

Unlocking the Future: Pluripotent Stem Cell-Based Lung Repair

Abstract
:1. Introduction
2. Pathogenesis of Life-Threatening Respiratory Diseases, Current Therapeutic Limitations and Disease-Specific Strategies for iPSC-Based Therapies
2.1. Pulmonary Fibrosis (PF)
2.2. Chronic Obstructive Pulmonary Disease (COPD)
2.3. Pulmonary Arterial Hypertension (PAH)
2.4. Cystic Fibrosis Lung Disease (CF)
2.5. Other Genetic Lung Diseases
3. Current State and Limitations of Human Pluripotent Stem Cell Technologies
4. Targeted Production of PSC-Derived Cell Lineages Relevant for Respiratory Diseases
5. Preclinical Models for Evaluating Novel Cell Therapies in Lung Diseases
6. Perspectives for Clinical Trials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, W.; Zeng, Y.; Wang, D. Stem cell-based therapy for pulmonary fibrosis. Stem Cell Res. Ther. 2022, 13, 492. [Google Scholar] [CrossRef] [PubMed]
- Kadota, T.; Fujita, Y.; Araya, J.; Watanabe, N.; Fujimoto, S.; Kawamoto, H.; Minagawa, S.; Hara, H.; Ohtsuka, T.; Yamamoto, Y.; et al. Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-beta-WNT crosstalk. J. Extracell. Vesicles 2021, 10, e12124. [Google Scholar] [CrossRef]
- Bonella, F.; Spagnolo, P.; Ryerson, C. Current and Future Treatment Landscape for Idiopathic Pulmonary Fibrosis. Drugs 2023, 83, 1581–1593. [Google Scholar] [CrossRef]
- Spagnolo, P.; Kropski, J.A.; Jones, M.G.; Lee, J.S.; Rossi, G.; Karampitsakos, T.; Maher, T.M.; Tzouvelekis, A.; Ryerson, C.J. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol. Ther. 2021, 222, 107798. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Guo, X.; Fan, H.; Shi, J.; Hou, S.; Lv, Q. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Idiopathic Pulmonary Fibrosis Microenvironment Targeted Delivery. Cells 2022, 11, 2322. [Google Scholar] [CrossRef]
- Suezawa, T.; Kanagaki, S.; Moriguchi, K.; Masui, A.; Nakao, K.; Toyomoto, M.; Tamai, K.; Mikawa, R.; Hirai, T.; Murakami, K.; et al. Disease modeling of pulmonary fibrosis using human pluripotent stem cell-derived alveolar organoids. Stem Cell Rep. 2021, 16, 2973–2987. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e117. [Google Scholar] [CrossRef]
- Dinh, P.C.; Paudel, D.; Brochu, H.; Popowski, K.D.; Gracieux, M.C.; Cores, J.; Huang, K.; Hensley, M.T.; Harrell, E.; Vandergriff, A.C.; et al. Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis. Nat. Commun. 2020, 11, 1064. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Calzetta, L.; Aiello, M.; Frizzelli, A.; Camardelli, F.; Cazzola, M.; Rogliani, P.; Chetta, A. Stem Cell-Based Regenerative Therapy and Derived Products in COPD: A Systematic Review and Meta-Analysis. Cells 2022, 11, 1797. [Google Scholar] [CrossRef]
- Chen, Y.T.; Miao, K.; Zhou, L.; Xiong, W.N. Stem cell therapy for chronic obstructive pulmonary disease. Chin. Med. J. 2021, 134, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Easter, M.; Bollenbecker, S.; Barnes, J.W.; Krick, S. Targeting Aging Pathways in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 6924. [Google Scholar] [CrossRef] [PubMed]
- Ridzuan, N.; Zakaria, N.; Widera, D.; Sheard, J.; Morimoto, M.; Kiyokawa, H.; Mohd Isa, S.A.; Chatar Singh, G.K.; Then, K.Y.; Ooi, G.C.; et al. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles ameliorate airway inflammation in a rat model of chronic obstructive pulmonary disease (COPD). Stem Cell Res. Ther. 2021, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Glassberg, M.K.; Csete, I.; Simonet, E.; Elliot, S.J. Stem Cell Therapy for COPD: Hope and Exploitation. Chest 2021, 160, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Hisata, S.; Racanelli, A.C.; Kermani, P.; Schreiner, R.; Houghton, S.; Palikuqi, B.; Kunar, B.; Zhou, A.; McConn, K.; Capili, A.; et al. Reversal of emphysema by restoration of pulmonary endothelial cells. J. Exp. Med. 2021, 218, e20200938. [Google Scholar] [CrossRef]
