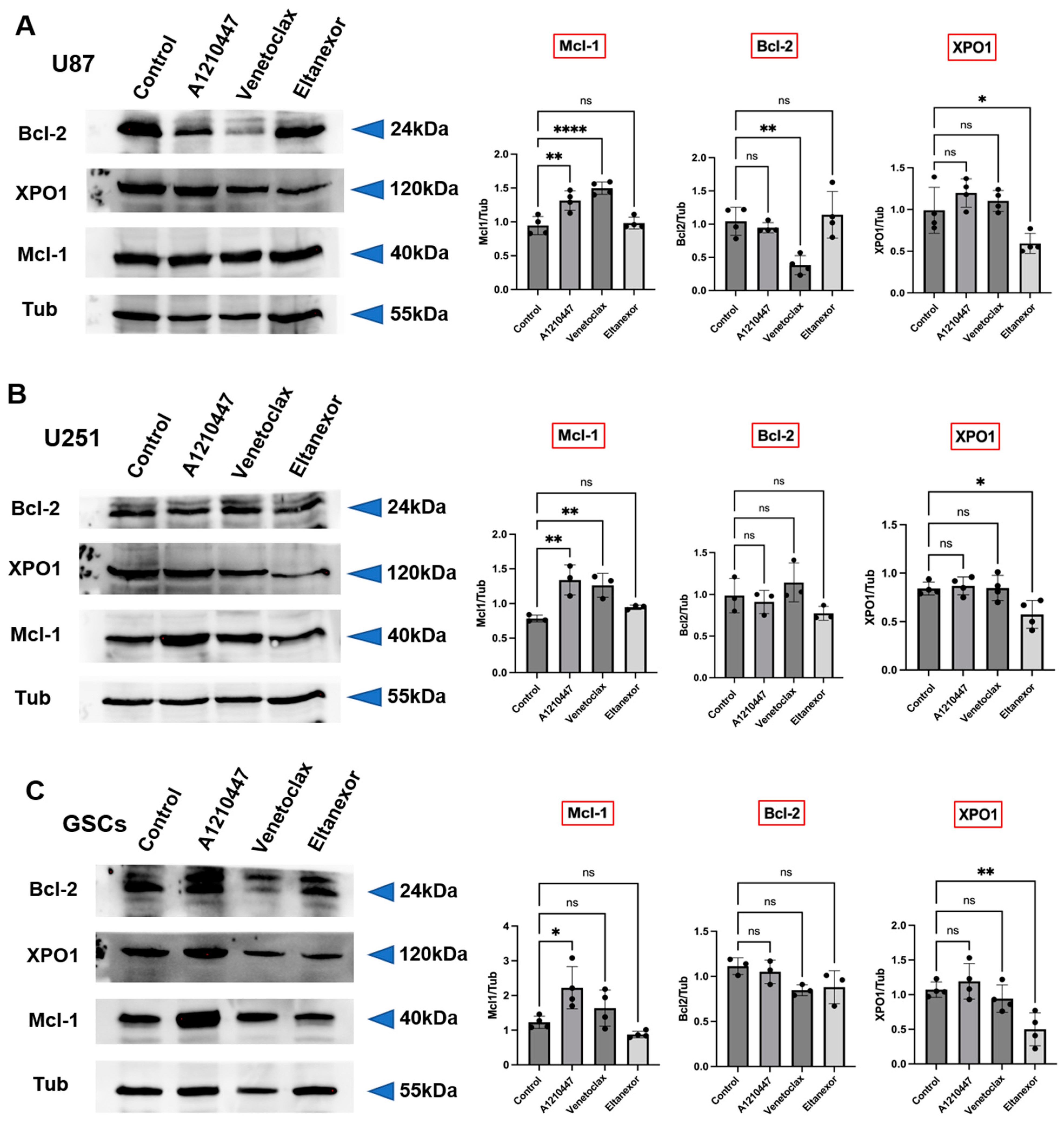
3.2. Expression Levels of Bcl-2, Mcl-1, and XPO1 in GB Cells, GSCs, and GB Patients
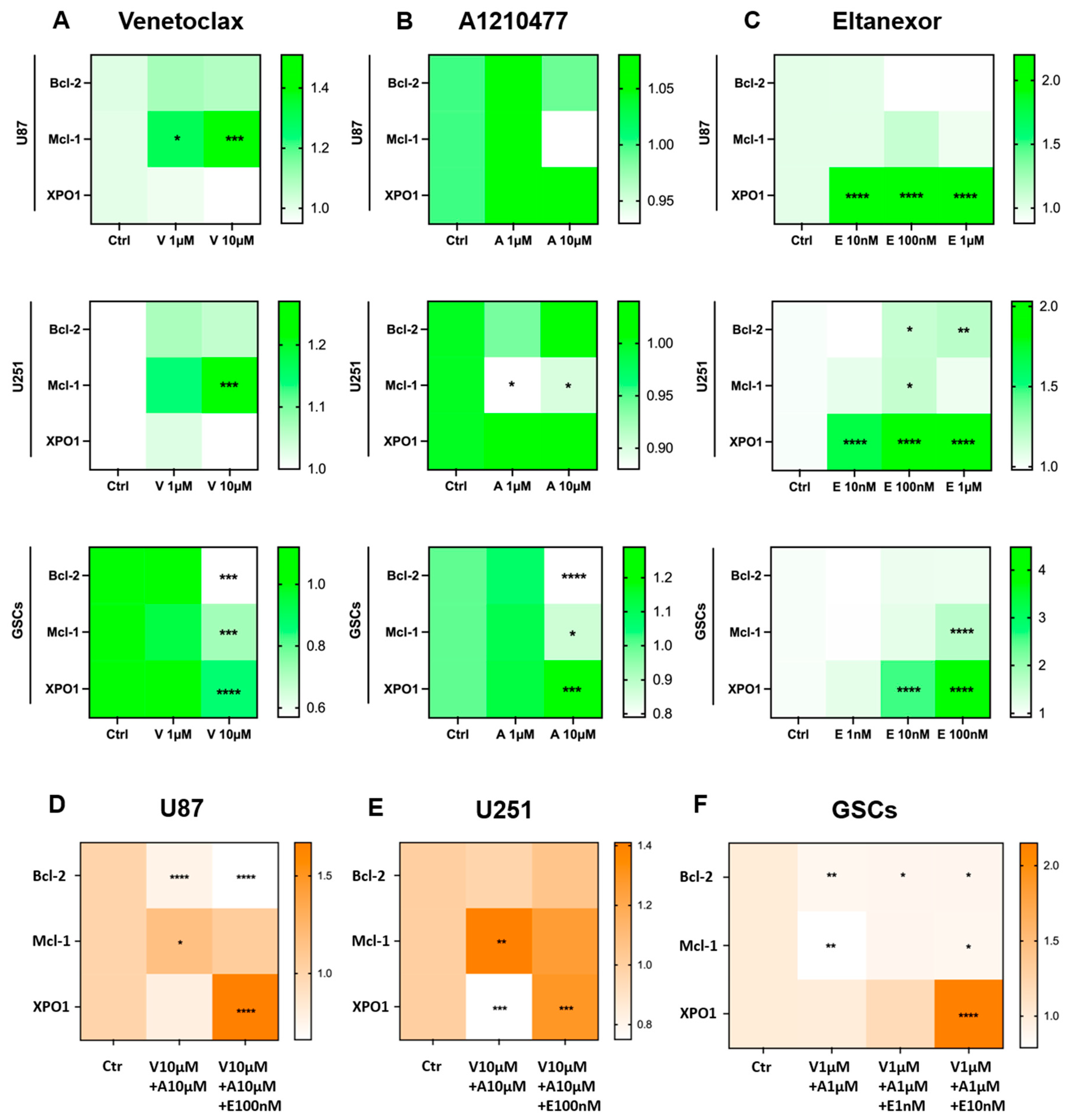
3.3. Treatment with E., V., and A. Results in Alterations of Bcl-2, Mcl-1, XPO1 Gene Expression at Both the Transcriptional and Translational Levels
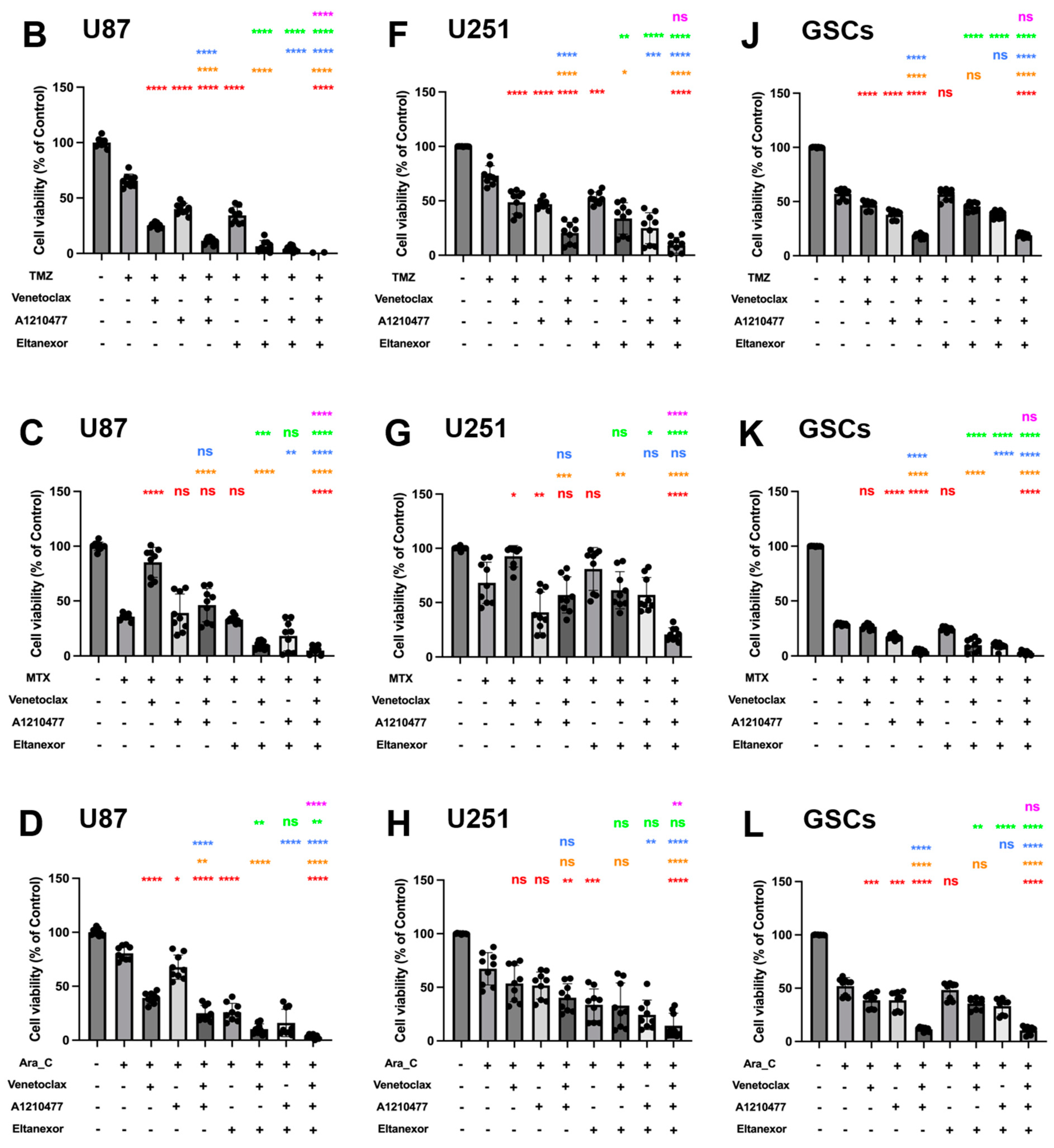
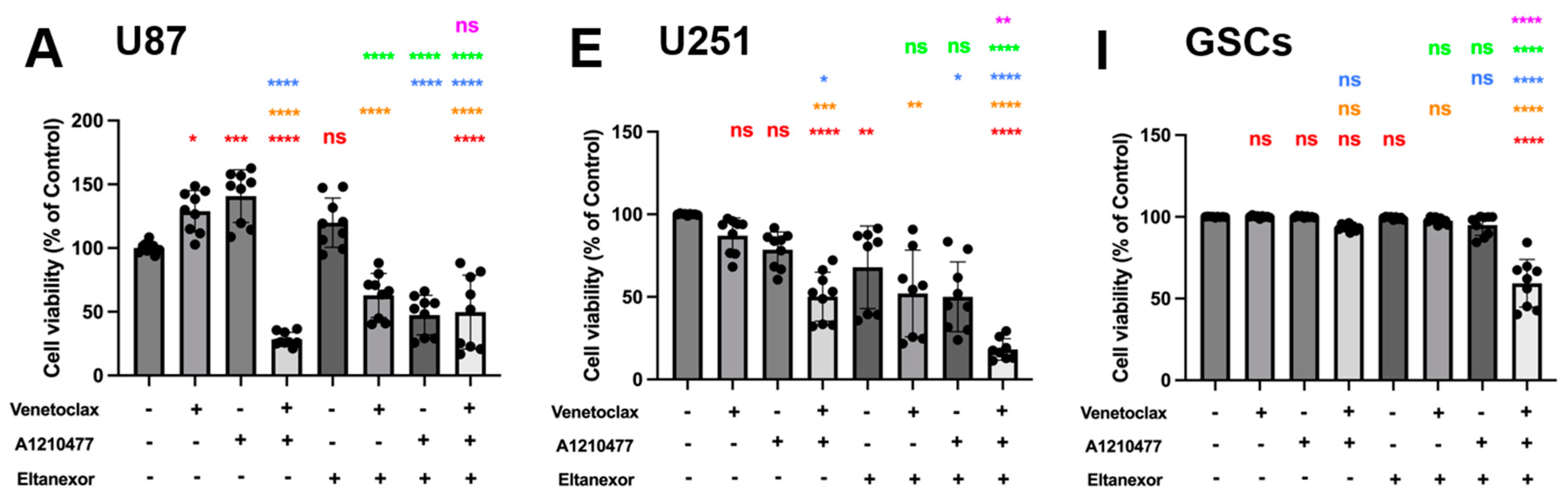
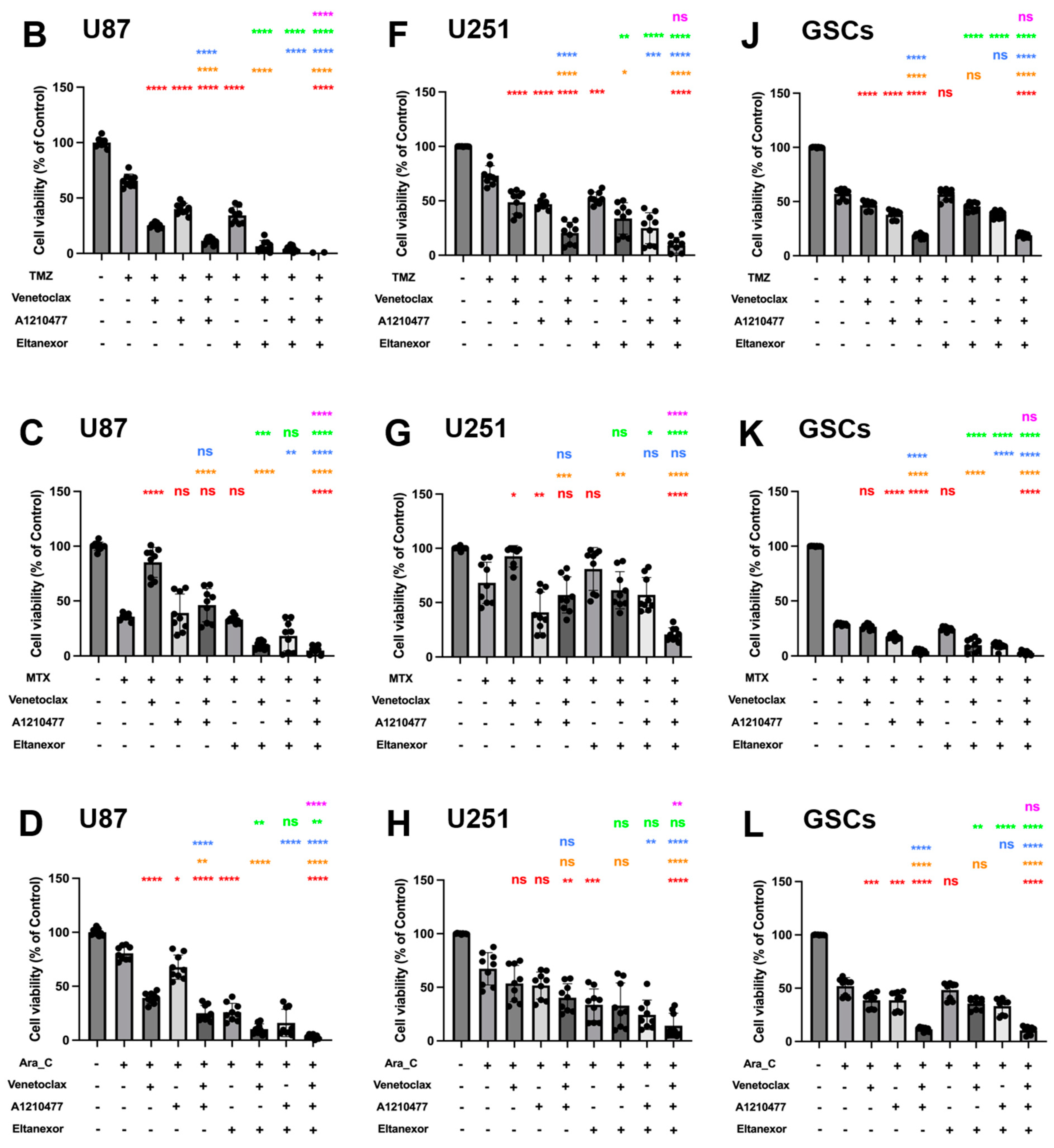
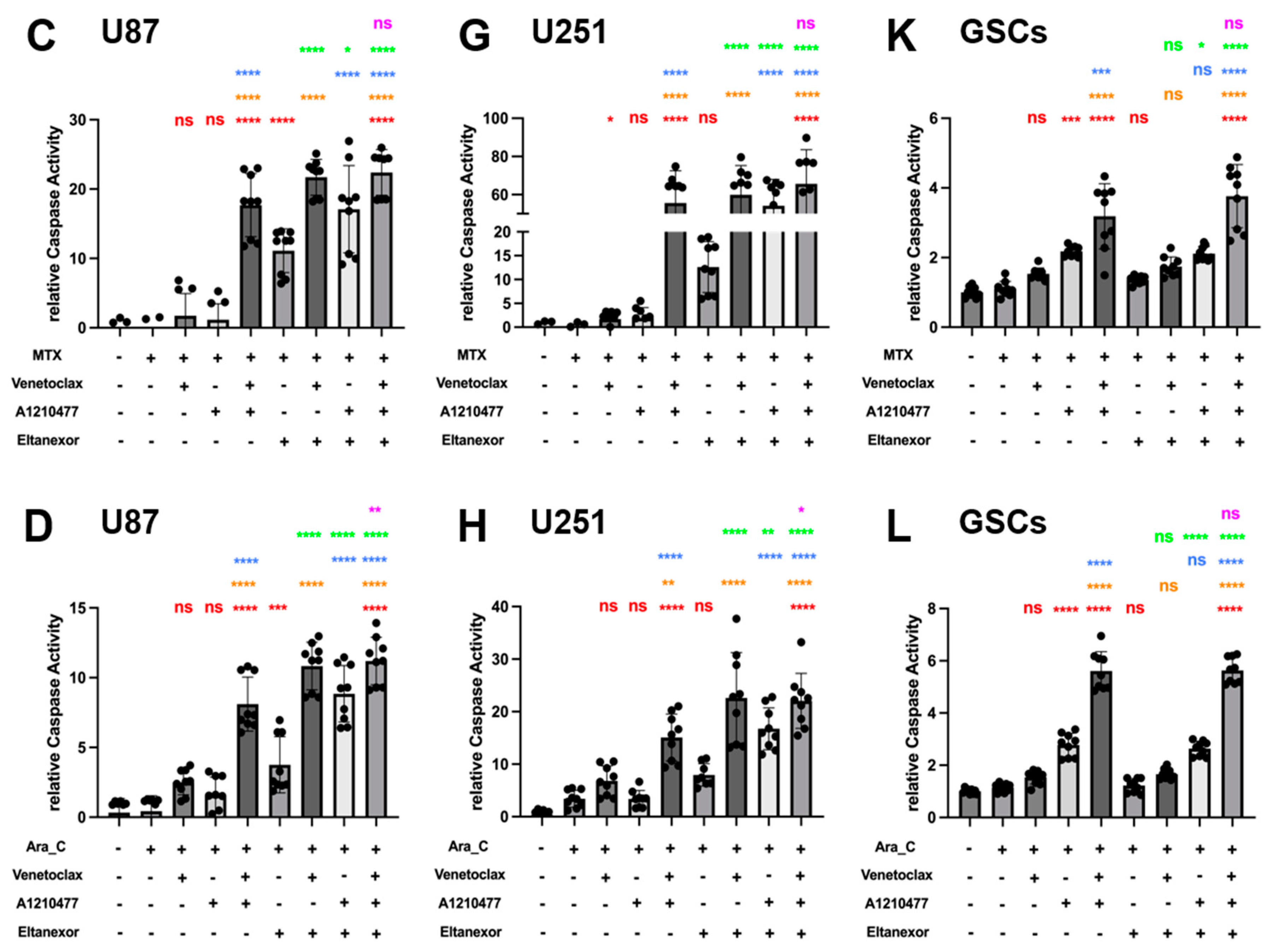
3.4. Evaluation of Cell Viability upon Combinatorial Treatment with E., V., A. and Chemotherapeutic Drugs in U87 and U251 Cells, as Well as GSCs
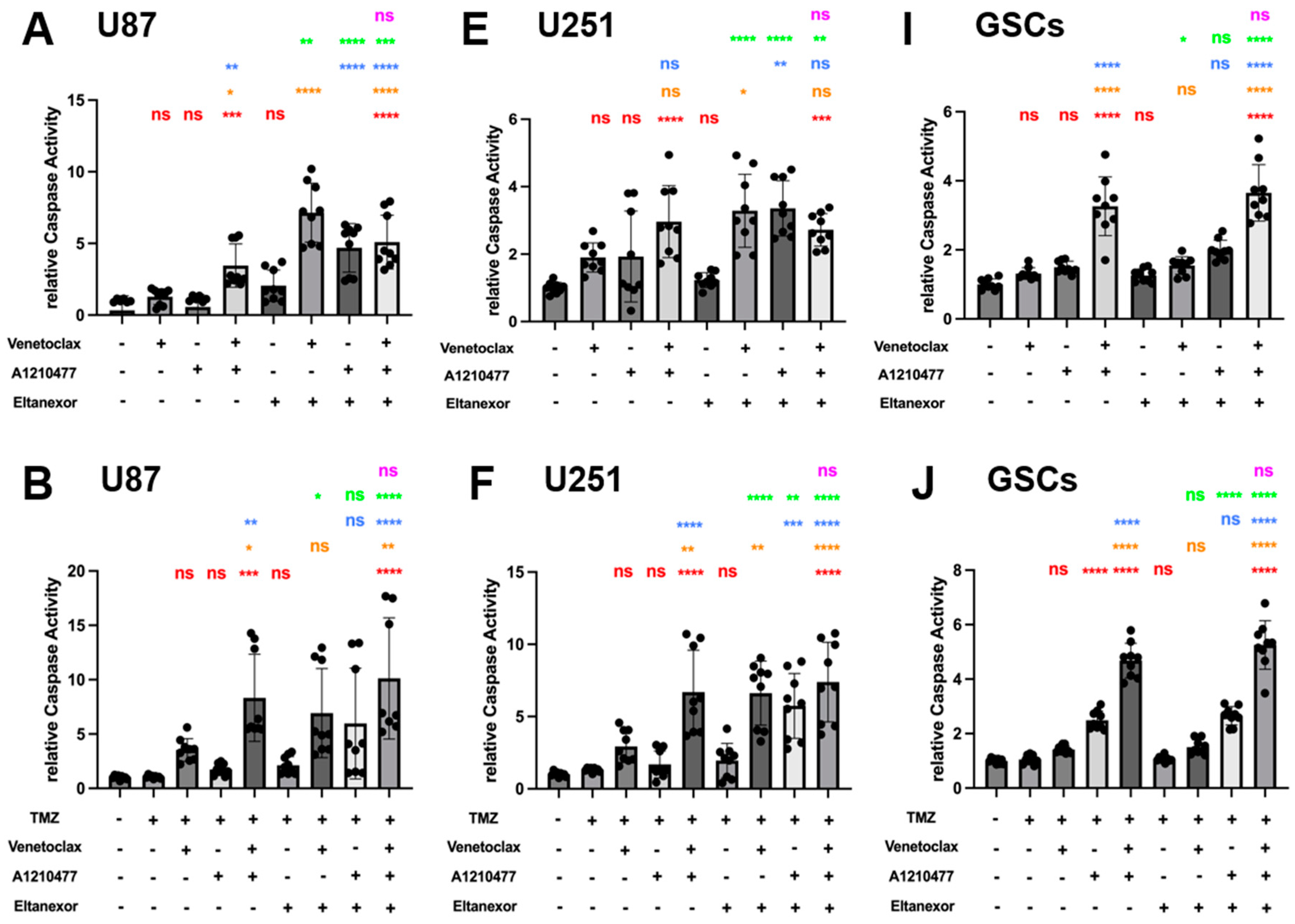
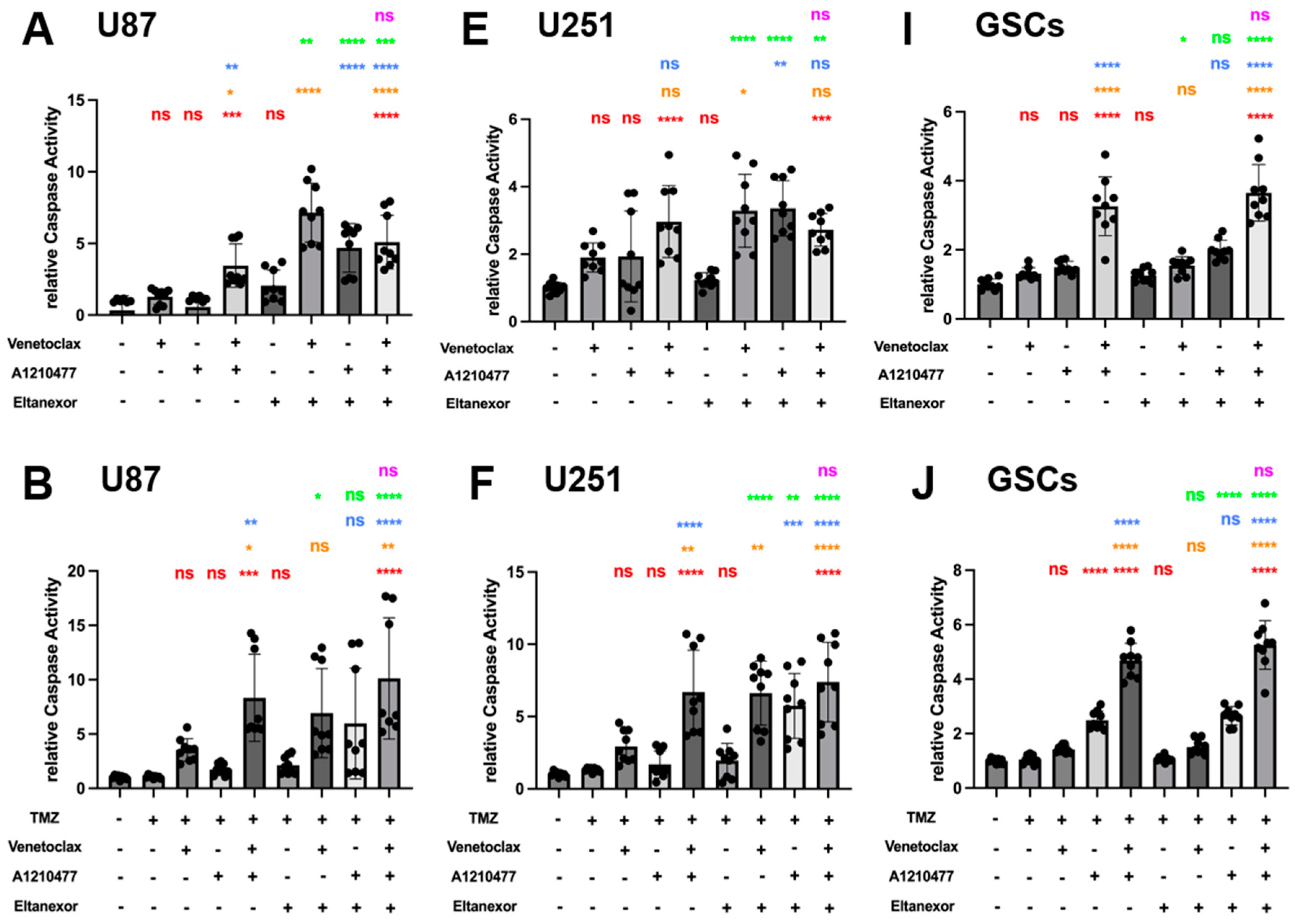
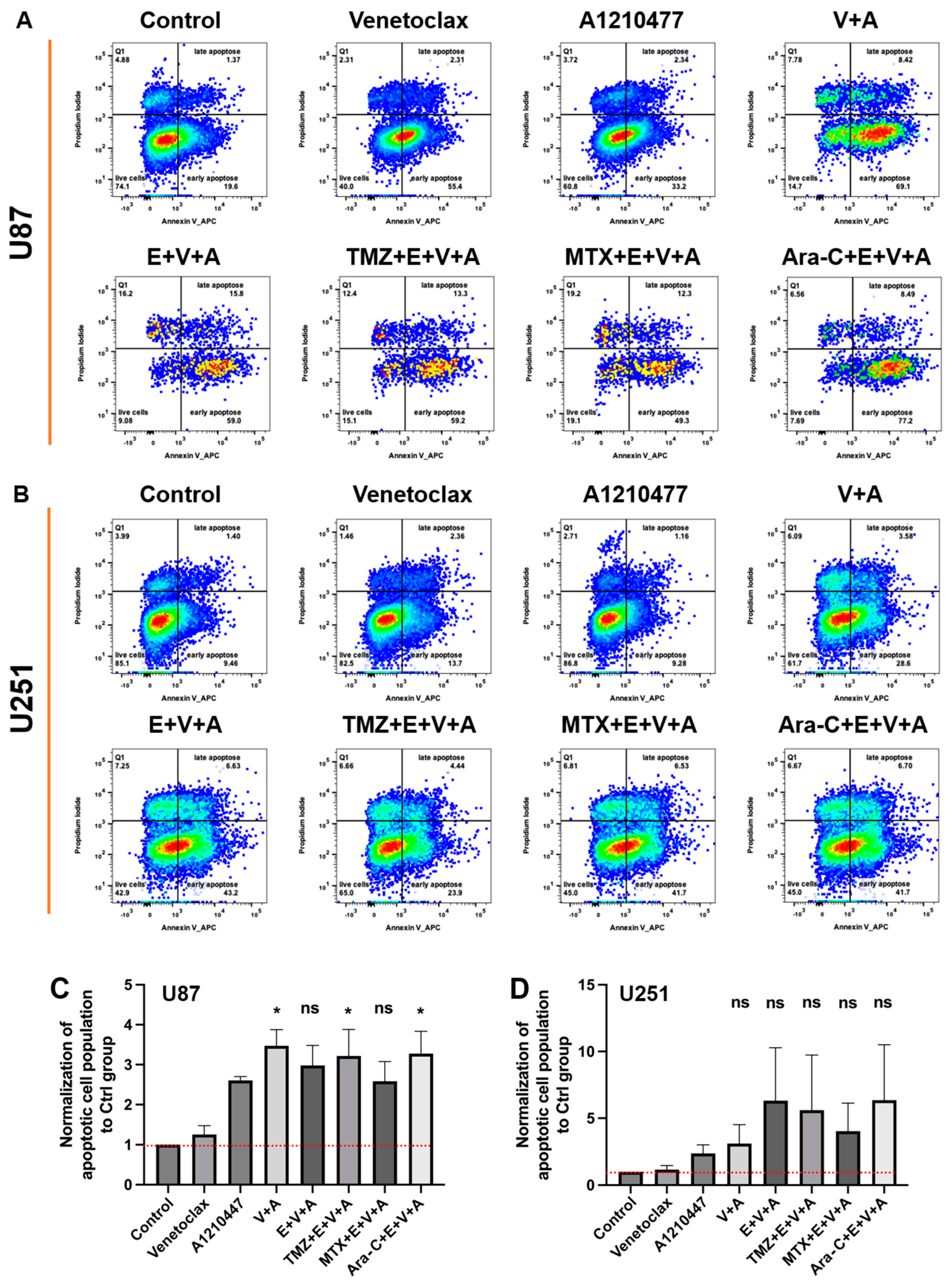
3.5. Evaluation of Cell Apoptosis upon Combinatorial Treatment with E., V., A. and Chemotherapeutic Drugs in U87 and U251 Cells, as Well as GSCs
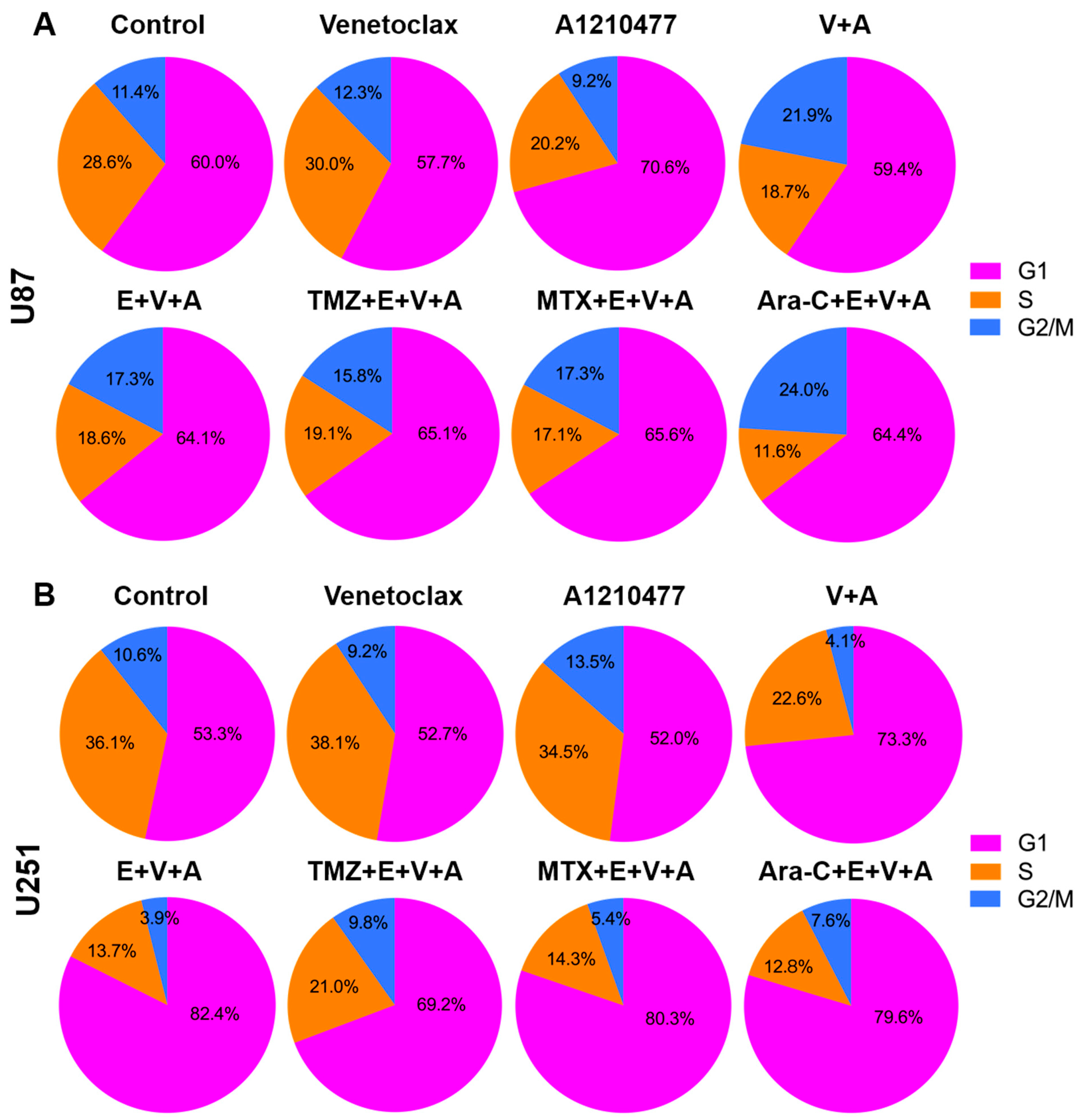
3.6. Detection of Cell Cycle Arrest and Apoptosis by Flow Cytometry
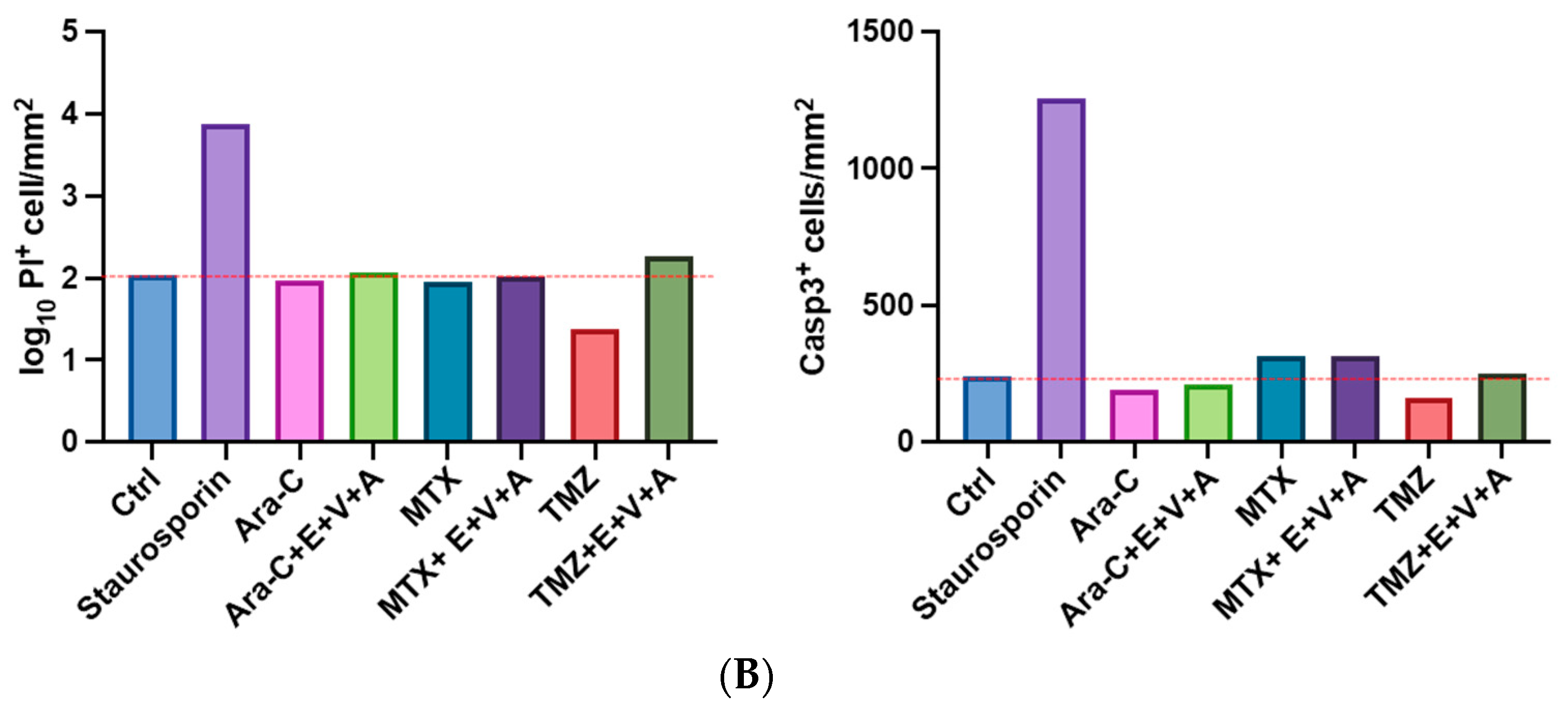
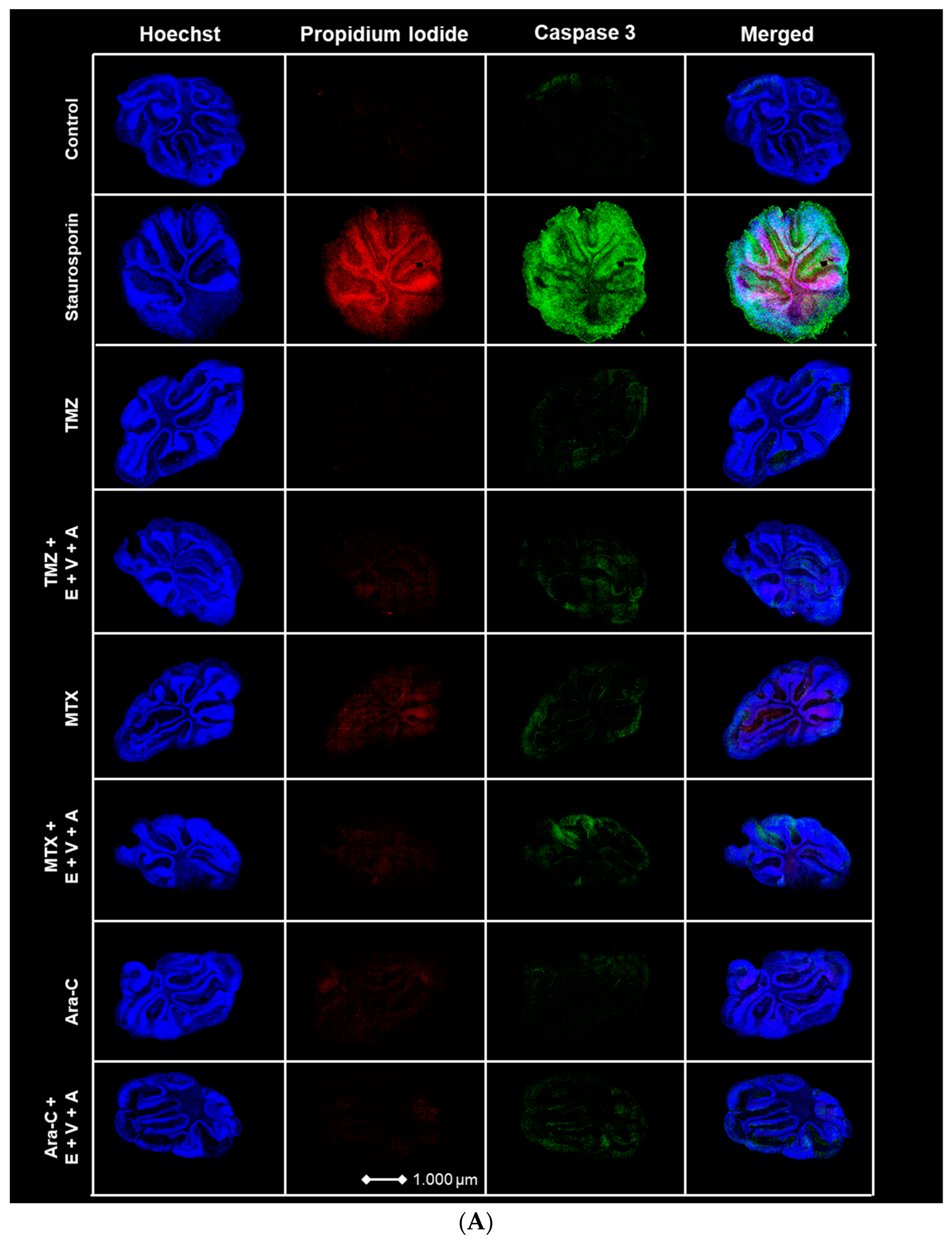
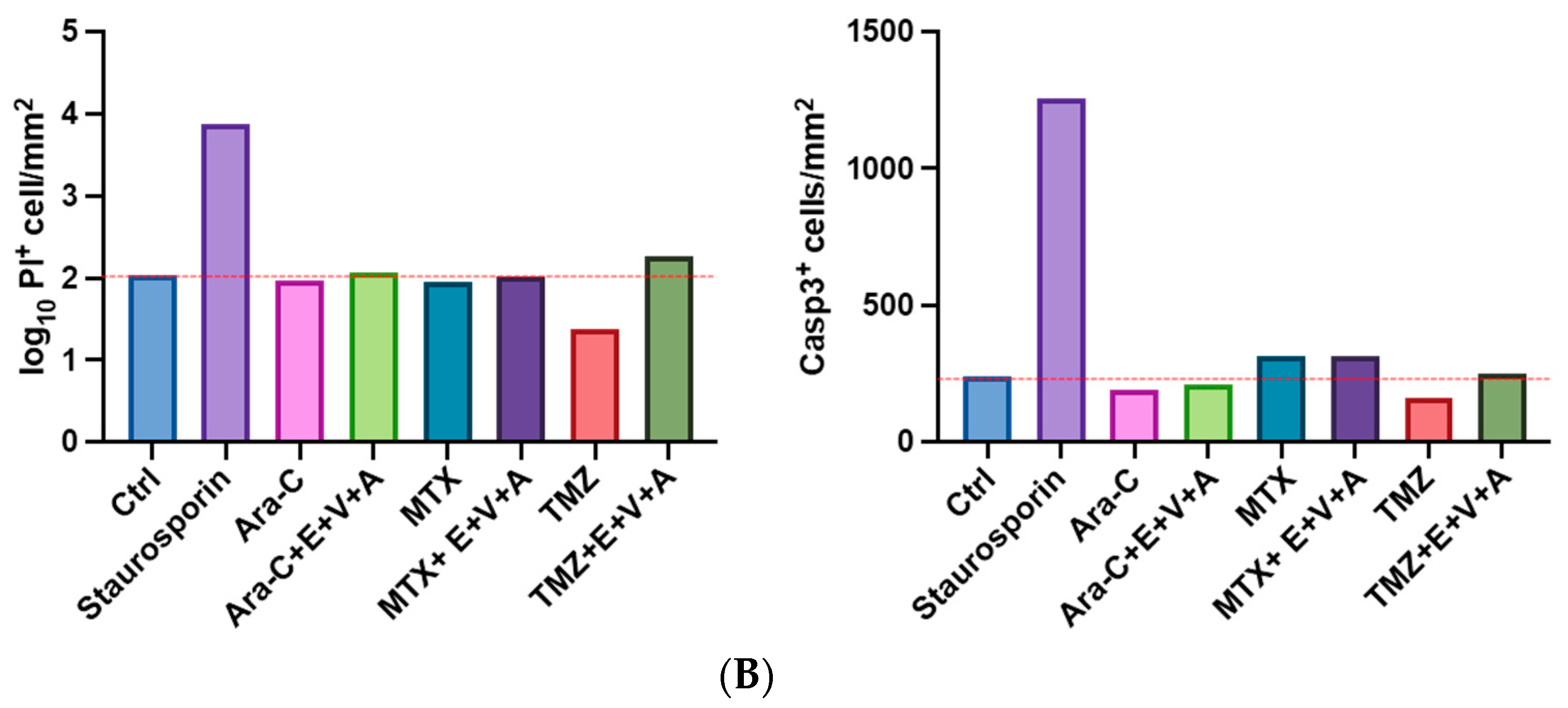
3.7. Apoptosis and Dead Cells Detected by Immunofluorescence Staining in Brain Slice Culture
3.8. Distribution of Venetoclax in CSF and Plasma of Patients after Oral Administration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef] [PubMed]
- King, J.L.; Benhabbour, S.R. Glioblastoma Multiforme-A Look at the Past and a Glance at the Future. Pharmaceutics 2021, 13, 1053. [Google Scholar] [CrossRef] [PubMed]
- Helseth, R.; Helseth, E.; Johannesen, T.B.; Langberg, C.W.; Lote, K.; Rønning, P.; Scheie, D.; Vik, A.; Meling, T.R. Overall survival, prognostic factors, and repeated surgery in a consecutive series of 516 patients with glioblastoma multiforme. Acta Neurol. Scand. 2010, 122, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Dinesan, M.; Ajayakumar, T. Survival and quality of life analysis in glioblastoma multiforme with adjuvant chemoradiotherapy: A retrospective study. Rep. Pract. Oncol. Radiother. 