

ZO-1 Regulates Hippo-Independent YAP Activity and Cell Proliferation via a GEF-H1- and TBK1-Regulated Signalling Network

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Experimental Setups
2.3. siRNA Transfections
2.4. Reporter Gene Assays
2.5. Antibodies and Immunological Methods
2.6. Cell Proliferation, Necrosis, and Apoptosis Assays
2.7. Light Microscopy
2.8. Image Processing and Quantifications
2.9. Statistics and Reproducibility
3. Results
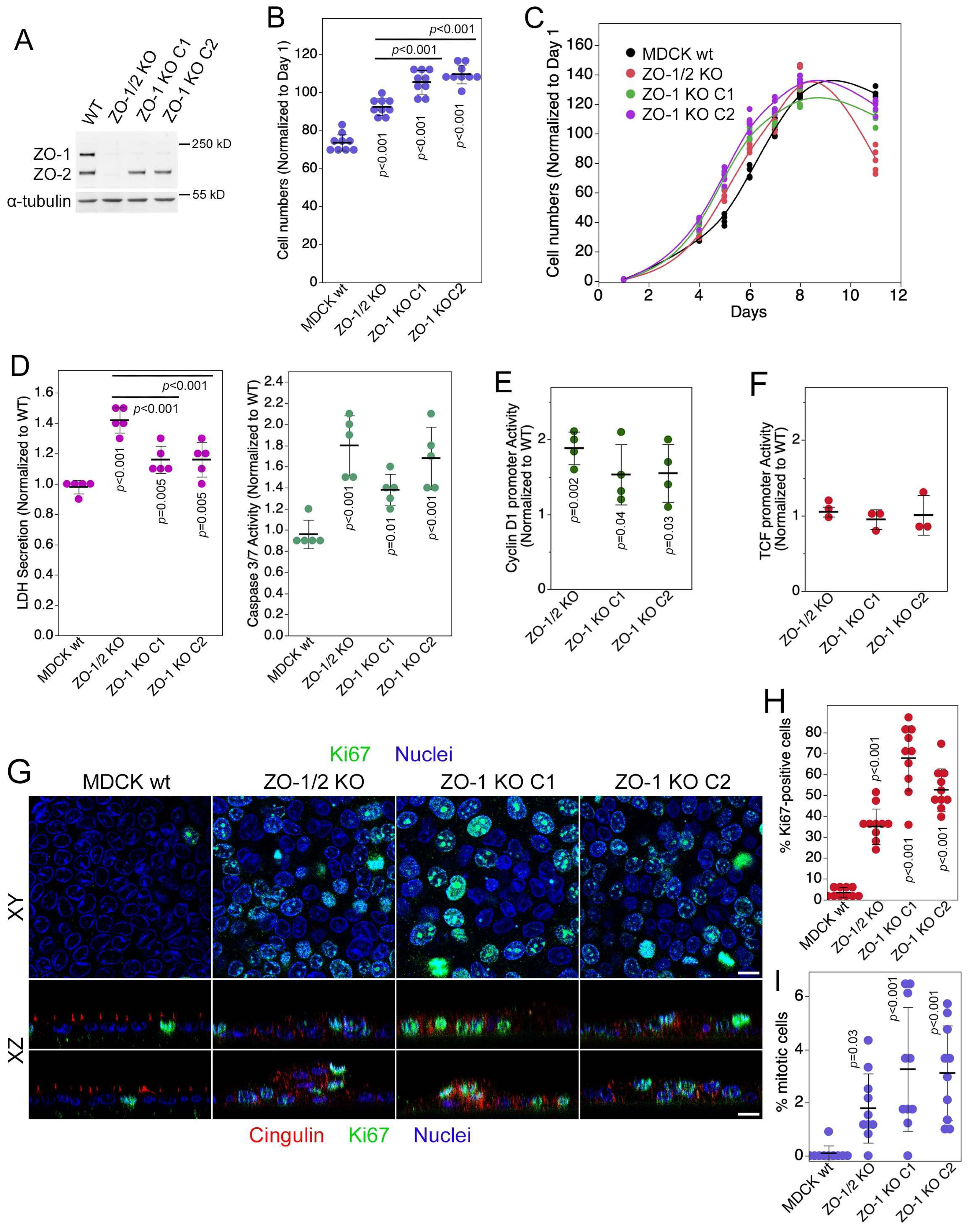
3.1. ZO-1 Regulates Cell Proliferation and Death
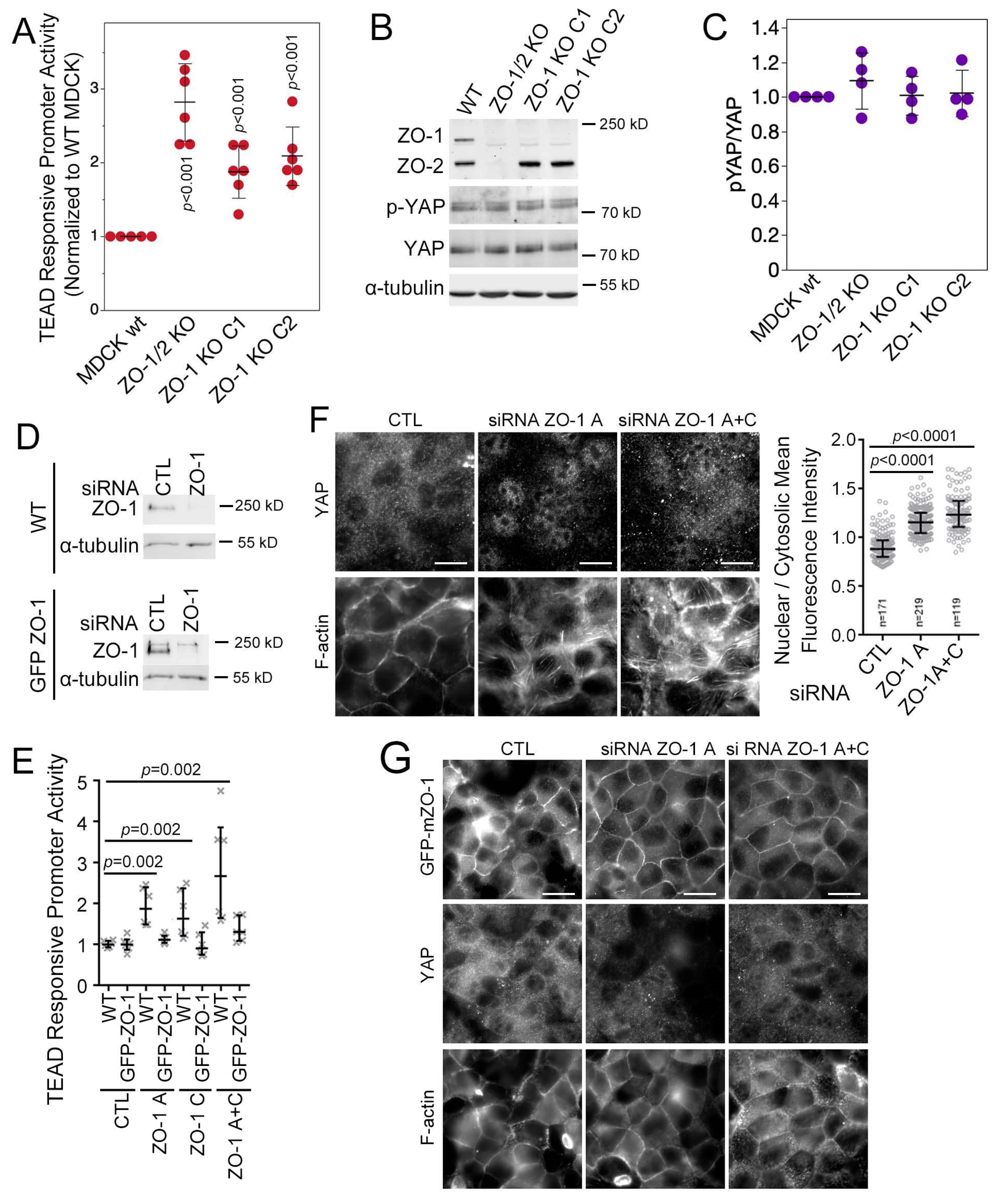
3.2. ZO-1 Regulates YAP/TEAD Activity
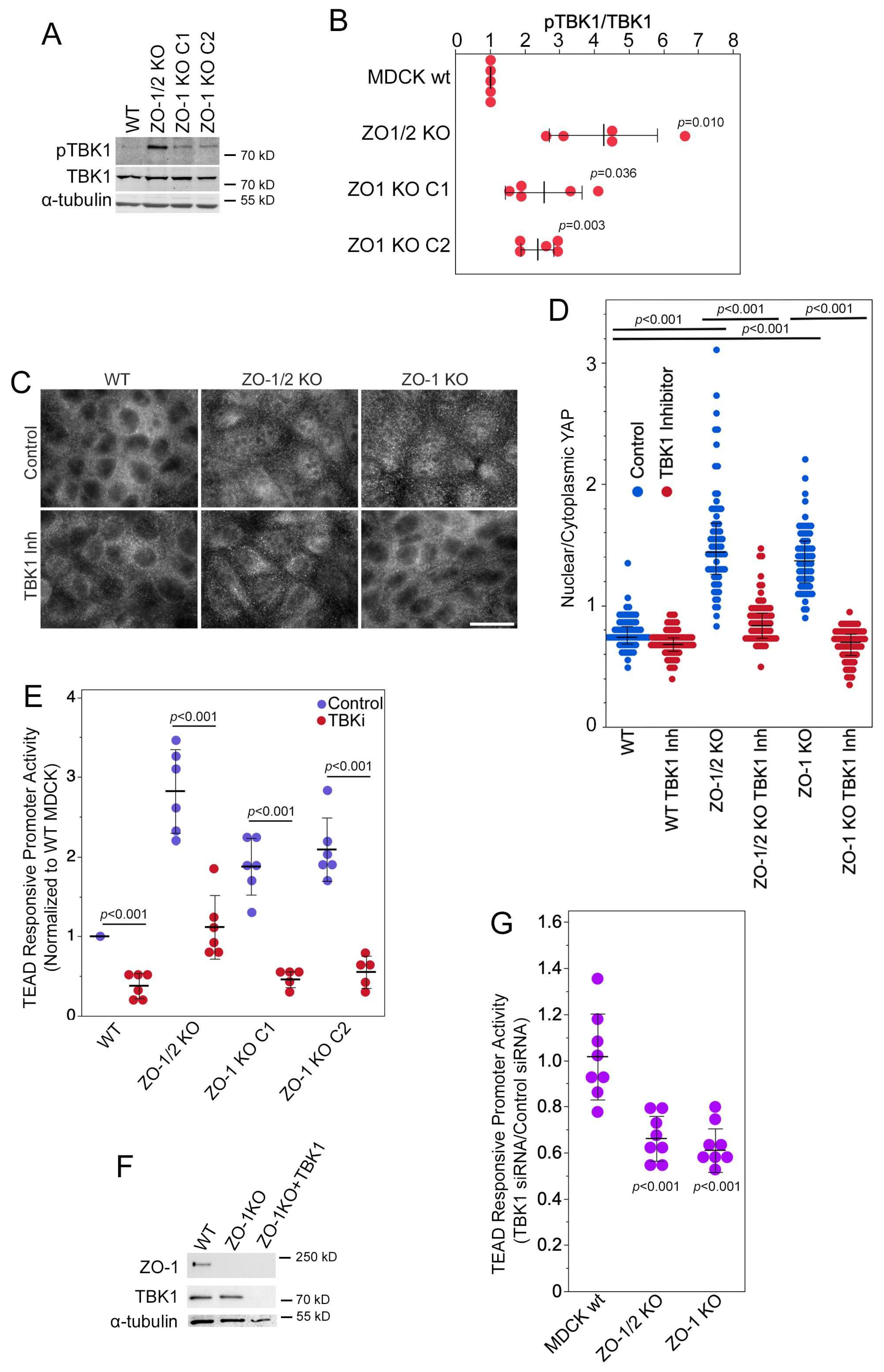
3.3. TBK1 Is Required for Regulation of YAP by ZO-1
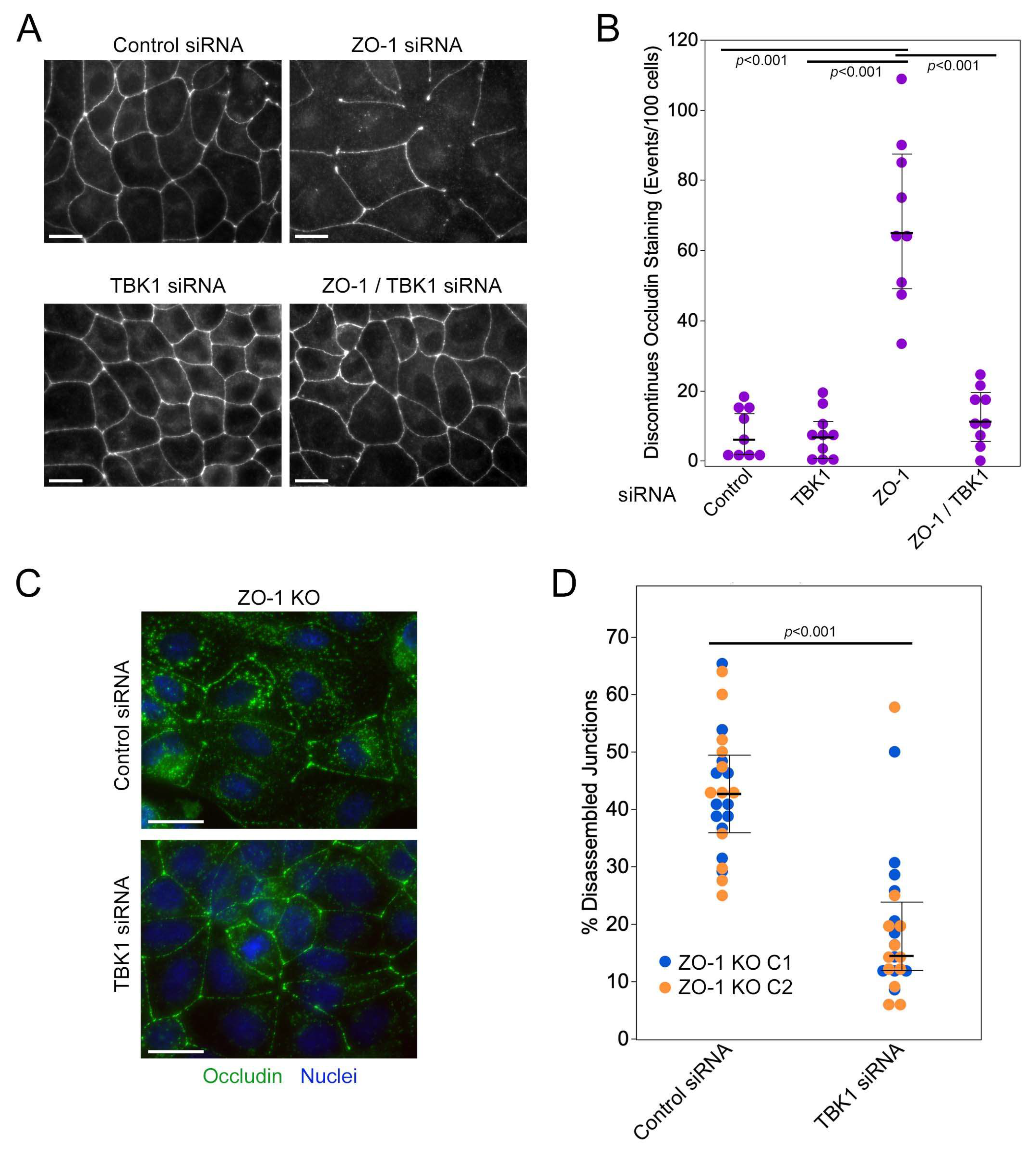
3.4. TBK1 Depletion Promotes Junction Formation by ZO-1-Deficient Cells
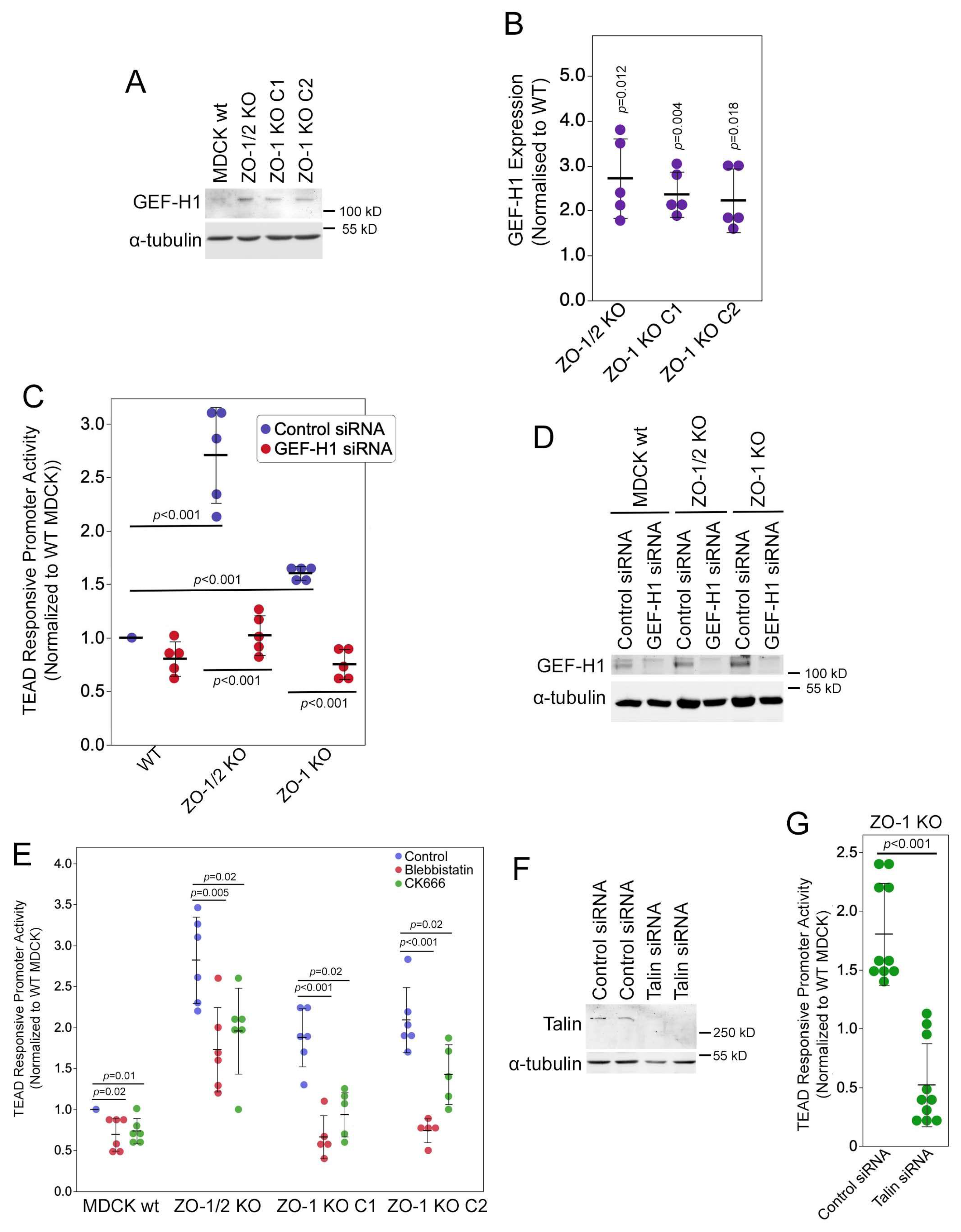