- Pu, X.; Du, L.; Hu, Y.; Fan, Y.; Xu, Q. Stem/Progenitor Cells and Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.W.; Sun, Z. Stem cell therapy for pulmonary arterial hypertension: An update. J. Heart Lung Transpl. 2022, 41, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Suen, C.M.; Stewart, D.J.; Montroy, J.; Welsh, C.; Levac, B.; Wesch, N.; Zhai, A.; Fergusson, D.; McIntyre, L.; Lalu, M.M. Regenerative cell therapy for pulmonary arterial hypertension in animal models: A systematic review. Stem Cell Res. Ther. 2019, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Dierick, F.; Solinc, J.; Bignard, J.; Soubrier, F.; Nadaud, S. Progenitor/Stem Cells in Vascular Remodeling during Pulmonary Arterial Hypertension. Cells 2021, 10, 1338. [Google Scholar] [CrossRef]
- Galkin, A.; Sitapara, R.; Clemons, B.; Garcia, E.; Kennedy, M.; Guimond, D.; Carter, L.L.; Douthitt, A.; Osterhout, R.; Gandjeva, A.; et al. Inhaled seralutinib exhibits potent efficacy in models of pulmonary arterial hypertension. Eur. Respir. J. 2022, 60, 2102356. [Google Scholar] [CrossRef]
- Zheng, R.; Xu, T.; Wang, X.; Yang, L.; Wang, J.; Huang, X. Stem cell therapy in pulmonary hypertension: Current practice and future opportunities. Eur. Respir. Rev. 2023, 32, 230112. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; O’Callaghan, D.S.; Savale, L.; Jais, X.; Yaici, A.; Maitre, S.; Dorfmuller, P.; Sitbon, O.; Simonneau, G.; Humbert, M. Pulmonary veno-occlusive disease: Recent progress and current challenges. Respir. Med. 2010, 104 (Suppl. S1), S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Duchesneau, P.; Waddell, T.K.; Karoubi, G. Cell-Based Therapeutic Approaches for Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 5219. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D., Jr.; Kopp, B.T.; Hill, C.L.; Lallier, S.W.; Schwartz, C.M.; Tadesse, M.; Alsudayri, A.; Reynolds, S.D. Cell Therapy for Cystic Fibrosis Lung Disease: Regenerative Basal Cell Amplification. Stem Cells Transl. Med. 2019, 8, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Cho, A.; Huang, E.N.; Xu, Y.; Quach, H.; Hu, J.; Wong, A.P. Gene therapy for cystic fibrosis: New tools for precision medicine. J. Transl. Med. 2021, 19, 452. [Google Scholar] [CrossRef] [PubMed]
- Allan, K.M.; Farrow, N.; Donnelley, M.; Jaffe, A.; Waters, S.A. Treatment of Cystic Fibrosis: From Gene- to Cell-Based Therapies. Front. Pharmacol. 2021, 12, 639475. [Google Scholar] [CrossRef]
- Roesch, E.A.; Bonfield, T.L.; Lazarus, H.M.; Reese, J.; Hilliard, K.; Hilliard, J.; Khan, U.; Heltshe, S.; Gluvna, A.; Dasenbrook, E.; et al. A phase I study assessing the safety and tolerability of allogeneic mesenchymal stem cell infusion in adults with cystic fibrosis. J. Cyst. Fibros. 2023, 22, 407–413. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Grasemann, H.; Ratjen, F. Cystic Fibrosis. N. Engl. J. Med. 2023, 389, 1693–1707. [Google Scholar] [CrossRef]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Lomas, D.A. New Therapeutic Targets for Alpha-1 Antitrypsin Deficiency. Chronic Obstr. Pulm Dis. 2018, 5, 233–243. [Google Scholar] [CrossRef]
- Stockley, R.A.; Turner, A.M. alpha-1-Antitrypsin deficiency: Clinical variability, assessment, and treatment. Trends Mol. Med. 2014, 20, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Torres-Duran, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 antitrypsin deficiency: Outstanding questions and future directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef]