2022, 27, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H. Epidemiology of brain tumors. Methods Mol. Biol. 2009, 472, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, G.S.; Lyutfi, E.; Georgieva, R.; Georgiev, R.; Dzhenkov, D.L.; Petkova, L.; Ivanov, B.D.; Kaprelyan, A.; Ghenev, P. Reclassification of Glioblastoma Multiforme According to the 2021 World Health Organization Classification of Central Nervous System Tumors: A Single Institution Report and Practical Significance. Cureus 2022, 14, e21822. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Ottaviani, D.; Tazare, J.; Gregson, J.; Kitchen, N.; Brandner, S.; Fersht, N.; Mulholland, P. Survival Outcomes and Prognostic Factors in Glioblastoma. Cancers 2022, 14, 3161. [Google Scholar] [CrossRef] [PubMed]
- Darefsky, A.S.; King, J.T., Jr.; Dubrow, R. Adult glioblastoma multiforme survival in the temozolomide era: A population-based analysis of Surveillance, Epidemiology, and End Results registries. Cancer 2012, 118, 2163–2172. [Google Scholar] [CrossRef]
- Taghavi, M.S.; Akbarzadeh, A.; Mahdian, R.; Azadmanesh, K.; Javadi, G. Cisplatin downregulates BCL2L12, a novel apoptosis-related gene, in glioblastoma cells. In Vitro Cell. Dev. Biol. Anim. 2013, 49, 465–472. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed Res. Int. 2017, 7403747. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Ishida, C.T.; Zhang, Y.; Halatsch, M.E.; Westhoff, M.A.; Siegelin, M.D. Targeting intrinsic apoptosis and other forms of cell death by BH3-mimetics in glioblastoma. Expert Opin. Drug Discov. 2017, 12, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Krawiec, K.; Strzałka, P.; Czemerska, M.; Wiśnik, A.; Zawlik, I.; Wierzbowska, A.; Pluta, A. Targeting Apoptosis in AML: Where Do We Stand? Cancers 2022, 14, 4995. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Antitumor imidazotetrazines--XV. Role of guanine O6 alkylation in the mechanism of cytotoxicity of imidazotetrazinones. Biochem. Pharmacol. 