3.5. ZO-1 Regulates YAP/TAZ Activity in a GEF-H1- and Focal Adhesion-Dependent Manner
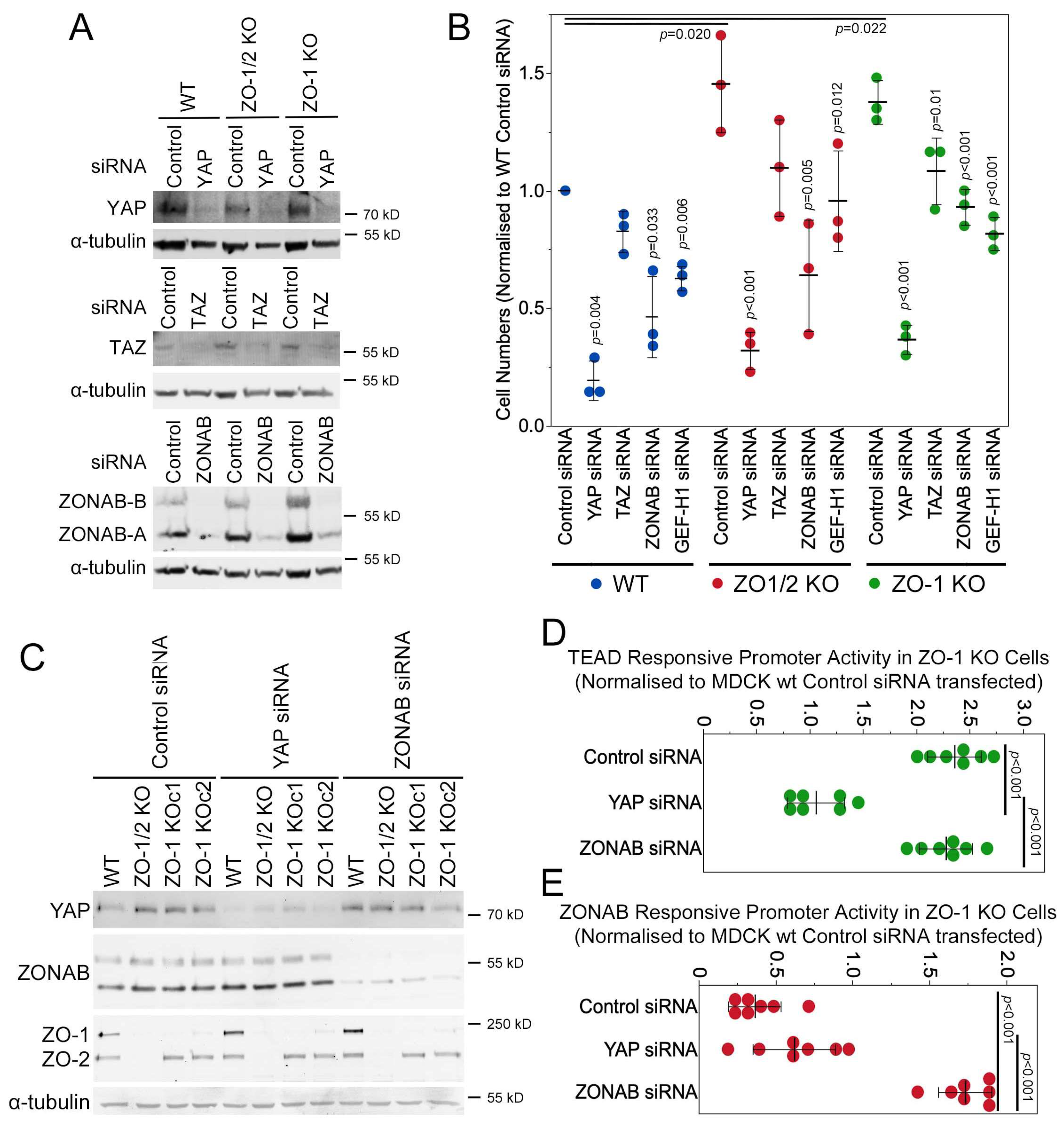
3.6. YAP and GEF-H1 Drive Proliferation of ZO-1 Knockout Cells
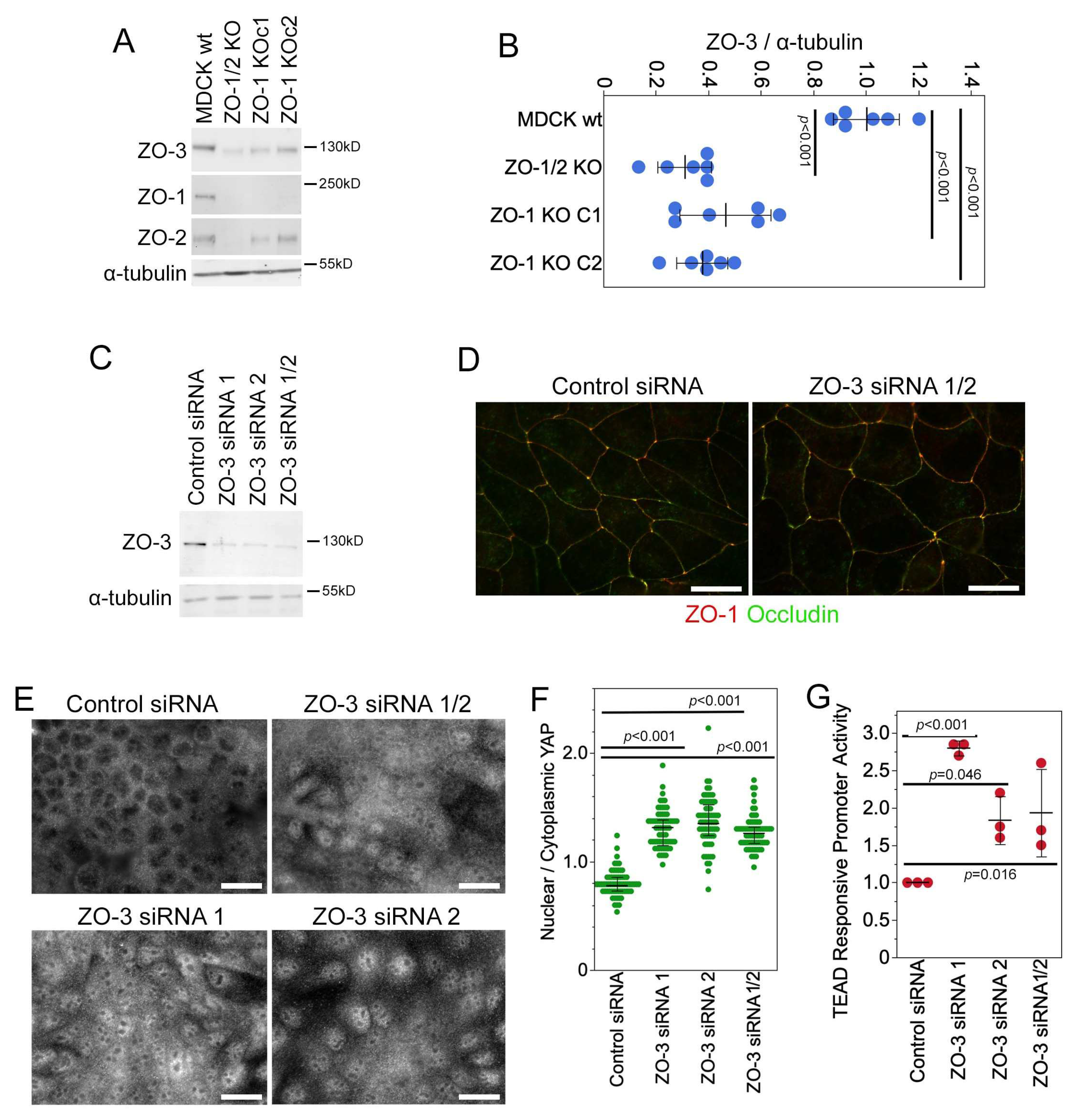
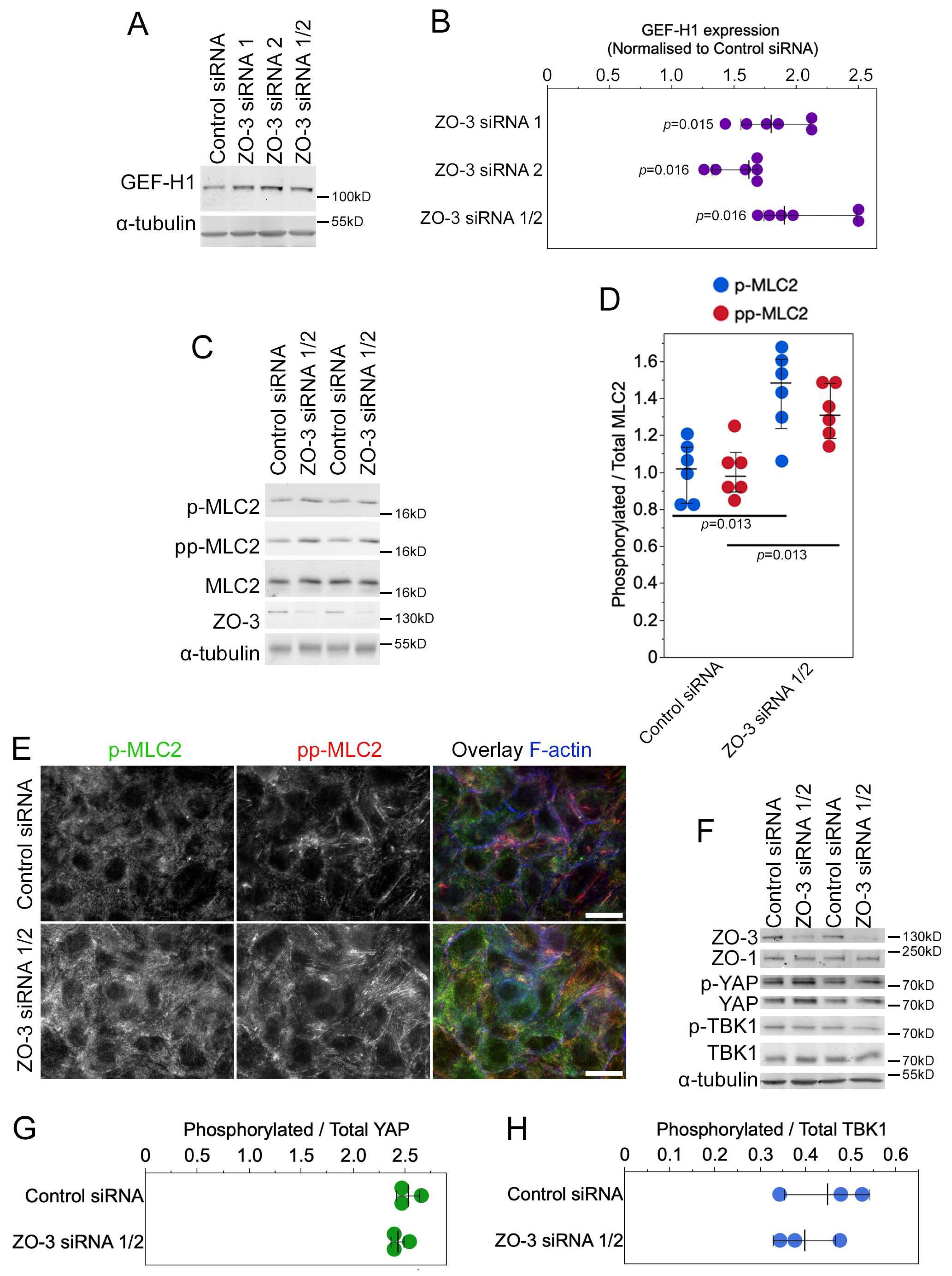
3.7. ZO-3 Regulates GEF-H1 Expression and YAP Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venhuizen, J.H.; Jacobs, F.J.C.; Span, P.N.; Zegers, M.M. P120 and E-cadherin: Double-edged swords in tumor metastasis. Semin. Cancer Biol. 2020, 60, 107–120. [Google Scholar] [CrossRef]
- McCrea, P.D.; Gottardi, C.J. Beyond beta-catenin: Prospects for a larger catenin network in the nucleus. Nat. Rev. Mol. Cell Biol. 2016, 17, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. Tight junctions. Curr. Biol. 2023, 33, R1135–R1140. [Google Scholar] [CrossRef]