- Mianne, J.; Ahmed, E.; Bourguignon, C.; Fieldes, M.; Vachier, I.; Bourdin, A.; Assou, S.; De Vos, J. Induced Pluripotent Stem Cells for Primary Ciliary Dyskinesia Modeling and Personalized Medicine. Am. J. Respir. Cell Mol. Biol. 2018, 59, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Paff, T.; Omran, H.; Nielsen, K.G.; Haarman, E.G. Current and Future Treatments in Primary Ciliary Dyskinesia. Int. J. Mol. Sci. 2021, 22, 9834. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Davis, S.D.; Polineni, D.; Manion, M.; Rosenfeld, M.; Dell, S.D.; Chilvers, M.A.; Ferkol, T.W.; Zariwala, M.A.; Sagel, S.D.; et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 197, e24–e39. [Google Scholar] [CrossRef]
- Magnani, J.E.; Donn, S.M. Persistent Respiratory Distress in the Term Neonate: Genetic Surfactant Deficiency Diseases. Curr. Pediatr. Rev. 2020, 16, 17–25. [Google Scholar] [CrossRef]
- Jehn, L.B.; Bonella, F. Pulmonary alveolar proteinosis-current and future therapeutical strategies. Curr. Opin. Pulm Med. 2023, 29, 465–474. [Google Scholar] [CrossRef]
- Trapnell, B.C.; Nakata, K.; Bonella, F.; Campo, I.; Griese, M.; Hamilton, J.; Wang, T.; Morgan, C.; Cottin, V.; McCarthy, C. Pulmonary alveolar proteinosis. Nat. Rev. Dis. Primers 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.; Glienke, W.; Engels, L.; Gohring, G.; Esser, R.; Arseniev, L.; Martin, U. GMP-compatible manufacturing of three iPS cell lines from human peripheral blood. Stem Cell Res. 2019, 35, 101394. [Google Scholar] [CrossRef] [PubMed]
- Merkert, S.; Martin, U. Targeted genome engineering using designer nucleases: State of the art and practical guidance for application in human pluripotent stem cells. Stem Cell Res. 2016, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Merkert, S.; Bednarski, C.; Göhring, G.; Cathomen, T.; Martin, U. Generation of a gene-corrected isogenic control iPSC line from cystic fibrosis patient-specific iPSCs homozygous for p.Phe508del mutation mediated by TALENs and ssODN. Stem Cell Res. 2017, 23, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Templin, C.; Zweigerdt, R.; Schwanke, K.; Olmer, R.; Ghadri, J.R.; Emmert, M.Y.; Muller, E.; Kuest, S.M.; Cohrs, S.; Schibli, R.; et al. Transplantation and tracking of human-induced pluripotent stem cells in a pig model of myocardial infarction: Assessment of cell survival, engraftment, and distribution by hybrid single photon emission computed tomography/computed tomography of sodium iodide symporter transgene expression. Circulation 2012, 126, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Merkert, S.; Wunderlich, S.; Bednarski, C.; Beier, J.; Haase, A.; Dreyer, A.K.; Schwanke, K.; Meyer, J.; Gohring, G.; Cathomen, T.; et al. Efficient designer nuclease-based homologous recombination enables direct PCR screening for footprintless targeted human pluripotent stem cells. Stem Cell Rep. 2014, 2, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, S.; Haase, A.; Merkert, S.; Jahn, K.; Deest, M.; Frieling, H.; Glage, S.; Korte, W.; Martens, A.; Kirschning, A.; et al. Targeted biallelic integration of an inducible Caspase 9 suicide gene in iPSCs for safer therapies. Mol. Ther. Methods Clin. Dev. 2022, 26, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Zweigerdt, R.; Olmer, R.; Singh, H.; Haverich, A.; Martin, U. Scalable expansion of human pluripotent stem cells in suspension culture. Nat. Protoc. 2011, 6, 689–700. [Google Scholar] [CrossRef]
- Manstein, F.; Ullmann, K.; Kropp, C.; Halloin, C.; Triebert, W.; Franke, A.; Farr, C.M.; Sahabian, A.; Haase, A.; Breitkreuz, Y.; et al. High density bioprocessing of human pluripotent stem cells by metabolic control and in silico modeling. Stem Cells Transl. Med. 2021, 10, 1063–1080. [Google Scholar] [CrossRef]