1987, 36, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Short, N.J.; Nasr, L.; Dabaja, B.S.; Fang, P.Q. Central Nervous System Prophylaxis and Treatment in Acute Leukemias. Curr. Treat. Options Oncol. 2022, 23, 1829–1844. [Google Scholar] [CrossRef]
- Djukic, M.; Trimmel, R.; Nagel, I.; Spreer, A.; Lange, P.; Stadelmann, C.; Nau, R. Cerebrospinal fluid abnormalities in meningeosis neoplastica: A retrospective 12-year analysis. Fluids Barriers CNS 2017, 14, 7. [Google Scholar] [CrossRef]
- Morikawa, N.; Mori, T.; Kawashima, H.; Fujiki, M.; Abe, T.; Kaku, T.; Konisi, Y.; Takeyama, M.; Hori, S. Pharmacokinetics of nimustine, methotrexate, and cytosine arabinoside during cerebrospinal fluid perfusion chemotherapy in patients with disseminated brain tumors. Eur. J. Clin. Pharmacol. 1998, 54, 415–420. [Google Scholar] [CrossRef]
- Rajagopalan, P.T.; Zhang, Z.; McCourt, L.; Dwyer, M.; Benkovic, S.J.; Hammes, G.G. Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 13481–13486. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, A.; Tadi, P. Cytarabine; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Townsend, P.A.; Kozhevnikova, M.V.; Cexus, O.N.F.; Zamyatnin, A.A., Jr.; Soond, S.M. BH3-mimetics: Recent developments in cancer therapy. J. Exp. Clin. Cancer Res. 2021, 40, 355. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Uren, R.T.; Iyer, S.; Kluck, R.M. Pore formation by dimeric Bak and Bax: An unusual pore? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160218. [Google Scholar] [CrossRef]
- Green, A.L.; Ramkissoon, S.H.; McCauley, D.; Jones, K.; Perry, J.A.; Hsu, J.H.; Ramkissoon, L.A.; Maire, C.L.; Hubbell-Engler, B.; Knoff, D.S.; et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol. 2015, 17, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Pareja, F.; Macleod, D.; Shu, C.; Crary, J.F.; Canoll, P.D.; Ross, A.H.; Siegelin, M.D. PI3K and Bcl-2 inhibition primes glioblastoma cells to apoptosis through downregulation of Mcl-1 and Phospho-BAD. Mol. Cancer Res. 2014, 12, 987–1001. [Google Scholar] [CrossRef]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef]
