
Leflunomide Treatment Does Not Protect Neural Cells following Oxygen-Glucose Deprivation (OGD) In Vitro

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
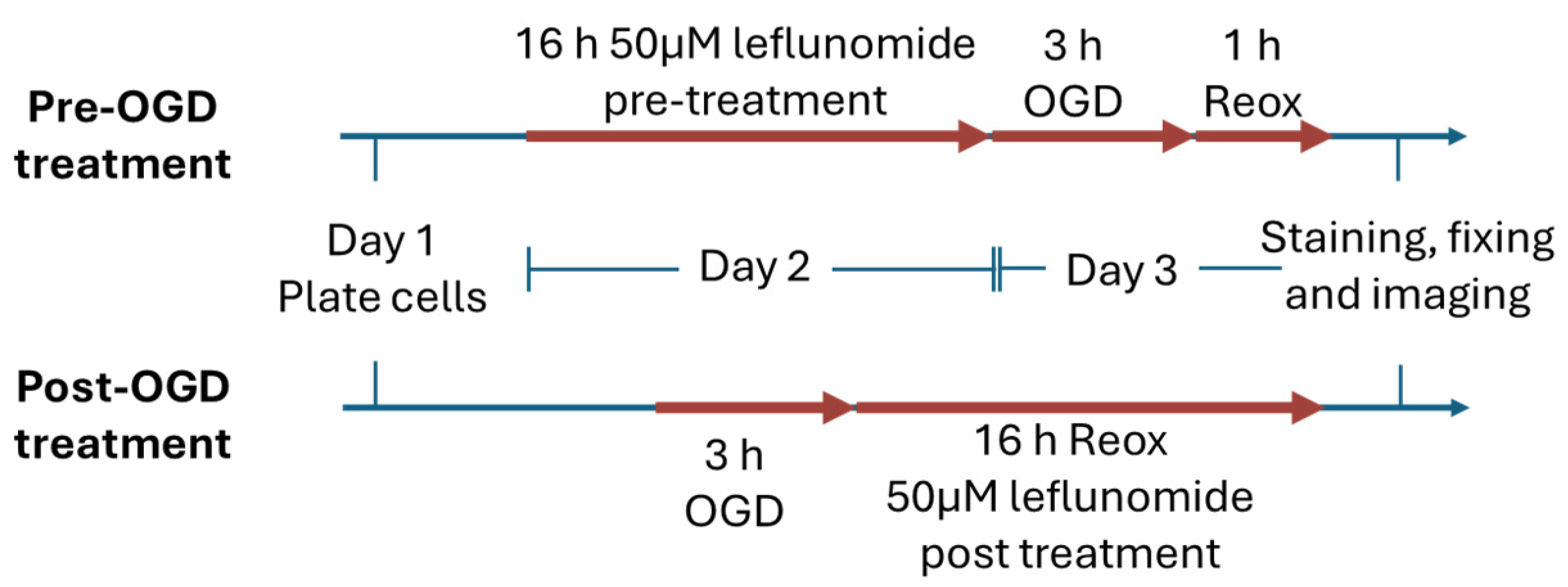
2.2. Oxygen-Glucose Deprivation (OGD)
2.3. Nuclear and Mitochondrial Staining in Live Cells
2.4. Leflunomide Treatment
2.5. Mitochondrial Stress Test Assay (Seahorse)
2.6. Western Blot
2.7. Image J Analyses
2.8. Statistical Analysis
3. Results
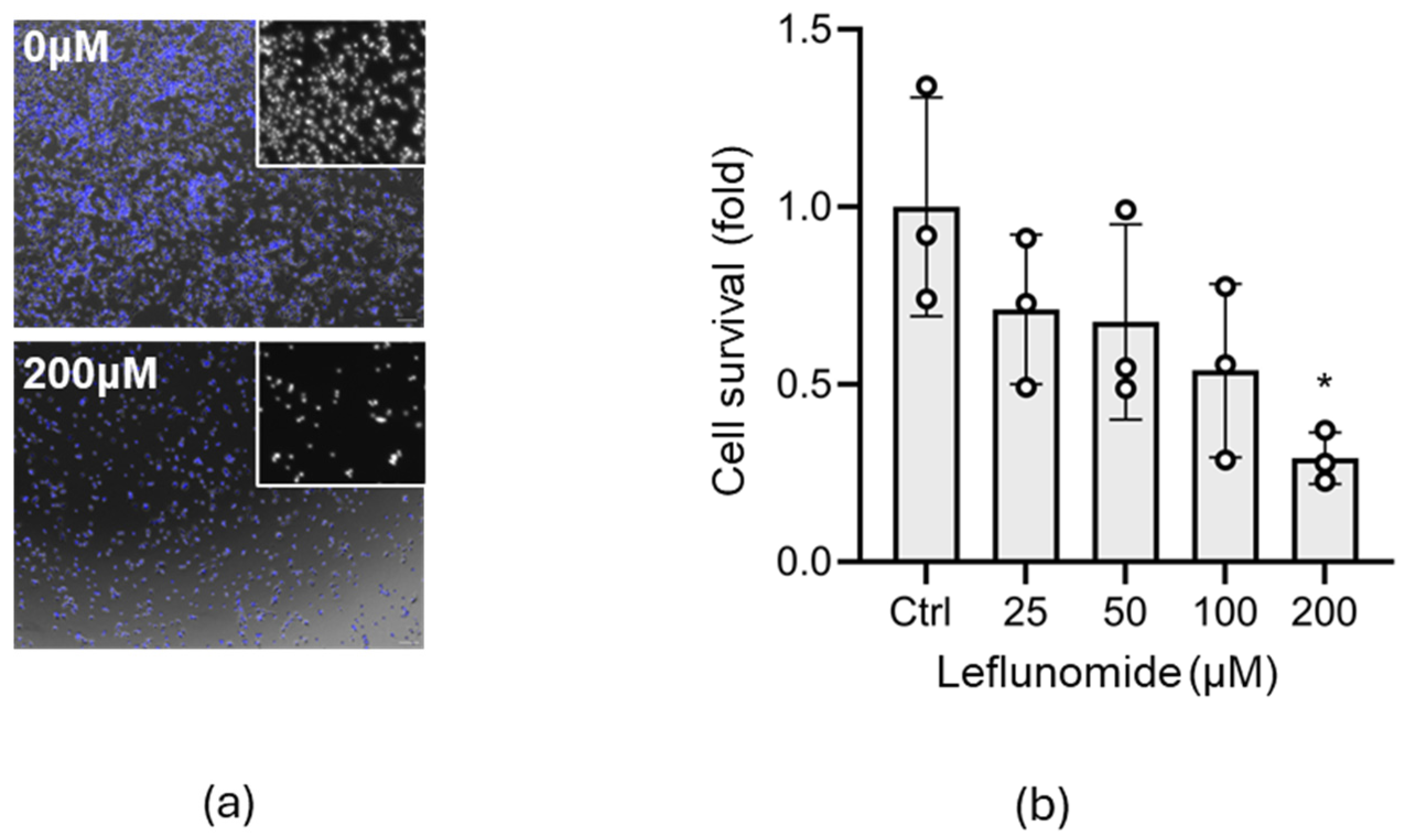
3.1. Identification of the Optimal Leflunomide Concentration for C17.2 Cells
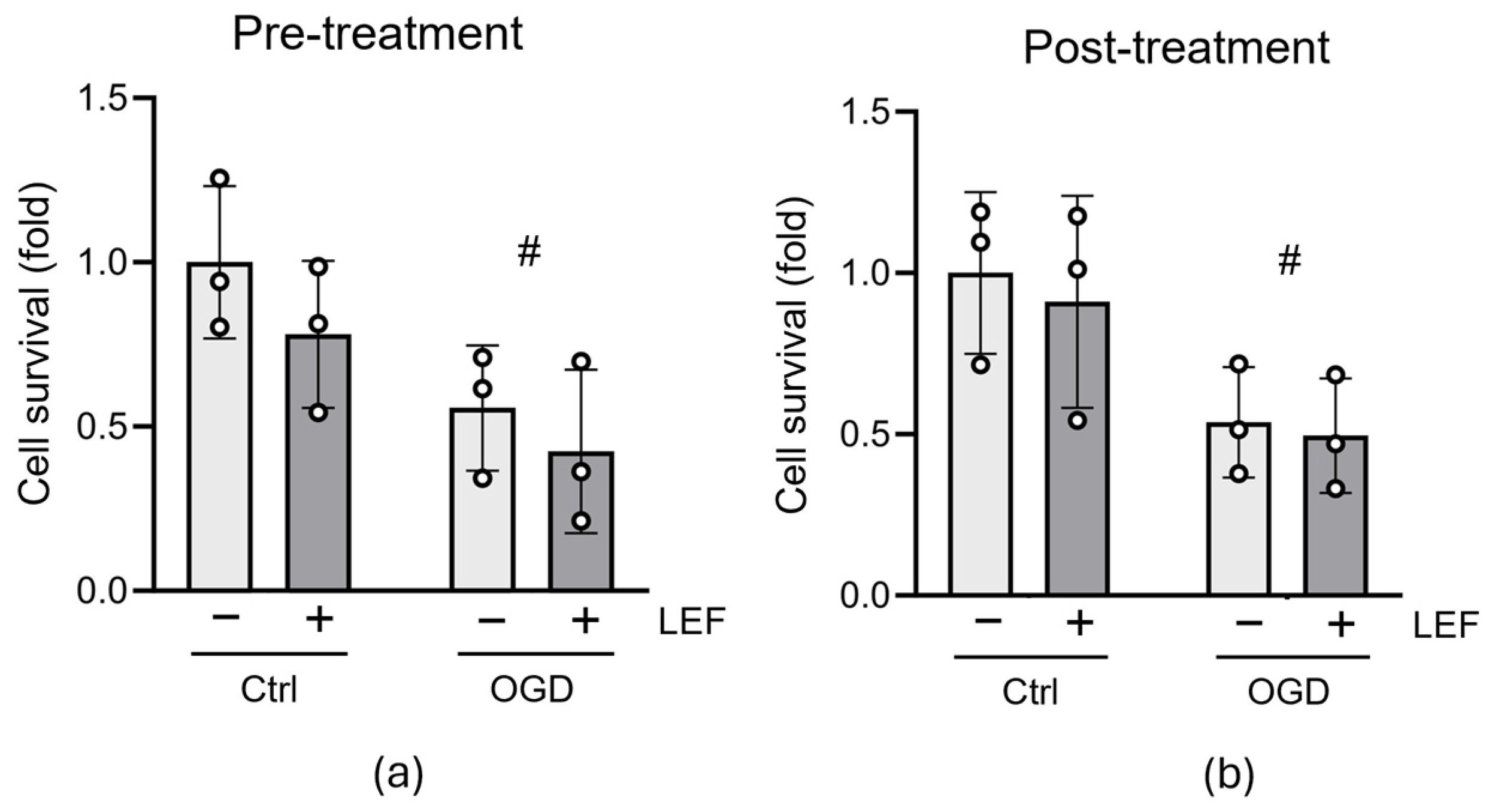
3.2. Leflunomide Treatment Does Not Alter Cell Survival following OGD
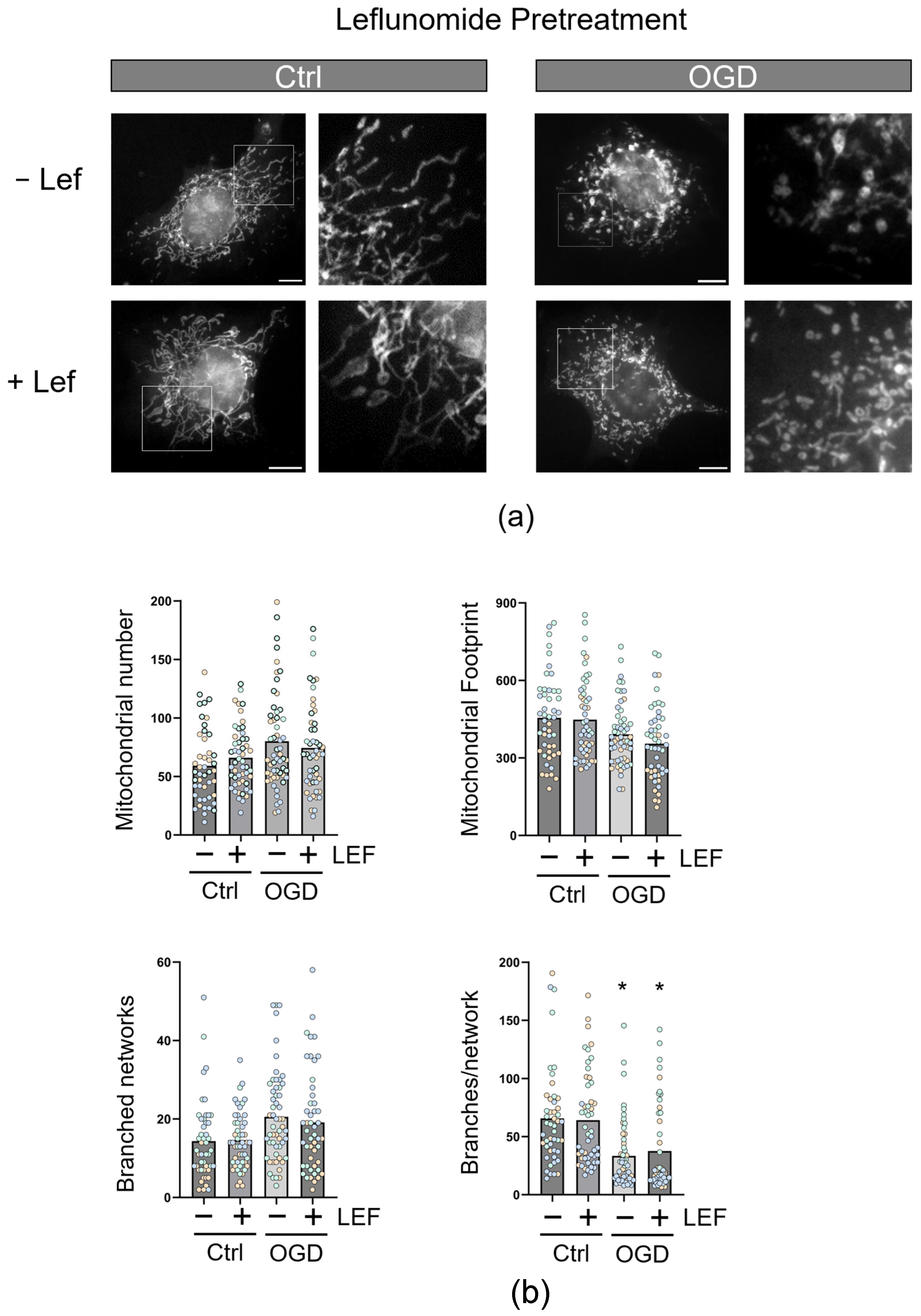
3.3. Leflunomide Treatment in C17.2 Cells Does Not Alter Mitochondrial Morphology
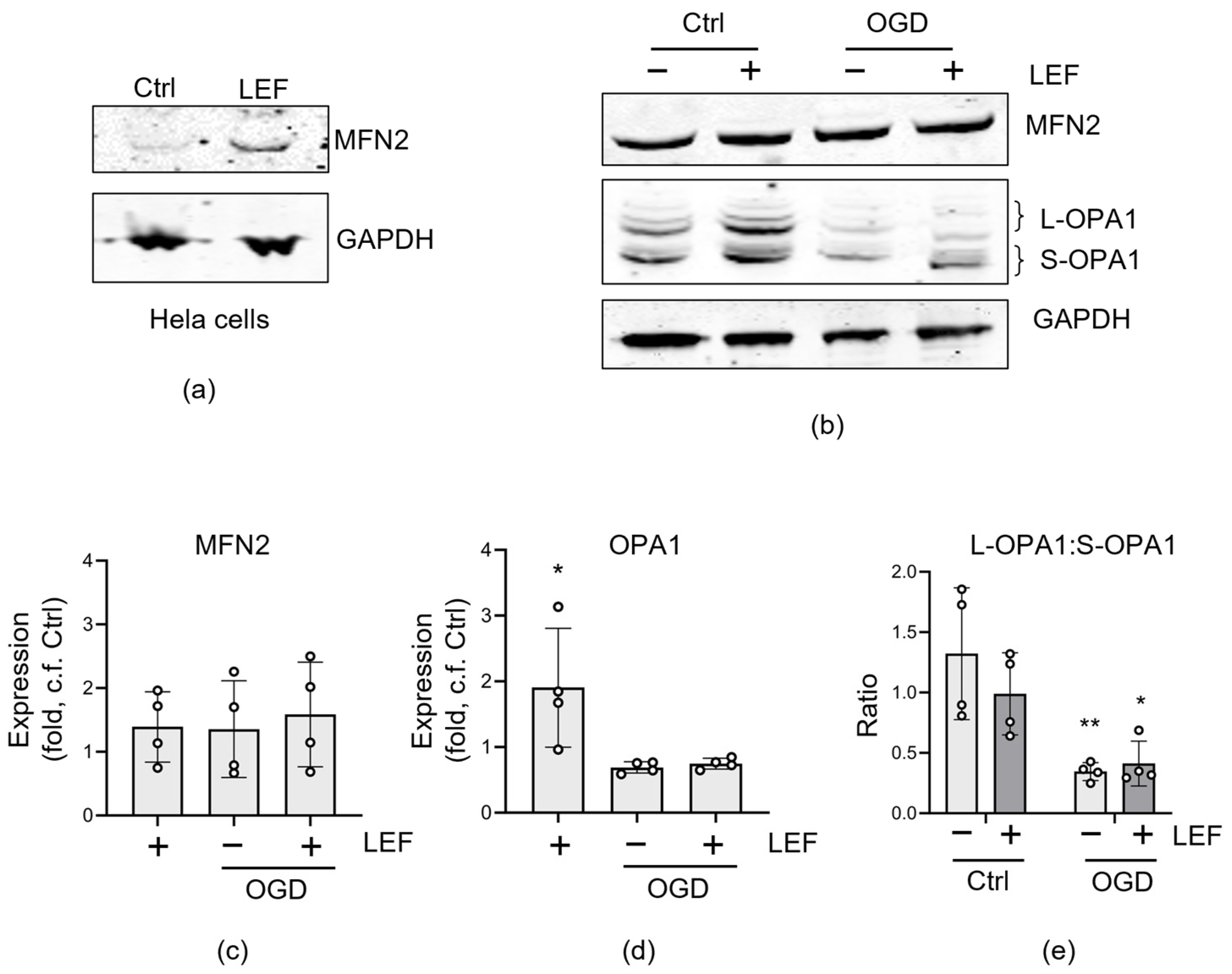
3.4. Effect of Leflunomide on Mitochondrial Fusion Proteins
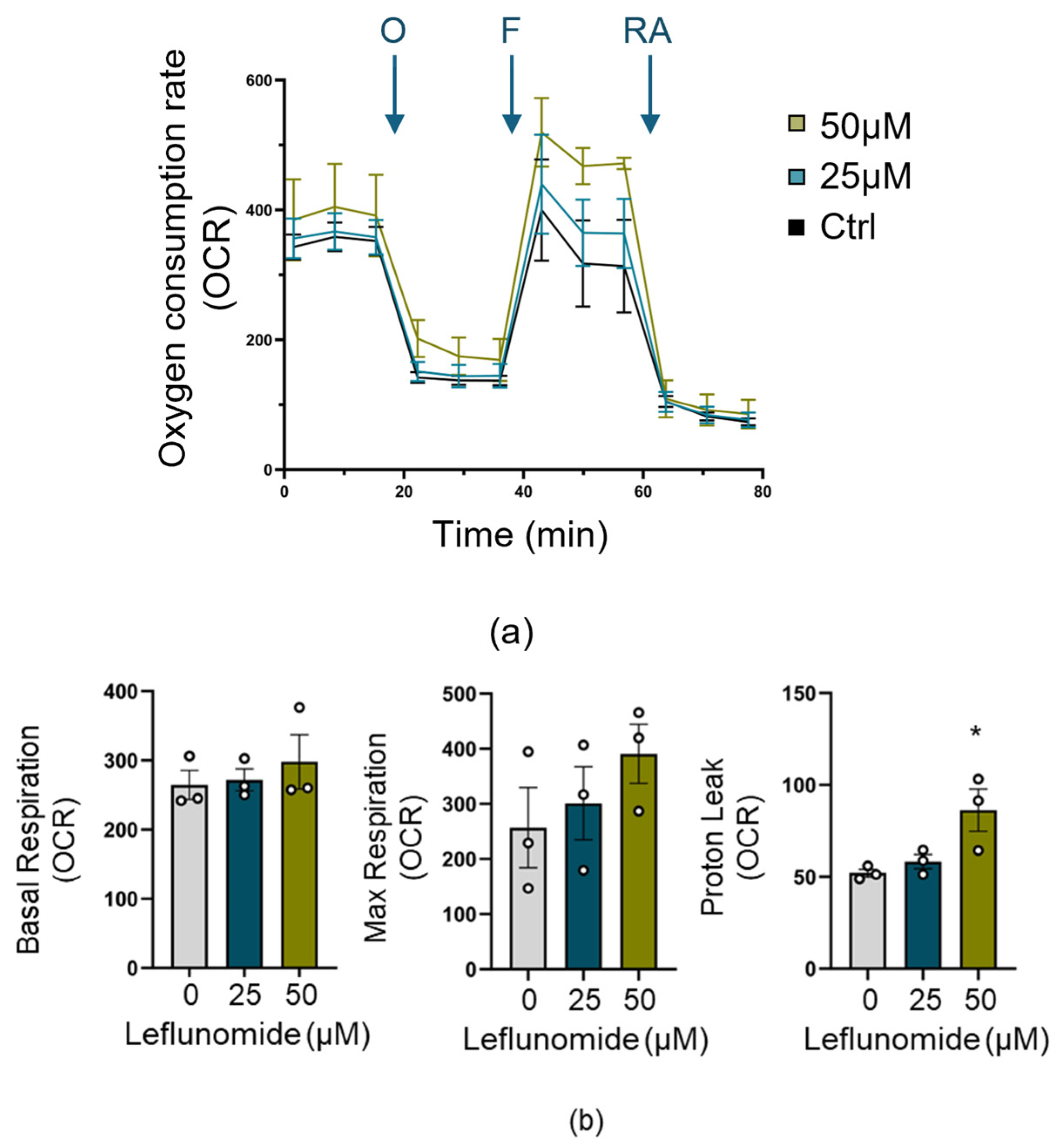
3.5. Leflunomide Pretreatment Increases Mitochondrial Proton Leak
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gale, C.; Statnikov, Y.; Jawad, S.; Uthaya, S.N.; Modi, N. Neonatal brain injuries in England: Population-based incidence derived from routinely recorded clinical data held in the National Neonatal Research Database. Arch. Dis. Child.-Fetal Neonatal Ed. 2018, 103, F301–F306. [Google Scholar] [CrossRef] [PubMed]
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Kumar, V.; Variane, G.F.T.; Carlo, W.A.; Bhutta, Z.A.; Sizonenko, S.; Hansen, A.; Shankaran, S.; Thayyil, S.; Newborn Brain Society, G.; et al. Need for more evidence in the prevention and management of perinatal asphyxia and neonatal encephalopathy in low and middle-income countries: A call for action. Semin. Fetal Neonatal Med. 2021, 26, 101271. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Ferreira, E.; Hristova, M. Plasticity in the Neonatal Brain following Hypoxic-Ischaemic Injury. Neural Plast. 2016, 2016, 4901014. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.; Rousset, C.I.; Kichev, A.; Miyakuni, Y.; Vontell, R.; Baburamani, A.A.; Fleiss, B.; Gressens, P.; Hagberg, H. Molecular mechanisms of neonatal brain injury. Neurol. Res. Int. 2012, 2012, 506320. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N. Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef]
- Bellos, I.; Devi, U.; Pandita, A. Therapeutic Hypothermia for Neonatal Encephalopathy in Low- and Middle-Income Countries: A Meta-Analysis. Neonatology 2022, 119, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Puka-Sundvall, M.; Gajkowska, B.; Cholewinski, M.; Blomgren, K.; Lazarewicz, J.W.; Hagberg, H. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain Res. Dev. Brain Res. 2000, 125, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Thornton, C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014, 13, 217–232. [Google Scholar] [CrossRef]
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, H.G.; Langer, T. The Good and the Bad of Mitochondrial Breakups. Trends Cell Biol. 2019, 29, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Leverin, A.L.; Rocha-Ferreira, E.; Sobotka, K.S.; Thornton, C.; Mallard, C.; Hagberg, H. Induction of Mitochondrial Fragmentation and Mitophagy after Neonatal Hypoxia-Ischemia. Cells 2022, 11, 1193. [Google Scholar] [CrossRef]