- Olmer, R.; Engels, L.; Usman, A.; Menke, S.; Malik, M.N.H.; Pessler, F.; Gohring, G.; Bornhorst, D.; Bolten, S.; Abdelilah-Seyfried, S.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Endothelial Cells in Scalable Suspension Culture. Stem Cell Rep. 2018, 10, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Halloin, C.; Schwanke, K.; Lobel, W.; Franke, A.; Szepes, M.; Biswanath, S.; Wunderlich, S.; Merkert, S.; Weber, N.; Osten, F.; et al. Continuous WNT Control Enables Advanced hPSC Cardiac Processing and Prognostic Surface Marker Identification in Chemically Defined Suspension Culture. Stem Cell Rep. 2019, 13, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Rafiei Hashtchin, A.; Manstein, F.; Carvalho Oliveira, M.; Kempf, H.; Zweigerdt, R.; Lachmann, N. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors. Nat. Protoc. 2022, 17, 513–539. [Google Scholar] [CrossRef] [PubMed]
- Sahabian, A.; Dahlmann, J.; Martin, U.; Olmer, R. Production and cryopreservation of definitive endoderm from human pluripotent stem cells under defined and scalable culture conditions. Nat. Protoc. 2021, 16, 1581–1599. [Google Scholar] [CrossRef]
- Vosough, M.; Omidinia, E.; Kadivar, M.; Shokrgozar, M.A.; Pournasr, B.; Aghdami, N.; Baharvand, H. Generation of functional hepatocyte-like cells from human pluripotent stem cells in a scalable suspension culture. Stem Cells Dev. 2013, 22, 2693–2705. [Google Scholar] [CrossRef]
- Yan, Y.; Song, L.; Tsai, A.C.; Ma, T.; Li, Y. Generation of Neural Progenitor Spheres from Human Pluripotent Stem Cells in a Suspension Bioreactor. Methods Mol. Biol. 2016, 1502, 119–128. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Zahmatkesh, E.; Ghanian, M.H.; Zarkesh, I.; Farzaneh, Z.; Halvaei, M.; Heydari, Z.; Moeinvaziri, F.; Othman, A.; Ruoss, M.; Piryaei, A.; et al. Tissue-Specific Microparticles Improve Organoid Microenvironment for Efficient Maturation of Pluripotent Stem-Cell-Derived Hepatocytes. Cells 2021, 10, 1274. [Google Scholar] [CrossRef]
- Ronen, D.; Benvenisty, N. Genomic stability in reprogramming. Curr. Opin. Genet. Dev. 2012, 22, 444–449. [Google Scholar] [CrossRef]
- Andrews, P.W.; Barbaric, I.; Benvenisty, N.; Draper, J.S.; Ludwig, T.; Merkle, F.T.; Sato, Y.; Spits, C.; Stacey, G.N.; Wang, H.; et al. The consequences of recurrent genetic and epigenetic variants in human pluripotent stem cells. Cell Stem Cell 2022, 29, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Martin, U. Genome stability of programmed stem cell products. Adv. Drug Deliv. Rev. 2017, 120, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Mariani, J.; Palejev, D.; Zhang, Y.; Haney, M.S.; Tomasini, L.; Ferrandino, A.F.; Rosenberg Belmaker, L.A.; Szekely, A.; Wilson, M.; et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 2012, 492, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Kosanke, M.; Osetek, K.; Haase, A.; Wiehlmann, L.; Davenport, C.; Schwarzer, A.; Adams, F.; Kleppa, M.J.; Schambach, A.; Merkert, S.; et al. Reprogramming enriches for somatic cell clones with small-scale mutations in cancer-associated genes. Mol. Ther. 2021, 29, 2535–2553. [Google Scholar] [CrossRef]
- International Stem Cell, I. Assessment of established techniques to determine developmental and malignant potential of human pluripotent stem cells. Nat. Commun. 2018, 9, 1925. [Google Scholar] [CrossRef]
- Andrews, P.W.; Ben-David, U.; Benvenisty, N.; Coffey, P.; Eggan, K.; Knowles, B.B.; Nagy, A.; Pera, M.; Reubinoff, B.; Rugg-Gunn, P.J.; et al. Assessing the Safety of Human Pluripotent Stem Cells and Their Derivatives for Clinical Applications. Stem Cell Rep. 2017, 9, 1–4. [Google Scholar] [CrossRef]