- Hutten, S.; Kehlenbach, R.H. CRM1-mediated nuclear export: To the pore and beyond. Trends Cell Biol. 2007, 17, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Ma, B.; Yang, Z.Y.; Li, O.; Liu, S.L.; Pan, L.J.; Gong, W.; Dong, P.; Shu, Y.J. Inhibition of XPO1 impairs cholangiocarcinoma cell proliferation by triggering p53 intranuclear accumulation. Cancer Med. 2023, 12, 5751–5763. [Google Scholar] [CrossRef] [PubMed]
- Otte, K.; Zhao, K.; Braun, M.; Neubauer, A.; Raifer, H.; Helmprobst, F.; Barrera, F.O.; Nimsky, C.; Bartsch, J.W.; Rusch, T. Eltanexor Effectively Reduces Viability of Glioblastoma and Glioblastoma Stem-Like Cells at Nano-Molar Concentrations and Sensitizes to Radiotherapy and Temozolomide. Biomedicines 2022, 10, 2145. [Google Scholar] [CrossRef] [PubMed]
- Karyopharm Therapeutics Inc. Press Release. Karyopharm Received Orphan Drug Designation from the FDA for Eltanexor for the Treatment of Myelodysplastic Syndromes. News Release 2022. Available online: https://bit.ly/3znInSG (accessed on 31 January 2024).
- Roberts, A.W.; Wei, A.H.; Huang, D.C.S. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood 2021, 138, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Widden, H.; Placzek, W.J. The multiple mechanisms of MCL1 in the regulation of cell fate. Commun. Biol. 2021, 4, 1029. [Google Scholar] [CrossRef] [PubMed]
- Mihalyova, J.; Jelinek, T.; Growkova, K.; Hrdinka, M.; Simicek, M.; Hajek, R. Venetoclax: A new wave in hematooncology. Exp. Hematol. 2018, 61, 10–25. [Google Scholar] [CrossRef]
- Tagscherer, K.E.; Fassl, A.; Campos, B.; Farhadi, M.; Kraemer, A.; Böck, B.C.; Macher-Goeppinger, S.; Radlwimmer, B.; Wiestler, O.D.; Herold-Mende, C.; et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 2008, 27, 6646–6656. [Google Scholar] [CrossRef]