- Baburamani, A.A.; Hurling, C.; Stolp, H.; Sobotka, K.; Gressens, P.; Hagberg, H.; Thornton, C. Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2015, 16, 22509–22526. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Neuronal hypoxia disrupts mitochondrial fusion. Neuroscience 2015, 301, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef]
- Wang, X.; Han, W.; Du, X.; Zhu, C.; Carlsson, Y.; Mallard, C.; Jacotot, E.; Hagberg, H. Neuroprotective effect of Bax-inhibiting peptide on neonatal brain injury. Stroke 2010, 41, 2050–2055. [Google Scholar] [CrossRef]
- Baburamani, A.A.; Miyakuni, Y.; Vontell, R.; Supramaniam, V.G.; Svedin, P.; Rutherford, M.; Gressens, P.; Mallard, C.; Takeda, S.; Thornton, C.; et al. Does Caspase-6 Have a Role in Perinatal Brain Injury? Dev. Neurosci. 2015, 37, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, Y.; Schwendimann, L.; Vontell, R.; Rousset, C.I.; Wang, X.; Lebon, S.; Charriaut-Marlangue, C.; Supramaniam, V.; Hagberg, H.; Gressens, P.; et al. Genetic inhibition of caspase-2 reduces hypoxic-ischemic and excitotoxic neonatal brain injury. Ann. Neurol. 2011, 70, 781–789. [Google Scholar] [CrossRef]
- Carlsson, Y.; Wang, X.; Schwendimann, L.; Rousset, C.I.; Jacotot, E.; Gressens, P.; Thoresen, M.; Mallard, C.; Hagberg, H. Combined effect of hypothermia and caspase-2 gene deficiency on neonatal hypoxic-ischemic brain injury. Pediatr. Res. 2012, 71, 566–572. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, X.; Huang, Z.; Qiu, L.; Xu, F.; Vahsen, N.; Nilsson, M.; Eriksson, P.S.; Hagberg, H.; Culmsee, C.; et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007, 14, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Xie, C.; Li, T.; Sun, Y.; Wang, Y.; Xu, Y.; Li, K.; Zhang, S.; Zhou, K.; Wang, Y.; et al. Inhibiting the interaction between apoptosis-inducing factor and cyclophilin A prevents brain injury in neonatal mice after hypoxia-ischemia. Neuropharmacology 2020, 171, 108088. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Miret-Casals, L.; Sebastian, D.; Brea, J.; Rico-Leo, E.M.; Palacin, M.; Fernandez-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278 e264. [Google Scholar] [CrossRef]
- Humphries, B.A.; Cutter, A.C.; Buschhaus, J.M.; Chen, Y.C.; Qyli, T.; Palagama, D.S.W.; Eckley, S.; Robison, T.H.; Bevoor, A.; Chiang, B.; et al. Enhanced mitochondrial fission suppresses signaling and metastasis in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 60. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, Y.; Kose, H.; Cecen, S.; Ergin, K.; Demir, E.M.; Serter, M. Protective effects of leflunomide on intestinal ischemia-reperfusion injury: Leflunomide against intestinal ischemia-reperfusion. Dig. Dis. Sci. 2010, 55, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Karaman, A.; Fadillioglu, E.; Turkmen, E.; Tas, E.; Yilmaz, Z. Protective effects of leflunomide against ischemia-reperfusion injury of the rat liver. Pediatr. Surg. Int. 2006, 22, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Karaman, A.; Turkmen, E.; Gursul, C.; Tas, E.; Fadillioglu, E. Prevention of renal ischemia/reperfusion-induced injury in rats by leflunomide. Int. J. Urol. 2006, 13, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, J.; El Andaloussi-Lilja, J.; Svensson, C.; Gustafsson Dorfh, H.; Forsby, A. Optimisation of culture conditions for differentiation of C17.2 neural stem cells to be used for in vitro toxicity tests. Toxicol. In Vitro 2013, 27, 1565–1569. [Google Scholar] [CrossRef]
- Mittelman, D.; Wilson, J.H. The fractured genome of HeLa cells. Genome Biol. 2013, 14, 111. [Google Scholar] [CrossRef]