- Lovell-Badge, R.; Anthony, E.; Barker, R.A.; Bubela, T.; Brivanlou, A.H.; Carpenter, M.; Charo, R.A.; Clark, A.; Clayton, E.; Cong, Y.; et al. ISSCR Guidelines for Stem Cell Research and Clinical Translation: The 2021 update. Stem Cell Rep. 2021, 16, 1398–1408. [Google Scholar] [CrossRef]
- Liu, Y.W.; Chen, B.; Yang, X.; Fugate, J.A.; Kalucki, F.A.; Futakuchi-Tsuchida, A.; Couture, L.; Vogel, K.W.; Astley, C.A.; Baldessari, A.; et al. Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 2018, 36, 597–605. [Google Scholar] [CrossRef]
- Newcomb, G.; Farkas, L. Endothelial cell clonality, heterogeneity and dysfunction in pulmonary arterial hypertension. Front. Med. 2023, 10, 1304766. [Google Scholar] [CrossRef]
- Cuthbertson, I.; Morrell, N.W.; Caruso, P. BMPR2 Mutation and Metabolic Reprogramming in Pulmonary Arterial Hypertension. Circ. Res. 2023, 132, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.; Gupta, G.; Costanzo, L.; Ahmed, H.; Wyman, A.E.; Geraghty, P. Senescence: Pathogenic Driver in Chronic Obstructive Pulmonary Disease. Medicina 2022, 58, 817. [Google Scholar] [CrossRef] [PubMed]
- Bateman, G.; Guo-Parke, H.; Rodgers, A.M.; Linden, D.; Bailey, M.; Weldon, S.; Kidney, J.C.; Taggart, C.C. Airway Epithelium Senescence as a Driving Mechanism in COPD Pathogenesis. Biomedicines 2023, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yan, Z.; Engelhardt, J.F. Viral Vectors, Animal Models, and Cellular Targets for Gene Therapy of Cystic Fibrosis Lung Disease. Hum. Gene Ther. 2020, 31, 524–537. [Google Scholar] [CrossRef]
- Horani, A.; Ferkol, T.W. Understanding Primary Ciliary Dyskinesia and Other Ciliopathies. J. Pediatr. 2021, 230, 15–22.e11. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, N.; Happle, C.; Ackermann, M.; Luttge, D.; Wetzke, M.; Merkert, S.; Hetzel, M.; Kensah, G.; Jara-Avaca, M.; Mucci, A.; et al. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2014, 189, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Happle, C.; Lachmann, N.; Ackermann, M.; Mirenska, A.; Gohring, G.; Thomay, K.; Mucci, A.; Hetzel, M.; Glomb, T.; Suzuki, T.; et al. Pulmonary Transplantation of Human Induced Pluripotent Stem Cell-derived Macrophages Ameliorates Pulmonary Alveolar Proteinosis. Am. J. Respir. Crit. Care Med. 2018, 198, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kuhnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 2018, 9, 5088. [Google Scholar] [CrossRef]
- Liu, X.; Qi, J.; Xu, X.; Zeisberg, M.; Guan, K.; Zeisberg, E.M. Differentiation of functional endothelial cells from human induced pluripotent stem cells: A novel, highly efficient and cost effective method. Differentiation 2016, 92, 225–236. [Google Scholar] [CrossRef]
- Orlova, V.V.; van den Hil, F.E.; Petrus-Reurer, S.; Drabsch, Y.; Ten Dijke, P.; Mummery, C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014, 9, 1514–1531. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Alber, A.B.; Marquez, H.A.; Ma, L.; Kwong, G.; Thapa, B.R.; Villacorta-Martin, C.; Lindstrom-Vautrin, J.; Bawa, P.; Wang, F.; Luo, Y.; et al. Directed differentiation of mouse pluripotent stem cells into functional lung-specific mesenchyme. Nat. Commun. 2023, 14, 3488. [Google Scholar] [CrossRef]
- Wilson, H.K.; Canfield, S.G.; Shusta, E.V.; Palecek, S.P. Concise review: Tissue-specific microvascular endothelial cells derived from human pluripotent stem cells. Stem Cells 2014, 32, 3037–3045. [Google Scholar] [CrossRef]
- Park, T.S.; Hirday, R.; Ali, A.; Megersa, R.; Villasmil, R.; Nguyen, E.; Bharti, K. Protocol to generate endothelial cells, pericytes, and fibroblasts in one differentiation round from human-induced pluripotent stem cells. STAR Protoc. 2023, 4, 102292. [Google Scholar] [CrossRef] [PubMed]