- Shang, E.; Nguyen, T.T.T.; Shu, C.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. Epigenetic Targeting of Mcl-1 Is Synthetically Lethal with Bcl-xL/Bcl-2 Inhibition in Model Systems of Glioblastoma. Cancers 2020, 12, 2137. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Ayoub, E.; Ostermann, L.B.; Huang, X.; Loghavi, S.; Boettcher, S.; Nishida, Y.; Ruvolo, V.; et al. Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics. Blood Cancer J. 2023, 13, 57. [Google Scholar] [CrossRef]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Weng, S.Y.; Yang, C.Y.; Li, C.C.; Sun, T.P.; Tung, S.Y.; Yen, J.J.; Tsai, T.F.; Chen, C.M.; Chen, S.H.; Hsiao, M.; et al. Synergism between p53 and Mcl-1 in protecting from hepatic injury, fibrosis and cancer. J. Hepatol. 2011, 54, 685–694. [Google Scholar] [CrossRef]
- Upton, D.H.; Ung, C.; George, S.M.; Tsoli, M.; Kavallaris, M.; Ziegler, D.S. Challenges and opportunities to penetrate the blood-brain barrier for brain cancer therapy. Theranostics 2022, 12, 4734–4752. [Google Scholar] [CrossRef] [PubMed]
- Abrey, L.E.; Christodoulou, C. Temozolomide for treating brain metastases. Semin. Oncol. 2001, 28 (Suppl. S13), 34–42. [Google Scholar] [CrossRef]
- Tang, L.; Feng, Y.; Gao, S.; Mu, Q.; Liu, C. Nanotherapeutics Overcoming the Blood-Brain Barrier for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 786700. [Google Scholar] [CrossRef] [PubMed]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, F.; Wang, H.; Lu, F.; Xue, H. Treatment of central nervous system relapse in PLZF::RARA-positive acute promyelocytic leukemia by venetoclax combined with arubicin, cytarabine and intrathecal therapy: A case report. Oncologie 2023, 26, 337–342. [Google Scholar] [CrossRef]
- Reda, G.; Cassin, R.; Dovrtelova, G.; Matteo, C.; Giannotta, J.; D’Incalci, M.; Cortelezzi, A.; Zucchetti, M. Venetoclax penetrates in cerebrospinal fluid and may be effective in chronic lymphocytic leukemia with central nervous system involvement. Haematologica 2019, 104, e222–e223. [Google Scholar] [CrossRef] [PubMed]