- Kesner, E.E.; Saada-Reich, A.; Lorberboum-Galski, H. Characteristics of Mitochondrial Transformation into Human Cells. Sci. Rep. 2016, 6, 26057. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.Y.; Deitcher, D.L.; Walsh, C.; Arnold-Aldea, S.; Hartwieg, E.A.; Cepko, C.L. Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell 1992, 68, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Yan, X.; Xiang, Z.; Ding, H.F.; Cui, H. Leflunomide reduces proliferation and induces apoptosis in neuroblastoma cells in vitro and in vivo. PLoS ONE 2013, 8, e71555. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 4, e126915. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Alka, K. Mitochondrial Quality Control and Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. Int. J. Mol. Sci. 2023, 24, 8076. [Google Scholar] [CrossRef] [PubMed]
- Victor, S.; Rocha-Ferreira, E.; Rahim, A.; Hagberg, H.; Edwards, D. New possibilities for neuroprotection in neonatal hypoxic-ischemic encephalopathy. Eur. J. Pediatr. 2022, 181, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 2013, CD003311. [Google Scholar] [CrossRef]
- Wassink, G.; Davidson, J.O.; Dhillon, S.K.; Zhou, K.; Bennet, L.; Thoresen, M.; Gunn, A.J. Therapeutic Hypothermia in Neonatal Hypoxic-Ischemic Encephalopathy. Curr. Neurol. Neurosci. Rep. 2019, 19, 2. [Google Scholar] [CrossRef]
- Leaw, B.; Nair, S.; Lim, R.; Thornton, C.; Mallard, C.; Hagberg, H. Mitochondria, Bioenergetics and Excitotoxicity: New Therapeutic Targets in Perinatal Brain Injury. Front. Cell Neurosci. 2017, 11, 199. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, H.; Yan, C.; Shen, B.; Zhang, Y.; Guo, X.; Sun, S.; Yu, F.; Yan, J.; Liu, R.; et al. Small molecule agonist of mitochondrial fusion repairs mitochondrial dysfunction. Nat. Chem. Biol. 2023, 19, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.W.; Wang, J.K.; Wang, L.C.; Guo, Q.; Liu, T.T.; Wang, F.J.; Feng, N.; Zhang, X.W.; Liao, L.X.; Zhao, M.M.; et al. Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct. Target. Ther. 2021, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Thornton, C. Mitochondrial dynamics in the neonatal brain—A potential target following injury? Biosci. Rep. 2022, 42, BSR20211696. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hamid, N.M.; Abass, S.A.; Eldomany, R.A.; Abdel-Kareem, M.A.; Zakaria, S. Dual regulating of mitochondrial fusion and Timp-3 by leflunomide and diallyl disulfide combination suppresses diethylnitrosamine-induced hepatocellular tumorigenesis in rats. Life Sci. 2022, 294, 120369. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, R.; Kumar, S. Mfn2-mediated mitochondrial fusion promotes autophagy and suppresses ovarian cancer progression by reducing ROS through AMPK/mTOR/ERK signaling. Cell. Mol. Life Sci. 2022, 79, 573. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, Q.; Chang, Q.; Xie, M.; Weng, J.; Wang, X.; Li, M.; Chen, J.; Huang, Y.; Yang, X.; et al. Role of mitochondrial fusion proteins MFN2 and OPA1 on lung cellular senescence in chronic obstructive pulmonary disease. Respir. Res. 2023, 24, 319. [Google Scholar] [CrossRef] [PubMed]
- Pellattiero, A.; Scorrano, L. Flaming Mitochondria: The Anti-inflammatory Drug Leflunomide Boosts Mitofusins. Cell Chem. Biol. 2018, 25, 231–233. [Google Scholar] [CrossRef]