- Szepes, M.; Melchert, A.; Dahlmann, J.; Hegermann, J.; Werlein, C.; Jonigk, D.; Haverich, A.; Martin, U.; Olmer, R.; Gruh, I. Dual Function of iPSC-Derived Pericyte-Like Cells in Vascularization and Fibrosis-Related Cardiac Tissue Remodeling In Vitro. Int. J. Mol. Sci. 2020, 21, 8947. [Google Scholar] [CrossRef]
- Marchand, M.; Anderson, E.K.; Phadnis, S.M.; Longaker, M.T.; Cooke, J.P.; Chen, B.; Reijo Pera, R.A. Concurrent generation of functional smooth muscle and endothelial cells via a vascular progenitor. Stem Cells Transl. Med. 2014, 3, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.N.; Edgar, A.J.; Samadikuchaksaraei, A.; Timson, C.M.; Romanska, H.M.; Polak, J.M.; Bishop, A.E. Derivation of type II alveolar epithelial cells from murine embryonic stem cells. Tissue Eng. 2002, 8, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Coraux, C.; Nawrocki-Raby, B.; Hinnrasky, J.; Kileztky, C.; Gaillard, D.; Dani, C.; Puchelle, E. Embryonic stem cells generate airway epithelial tissue. Am. J. Respir. Cell Mol. Biol. 2005, 32, 87–92. [Google Scholar] [CrossRef]
- Schmeckebier, S.; Mauritz, C.; Katsirntaki, K.; Sgodda, M.; Puppe, V.; Duerr, J.; Schubert, S.C.; Schmiedl, A.; Lin, Q.; Palecek, J.; et al. Keratinocyte Growth Factor and Dexamethasone Plus Elevated cAMP Levels Synergistically Support Pluripotent Stem Cell Differentiation into Alveolar Epithelial Type II Cells. Tissue Eng. Part A 2013, 19, 938–951. [Google Scholar] [CrossRef]
- Katsirntaki, K.; Mauritz, C.; Olmer, R.; Schmeckebier, S.; Sgodda, M.; Puppe, V.; Eggenschwiler, R.; Duerr, J.; Schubert, S.C.; Schmiedl, A.; et al. Bronchoalveolar sublineage specification of pluripotent stem cells: Effect of dexamethasone plus cAMP-elevating agents and keratinocyte growth factor. Tissue Eng. Part A 2015, 21, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, H.W. Modeling human lung development and disease using pluripotent stem cells. Development 2015, 142, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Longmire, T.A.; Ikonomou, L.; Hawkins, F.; Christodoulou, C.; Cao, Y.; Jean, J.C.; Kwok, L.W.; Mou, H.; Rajagopal, J.; Shen, S.S.; et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 2012, 10, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Islam, M.N.; O’Neill, J.; Hu, Z.; Yang, Y.G.; Chen, Y.W.; Mumau, M.; Green, M.D.; Vunjak-Novakovic, G.; Bhattacharya, J.; et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Green, M.D.; de Carvalho, A.T.; Mumau, M.; Chen, Y.W.; D’Souza, S.L.; Snoeck, H.W. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 413–425. [Google Scholar] [CrossRef]
- Hawkins, F.; Kramer, P.; Jacob, A.; Driver, I.; Thomas, D.C.; McCauley, K.B.; Skvir, N.; Crane, A.M.; Kurmann, A.A.; Hollenberg, A.N.; et al. Prospective isolation of NKX2-1-expressing human lung progenitors derived from pluripotent stem cells. J. Clin. Investig. 2017, 127, 2277–2294. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell 2017, 20, 844–857.e846. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Kotton, D.N. Derivation of Epithelial-Only Airway Organoids from Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2018, 45, e51. [Google Scholar] [CrossRef]
- Hawkins, F.J.; Suzuki, S.; Beermann, M.L.; Barilla, C.; Wang, R.; Villacorta-Martin, C.; Berical, A.; Jean, J.C.; Le Suer, J.; Matte, T.; et al. Derivation of Airway Basal Stem Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2021, 28, 79–95.e78. [Google Scholar] [CrossRef]