- Hannen, R.; Selmansberger, M.; Hauswald, M.; Pagenstecher, A.; Nist, A.; Stiewe, T.; Acker, T.; Carl, B.; Nimsky, C.; Bartsch, J.W. Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers 2019, 11, 921. [Google Scholar] [CrossRef]
- Zhao, K.; Schäfer, A.; Zhang, Z.; Elsässer, K.; Culmsee, C.; Zhong, L.; Pagenstecher, A.; Nimsky, C.; Bartsch, J.W. Inhibition of Carbonic Anhydrase 2 Overcomes Temozolomide Resistance in Glioblastoma Cells. Int. J. Mol. Sci. 2021, 23, 157. [Google Scholar] [CrossRef]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Gähwiler, B.H.; Thompson, S.M.; Muller, D. Preparation and maintenance of organotypic slice cultures of CNS tissue. Curr. Protoc. Neurosci. 2001, 9, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; DiNardo, C.D.; Potluri, J.; Dunbar, M.; Kantarjian, H.M.; Humerickhouse, R.A.; Wong, S.L.; Menon, R.M.; Konopleva, M.Y.; Salem, A.H. Management of Venetoclax-Posaconazole Interaction in Acute Myeloid Leukemia Patients: Evaluation of Dose Adjustments. Clin. Ther. 2017, 39, 359–367. [Google Scholar] [CrossRef]
- Jones, A.K.; Freise, K.J.; Agarwal, S.K.; Humerickhouse, R.A.; Wong, S.L.; Salem, A.H. Clinical Predictors of Venetoclax Pharmacokinetics in Chronic Lymphocytic Leukemia and Non-Hodgkin’s Lymphoma Patients: A Pooled Population Pharmacokinetic Analysis. AAPS J. 2016, 18, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ye, H. Progress in understanding the mechanisms of resistance to BCL-2 inhibitors. Exp. Hematol. Oncol. 2022, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Montero, J.; Haq, R. Adapted to Survive: Targeting Cancer Cells with BH3 Mimetics. Cancer Discov. 2022, 12, 1217–1232. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- van Schaik, T.A.; Chen, K.S.; Shah, K. Therapy-Induced Tumor Cell Death: Friend or Foe of Immunotherapy? Front. Oncol. 2021, 11, 678562. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Zheng, H.L.; Wang, X.R.; Zheng, X.L.; Chen, Y.; Fei, W.D.; Zheng, Y.Q.; Wang, W.X.; Zheng, C.H. Enhanced Percutaneous Delivery of Methotrexate Using Micelles Prepared with Novel Cationic Amphipathic Material. Int. J. Nanomed. 2020, 15, 3539–3550. [Google Scholar] [CrossRef]