- Xuan, J.; Ren, Z.; Qing, T.; Couch, L.; Shi, L.; Tolleson, W.H.; Guo, L. Mitochondrial dysfunction induced by leflunomide and its active metabolite. Toxicology 2018, 396–397, 33–45. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, H.; Wang, Z.; Huang, H.; Zhuo, D.; Lin, J. Leflunomide Inhibits Proliferation and Induces Apoptosis via Suppressing Autophagy and PI3K/Akt Signaling Pathway in Human Bladder Cancer Cells. Drug Des. Devel Ther. 2020, 14, 1897–1908. [Google Scholar] [CrossRef]
- Maremanda, K.P.; Sundar, I.K.; Rahman, I. Role of inner mitochondrial protein OPA1 in mitochondrial dysfunction by tobacco smoking and in the pathogenesis of COPD. Redox Biol. 2021, 45, 102055. [Google Scholar] [CrossRef] [PubMed]
- Maiwald, C.A.; Annink, K.V.; Rüdiger, M.; Benders, M.; van Bel, F.; Allegaert, K.; Naulaers, G.; Bassler, D.; Klebermaß-Schrehof, K.; Vento, M.; et al. Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (ALBINO): Study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). BMC Pediatr. 2019, 19, 210. [Google Scholar] [CrossRef]
- Korf, J.M.; McCullough, L.D.; Caretti, V. A narrative review on treatment strategies for neonatal hypoxic ischemic encephalopathy. Transl. Pediatr. 2023, 12, 1552–1571. [Google Scholar] [CrossRef] [PubMed]
- Tetorou, K.; Sisa, C.; Iqbal, A.; Dhillon, K.; Hristova, M. Current Therapies for Neonatal Hypoxic-Ischaemic and Infection-Sensitised Hypoxic-Ischaemic Brain Damage. Front. Synaptic Neurosci. 2021, 13, 709301. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Mathur, A.M.; Chang, T.; McKinstry, R.C.; Mulkey, S.B.; Mayock, D.E.; Van Meurs, K.P.; Rogers, E.E.; Gonzalez, F.F.; Comstock, B.A.; et al. High-Dose Erythropoietin and Hypothermia for Hypoxic-Ischemic Encephalopathy: A Phase II Trial. Pediatrics 2016, 137, e20160191. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Comstock, B.A.; Gonzalez, F.F.; Mayock, D.E.; Goodman, A.M.; Maitre, N.L.; Chang, T.; Van Meurs, K.P.; Lampland, A.L.; Bendel-Stenzel, E.; et al. Trial of Erythropoietin for Hypoxic-Ischemic Encephalopathy in Newborns. N. Engl. J. Med. 2022, 387, 148–159. [Google Scholar] [CrossRef]