- Suzuki, S.; Hawkins, F.J.; Barilla, C.; Beermann, M.L.; Kotton, D.N.; Davis, B.R. Differentiation of human pluripotent stem cells into functional airway basal stem cells. STAR Protoc. 2021, 2, 100683. [Google Scholar] [CrossRef]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.C.; Heins, H.; Na, C.L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017, 21, 472–488.e410. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Vedaie, M.; Roberts, D.A.; Thomas, D.C.; Villacorta-Martin, C.; Alysandratos, K.D.; Hawkins, F.; Kotton, D.N. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat. Protoc. 2019, 14, 3303–3332. [Google Scholar] [CrossRef] [PubMed]
- Burgess, C.L.; Huang, J.; Bawa, P.; Alysandratos, K.D.; Minakin, K.; Morley, M.P.; Babu, A.; Villacorta-Martin, C.; Hinds, A.; Thapa, B.R.; et al. Generation of human alveolar epithelial type I cells from pluripotent stem cells. bioRxiv 2023. [Google Scholar] [CrossRef]
- Katsura, H.; Sontake, V.; Tata, A.; Kobayashi, Y.; Edwards, C.E.; Heaton, B.E.; Konkimalla, A.; Asakura, T.; Mikami, Y.; Fritch, E.J.; et al. Human Lung Stem Cell-Based Alveolospheres Provide Insights into SARS-CoV-2-Mediated Interferon Responses and Pneumocyte Dysfunction. Cell Stem Cell 2020, 27, 890–904.e898. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Choi, S.S.; Rustagi, A.; Zhu, J.; van Unen, V.; de la, O.S.; Flynn, R.A.; Margalef-Catala, M.; Santos, A.J.M.; Ju, J.; et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 2020, 588, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Thapa, B.R.; Le Suer, J.A.; Tilston-Lunel, A.; Herriges, M.J.; Berical, A.; Beermann, M.L.; Wang, F.; Bawa, P.S.; Kohn, A.; et al. Airway stem cell reconstitution by the transplantation of primary or pluripotent stem cell-derived basal cells. Cell Stem Cell 2023, 30, 1199–1216.e1197. [Google Scholar] [CrossRef] [PubMed]
- Herriges, M.J.; Yampolskaya, M.; Thapa, B.R.; Lindstrom-Vautrin, J.; Wang, F.; Huang, J.; Na, C.L.; Ma, L.; Montminy, M.M.; Bawa, P.; et al. Durable alveolar engraftment of PSC-derived lung epithelial cells into immunocompetent mice. Cell Stem Cell 2023, 30, 1217–1234.e1217. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.R.; Russell, M.L.; Roggli, V.L.; Crapo, J.D. Cell number and distribution in human and rat airways. Am. J. Respir. Cell Mol. Biol. 1994, 10, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Danopoulos, S.; Shiosaki, J.; Al Alam, D. FGF Signaling in Lung Development and Disease: Human Versus Mouse. Front. Genet. 2019, 10, 170. [Google Scholar] [CrossRef]
- Rosen, C.; Shezen, E.; Aronovich, A.; Klionsky, Y.Z.; Yaakov, Y.; Assayag, M.; Biton, I.E.; Tal, O.; Shakhar, G.; Ben-Hur, H.; et al. Preconditioning allows engraftment of mouse and human embryonic lung cells, enabling lung repair in mice. Nat. Med. 2015, 21, 869–879. [Google Scholar] [CrossRef]
- Kadam, A.H.; Schnitzer, J.E. Characterization of acute lung injury in the bleomycin rat model. Physiol. Rep. 2023, 11, e15618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, Y.; Li, W.C.; Tang, B.X.; Li, J.; Zang, Y. Roxithromycin attenuates bleomycin-induced pulmonary fibrosis by targeting senescent cells. Acta Pharmacol. Sin. 2021, 42, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shi, J.; Tang, H. Animal models of drug-induced pulmonary fibrosis: An overview of molecular mechanisms and characteristics. Cell Biol. Toxicol. 2022, 38, 699–723. [Google Scholar] [CrossRef] [PubMed]