- Jaafar-Maalej, C.; Diab, R.; Andrieu, V.; Elaissari, A.; Fessi, H. Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J. Liposome Res. 2010, 20, 228–243. [Google Scholar] [CrossRef]
- Bazan, F.; Dobi, E.; Royer, B.; Curtit, E.; Mansi, L.; Menneveau, N.; Paillard, M.J.; Meynard, G.; Villanueva, C.; Pivot, X.; et al. Systemic high-dose intravenous methotrexate in patients with central nervous system metastatic breast cancer. BMC Cancer 2019, 19, 1029. [Google Scholar] [CrossRef]
- Lassman, A.B.; Abrey, L.E.; Shah, G.D.; Panageas, K.S.; Begemann, M.; Malkin, M.G.; Raizer, J.J. Systemic high-dose intravenous methotrexate for central nervous system metastases. J. Neurooncol. 2006, 78, 255–260. [Google Scholar] [CrossRef]
- Zhao, K.H.; Zhang, C.; Bai, Y.; Li, Y.; Kang, X.; Chen, J.X.; Yao, K.; Jiang, T.; Zhong, X.S.; Li, W.B. Antiglioma effects of cytarabine on leptomeningeal metastasis of high-grade glioma by targeting the PI3K/Akt/mTOR pathway. Drug Des. Devel Ther. 2017, 11, 1905–1915. [Google Scholar] [CrossRef]
- Fiveash, J.B.; Ye, X.; Peerboom, D.M.; Mikkelsen, T.; Chowdhary, S.; Rosenfeld, M.; Lesser, G.J.; Fisher, J.; Desideri, S.; Grossman, S.; et al. Clinical trials of R-(-)-gossypol (AT-101) in newly diagnosed and recurrent glioblastoma: NABTT 0602 and NABTT 0702. PLoS ONE 2024, 19, e0291128. [Google Scholar] [CrossRef]
- Yang, X.; Mei, C.; He, X.; He, L.; Lu, X.; Tong, H.; Lou, Y. Quantification of Venetoclax for Therapeutic Drug Monitoring in Chinese Acute Myeloid Leukemia Patients by a Validated UPLC-MS/MS Method. Molecules 2022, 27, 1607. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | IC50 (nM) | ||
|---|---|---|---|
| TMZ | MTX | Ara-C | |
| U87 | 671.3 × 103 | 59.87 | 4886 |