- Juul, S.E.; Comstock, B.A.; Cornet, M.C.; Gonzalez, F.F.; Mayock, D.E.; Glass, H.C.; Schreiber, M.D.; Heagerty, P.J.; Wu, Y.W. Safety of High Dose Erythropoietin Used with Therapeutic Hypothermia as Treatment for Newborn Hypoxic-Ischemic Encephalopathy: Secondary Analysis of the HEAL Randomized Controlled Trial. J. Pediatr. 2023, 258, 113400. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Final Conc | Target |
|---|---|---|
| Oligomycin | 1.5 | ATP synthase |
| FCCP | 1.0 | Uncoupler |
| Rotenone/ Antimycin A | 0.5 | Complex I/ Complex III |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curel, C.J.M.; Nobeli, I.; Thornton, C. Leflunomide Treatment Does Not Protect Neural Cells following Oxygen-Glucose Deprivation (OGD) In Vitro. Cells 2024, 13, 631. https://doi.org/10.3390/cells13070631
Curel CJM, Nobeli I, Thornton C. Leflunomide Treatment Does Not Protect Neural Cells following Oxygen-Glucose Deprivation (OGD) In Vitro. Cells. 2024; 13(7):631. https://doi.org/10.3390/cells13070631
Chicago/Turabian StyleCurel, Claire J. M., Irene Nobeli, and Claire Thornton. 2024. "Leflunomide Treatment Does Not Protect Neural Cells following Oxygen-Glucose Deprivation (OGD) In Vitro" Cells 13, no. 7: 631. https://doi.org/10.3390/cells13070631
APA StyleCurel, C. J. M., Nobeli, I., & Thornton, C. (2024). Leflunomide Treatment Does Not Protect Neural Cells following Oxygen-Glucose Deprivation (OGD) In Vitro. Cells, 13(7), 631. https://doi.org/10.3390/cells13070631