- Vats, A.; Chaturvedi, P. The Regenerative Power of Stem Cells: Treating Bleomycin-Induced Lung Fibrosis. Stem Cells Cloning 2023, 16, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Yasutomo, K. Genetics and animal models of familial pulmonary fibrosis. Int. Immunol. 2021, 33, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Gazdhar, A.; Grad, I.; Tamo, L.; Gugger, M.; Feki, A.; Geiser, T. The secretome of induced pluripotent stem cells reduces lung fibrosis in part by hepatocyte growth factor. Stem Cell Res. Ther. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.; Schmidt, M.; Mattoli, S. Stem Cell-Based Therapy in Idiopathic Pulmonary Fibrosis. Stem Cell Rev. Rep. 2015, 11, 598–620. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Quan, Y.; Sun, H.; Peng, X.; Zou, Z.; Alcorn, J.L.; Wetsel, R.A.; Wang, D. A site-specific genetic modification for induction of pluripotency and subsequent isolation of derived lung alveolar epithelial type II cells. Stem Cells 2014, 32, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Palomo, B.; Sanchez-Lopez, L.I.; Moodley, Y.; Edel, M.J.; Serrano-Mollar, A. Induced pluripotent stem cell-derived lung alveolar epithelial type II cells reduce damage in bleomycin-induced lung fibrosis. Stem Cell Res. Ther. 2020, 11, 213. [Google Scholar] [CrossRef]
- Upadhyay, P.; Wu, C.W.; Pham, A.; Zeki, A.A.; Royer, C.M.; Kodavanti, U.P.; Takeuchi, M.; Bayram, H.; Pinkerton, K.E. Animal models and mechanisms of tobacco smoke-induced chronic obstructive pulmonary disease (COPD). J. Toxicol. Environ. Health B Crit. Rev. 2023, 26, 275–305. [Google Scholar] [CrossRef]
- Smith, P.; Kay, J.M.; Heath, D. Hypertensive pulmonary vascular disease in rats after prolonged feeding with Crotalaria spectabilis seeds. J. Pathol. 1970, 102, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Agrawal, V.; Lawrie, A.; Bonnet, S. The Latest in Animal Models of Pulmonary Hypertension and Right Ventricular Failure. Circ. Res. 2022, 130, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Hautefort, A.; Mendes-Ferreira, P.; Sabourin, J.; Manaud, G.; Bertero, T.; Rucker-Martin, C.; Riou, M.; Adao, R.; Manoury, B.; Lambert, M.; et al. Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension. Circulation 2019, 139, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, X.; Sung, Y.K.; Shuffle, E.; Wu, T.H.; Kao, P.N.; Tu, A.B.; Dorfmuller, P.; Cao, A.; Wang, L.; et al. Phenotypically Silent Bone Morphogenetic Protein Receptor 2 Mutations Predispose Rats to Inflammation-Induced Pulmonary Arterial Hypertension by Enhancing the Risk for Neointimal Transformation. Circulation 2019, 140, 1409–1425. [Google Scholar] [CrossRef]
- Huang, W.C.; Ke, M.W.; Cheng, C.C.; Chiou, S.H.; Wann, S.R.; Shu, C.W.; Chiou, K.R.; Tseng, C.J.; Pan, H.W.; Mar, G.Y.; et al. Therapeutic Benefits of Induced Pluripotent Stem Cells in Monocrotaline-Induced Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0142476. [Google Scholar] [CrossRef]
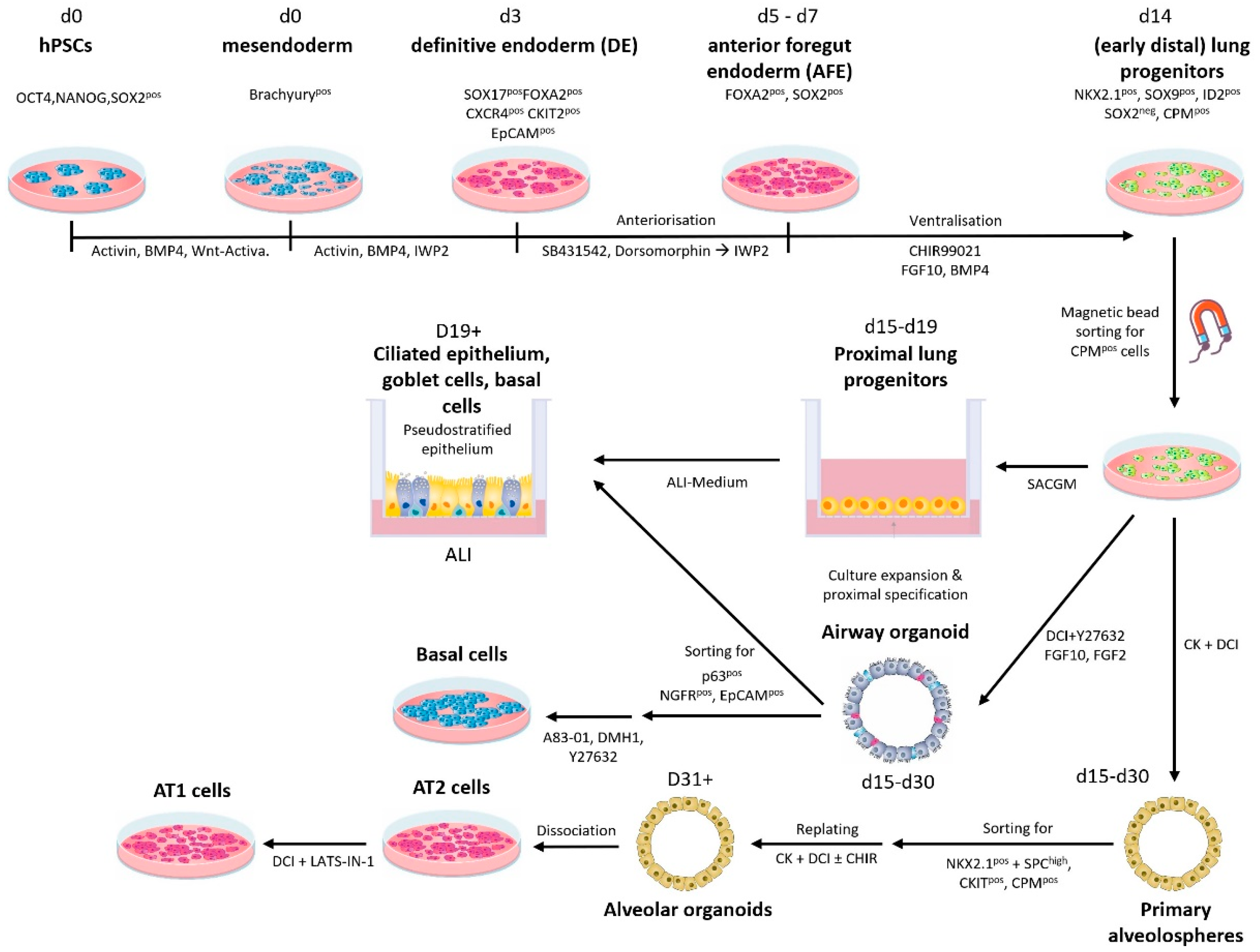

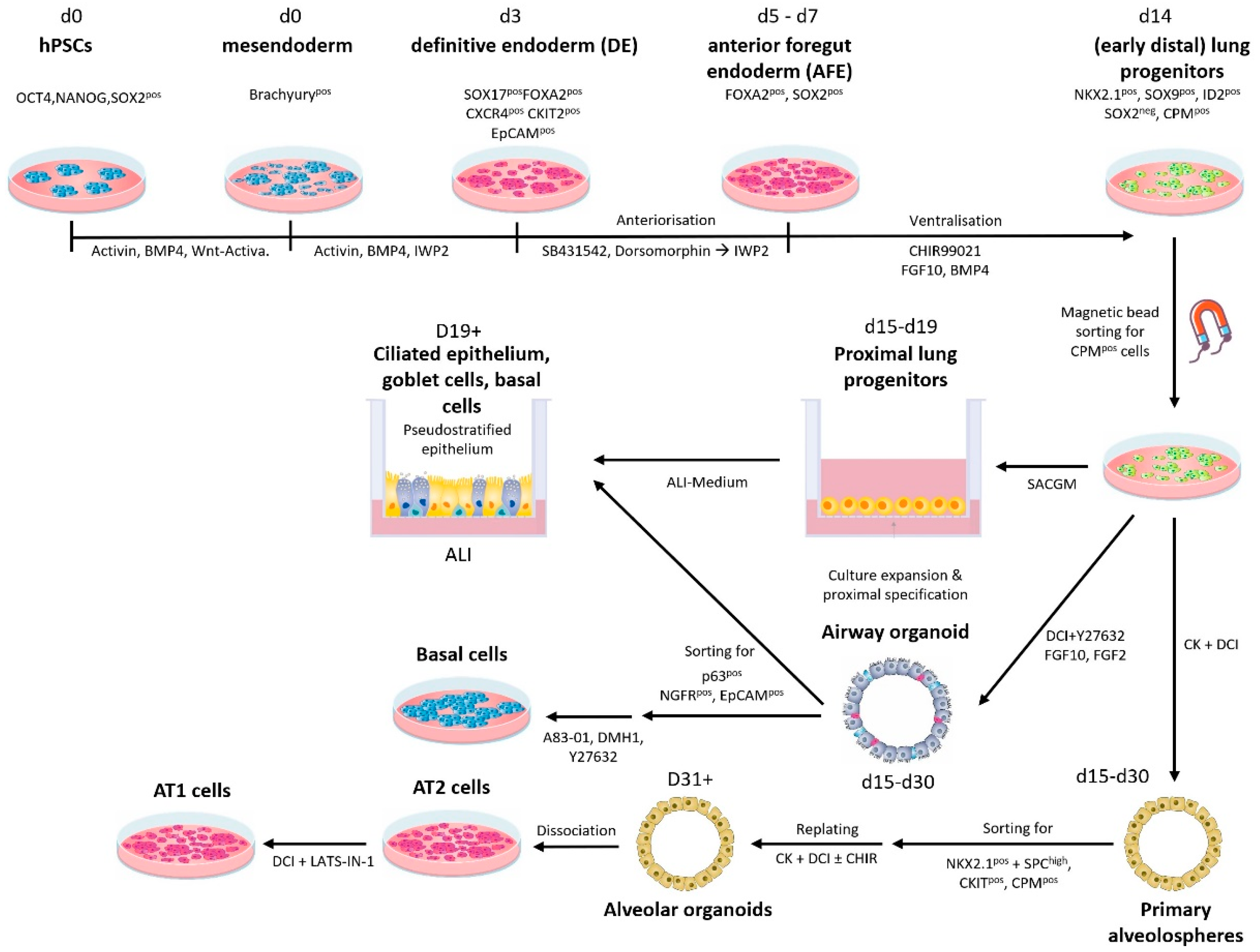{kind=link}
| Cell Type | ECs | Basal Cells | AT2 Cells | Macrophages | ||||
|---|---|---|---|---|---|---|---|---|
| Lung Disease | Allo | Auto | Allo | Auto | Allo | Auto | Allo | Auto |
| PAH | X | |||||||
| COPD | X | X | ||||||
| Fibrosis | X | |||||||
| Cystic Fibrosis | X | |||||||
| PCDs | X | |||||||
| SDs | X | |||||||
| PAP | X | |||||||
| Bacterial Inf. | X | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goecke, T.; Ius, F.; Ruhparwar, A.; Martin, U. Unlocking the Future: Pluripotent Stem Cell-Based Lung Repair. Cells 2024, 13, 635. https://doi.org/10.3390/cells13070635
Goecke T, Ius F, Ruhparwar A, Martin U. Unlocking the Future: Pluripotent Stem Cell-Based Lung Repair. Cells. 2024; 13(7):635. https://doi.org/10.3390/cells13070635
Chicago/Turabian StyleGoecke, Tobias, Fabio Ius, Arjang Ruhparwar, and Ulrich Martin. 2024. "Unlocking the Future: Pluripotent Stem Cell-Based Lung Repair" Cells 13, no. 7: 635. https://doi.org/10.3390/cells13070635
APA StyleGoecke, T., Ius, F., Ruhparwar, A., & Martin, U. (2024). Unlocking the Future: Pluripotent Stem Cell-Based Lung Repair. Cells, 13(7), 635. https://doi.org/10.3390/cells13070635