| U251 | 48.22 × 103 | 30.56 | 1748 |
| GSCs | 68.86 × 103 | 123.0 | 367.7 |
| Daily Dose (p.o.) | Serum | CSF | Ratio (⌀) | |
|---|---|---|---|---|
| Patient 1 | 100 mg | 230 ng/mL | 340:1 | |
| Patient 2 | 200 mg | 2200 ng/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, K.; Braun, M.; Meyer, L.; Otte, K.; Raifer, H.; Helmprobst, F.; Möschl, V.; Pagenstecher, A.; Urban, H.; Ronellenfitsch, M.W.; et al. A Novel Approach for Glioblastoma Treatment by Combining Apoptosis Inducers (TMZ, MTX, and Cytarabine) with E.V.A. (Eltanexor, Venetoclax, and A1210477) Inhibiting XPO1, Bcl-2, and Mcl-1. Cells 2024, 13, 632. https://doi.org/10.3390/cells13070632
Zhao K, Braun M, Meyer L, Otte K, Raifer H, Helmprobst F, Möschl V, Pagenstecher A, Urban H, Ronellenfitsch MW, et al. A Novel Approach for Glioblastoma Treatment by Combining Apoptosis Inducers (TMZ, MTX, and Cytarabine) with E.V.A. (Eltanexor, Venetoclax, and A1210477) Inhibiting XPO1, Bcl-2, and Mcl-1. Cells. 2024; 13(7):632. https://doi.org/10.3390/cells13070632
Chicago/Turabian StyleZhao, Kai, Madita Braun, Leonie Meyer, Katharina Otte, Hartmann Raifer, Frederik Helmprobst, Vincent Möschl, Axel Pagenstecher, Hans Urban, Michael W. Ronellenfitsch, and et al. 2024. "A Novel Approach for Glioblastoma Treatment by Combining Apoptosis Inducers (TMZ, MTX, and Cytarabine) with E.V.A. (Eltanexor, Venetoclax, and A1210477) Inhibiting XPO1, Bcl-2, and Mcl-1" Cells 13, no. 7: 632. https://doi.org/10.3390/cells13070632
APA StyleZhao, K., Braun, M., Meyer, L., Otte, K., Raifer, H., Helmprobst, F., Möschl, V., Pagenstecher, A., Urban, H., Ronellenfitsch, M. W., Steinbach, J. P., Pesek, J., Watzer, B., Nockher, W. A., Taudte, R. V., Neubauer, A., Nimsky, C., Bartsch, J. W., & Rusch, T. (2024). A Novel Approach for Glioblastoma Treatment by Combining Apoptosis Inducers (TMZ, MTX, and Cytarabine) with E.V.A. (Eltanexor, Venetoclax, and A1210477) Inhibiting XPO1, Bcl-2, and Mcl-1. Cells, 13(7), 632. https://doi.org/10.3390/cells13070632