- Balda, M.S.; Matter, K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 2000, 19, 2024–2033. [Google Scholar] [CrossRef]
- Balda, M.S.; Garrett, M.D.; Matter, K. The ZO-1–associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003, 160, 423–432. [Google Scholar] [CrossRef] [PubMed]
- González-Mariscal, L.; Domínguez-Calderón, A.; Raya-Sandino, A.; Ortega-Olvera, J.M.; Vargas-Sierra, O.; Martínez-Revollar, G. Tight junctions and the regulation of gene expression. Semin. Cell Dev. Biol. 2014, 36, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.J.; Zihni, C.; Ruppel, A.; Hartmann, C.; Ebnet, K.; Tada, M.; Balda, M.S.; Matter, K. Interplay between Extracellular Matrix Stiffness and JAM-A Regulates Mechanical Load on ZO-1 and Tight Junction Assembly. Cell Rep. 2020, 32, 107924. [Google Scholar] [CrossRef]
- Haas, A.J.; Zihni, C.; Krug, S.M.; Maraspini, R.; Otani, T.; Furuse, M.; Honigmann, A.; Balda, M.; Matter, K. ZO-1 guides tight junction assembly and epithelial morphogenesis via cytoskeletal tension-dependent and -independent functions. Cells 2022, 11, 3775. [Google Scholar] [CrossRef]
- Martin, T.A. The role of tight junctions in cancer metastasis. Semin. Cell Dev. Biol. 2014, 36, 224–231. [Google Scholar] [CrossRef]
- Georgiadis, A.; Tschernutter, M.; Bainbridge, J.W.; Balaggan, K.S.; Mowat, F.; West, E.L.; Munro, P.M.; Thrasher, A.J.; Matter, K.; Balda, M.S.; et al. The tight junction associated signalling proteins ZO-1 and ZONAB regulate retinal pigment epithelium homeostasis in mice. PLoS ONE 2010, 5, e15730. [Google Scholar] [CrossRef]
- Katsuno, T.; Umeda, K.; Matsui, T.; Hata, M.; Tamura, A.; Itoh, M.; Takeuchi, K.; Fujimori, T.; Nabeshima, Y.; Noda, T.; et al. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol. Biol. Cell 2008, 19, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, D.; Tapia, R.; Jond, L.; Sudol, M.; Fanning, A.S.; Citi, S. ZO Proteins Redundantly Regulate the Transcription Factor DbpA/ZONAB. J. Biol. Chem. 2014, 289, 22500–22511. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef]
- Karaman, R.; Halder, G. Cell Junctions in Hippo Signaling. Cold Spring Harb. Perspect. Biol. 2018, 10, a028753. [Google Scholar] [CrossRef]
- Fan, S.; Smith, M.S.; Keeney, J.; O’Leary, M.N.; Nusrat, A.; Parkos, C.A. JAM-A signals through the Hippo pathway to regulate intestinal epithelial proliferation. iScience 2022, 25, 104316. [Google Scholar] [CrossRef] [PubMed]
- Benham-Pyle, B.W.; Pruitt, B.L.; Nelson, W.J. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and beta-catenin activation to drive cell cycle entry. Science 2015, 348, 1024–1027. [Google Scholar] [CrossRef]
- Dominguez-Calderon, A.; Avila-Flores, A.; Ponce, A.; Lopez-Bayghen, E.; Calderon-Salinas, J.V.; Luis Reyes, J.; Chavez-Munguia, B.; Segovia, J.; Angulo, C.; Ramirez, L.; et al. ZO-2 silencing induces renal hypertrophy through a cell cycle mechanism and the activation of YAP and the mTOR pathway. Mol. Biol. Cell 2016, 27, 1581–1595. [Google Scholar] [CrossRef]
- Wittchen, E.S.; Haskins, J.; Stevenson, B.R. Protein interactions at the tight junction. Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J. Biol. Chem. 1999, 274, 35179–35185. [Google Scholar] [CrossRef]
- Balda, M.S.; Gonzalez-Mariscal, L.; Matter, K.; Cereijido, M.; Anderson, J.M. Assembly of the tight junction: The role of diacylglycerol. J. Cell Biol. 1993, 123, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Haskins, J.; Gu, L.; Wittchen, E.S.; Hibbard, J.; Stevenson, B.R. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J. Cell Biol. 1998, 141, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Wittchen, E.S.; Haskins, J.; Stevenson, B.R. NZO-3 expression causes global changes to actin cytoskeleton in Madin-Darby canine kidney cells: Linking a tight junction protein to Rho GTPases. Mol. Biol. Cell 2003, 14, 1757–1768. [Google Scholar] [CrossRef]
- Capaldo, C.T.; Koch, S.; Kwon, M.; Laur, O.; Parkos, C.A.; Nusrat, A. Tight function zonula occludens-3 regulates cyclin D1-dependent cell proliferation. Mol. Biol. Cell 2011, 22, 1677–1685. [Google Scholar] [CrossRef]
- Beutel, O.; Maraspini, R.; Pombo-García, K.; Martin-Lemaitre, C.; Honigmann, A. Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions. Cell 2019, 179, 923–936.e11. [Google Scholar] [CrossRef] [PubMed]
- Matter, K.; Whitney, J.A.; Yamamoto, E.M.; Mellman, I. Common signals control low density lipoprotein receptor sorting in endosomes and the Golgi complex of MDCK cells. Cell 1993, 74, 1053–1064. [Google Scholar] [CrossRef]
- Vitiello, E.; Moreau, P.; Nunes, V.; Mettouchi, A.; Maiato, H.; Ferreira, J.G.; Wang, I.; Balland, M. Acto-myosin force organization modulates centriole separation and PLK4 recruitment to ensure centriole fidelity. Nat. Commun. 2019, 10, 52. [Google Scholar] [CrossRef]
- Tse, J.R.; Engler, A.J. Preparation of hydrogel substrates with tunable mechanical properties. Curr. Protoc. Cell Biol. 2010, 47, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Sourisseau, T.; Georgiadis, A.; Tsapara, A.; Ali, R.R.; Pestell, R.; Matter, K.; Balda, M.S. Regulation of PCNA and Cyclin D1 Expression and Epithelial Morphogenesis by the ZO-1-Regulated Transcription Factor ZONAB/DbpA. Mol. Cell. Biol. 2006, 26, 2387–2398. [Google Scholar] [CrossRef]
- Balda, M.S.; Whitney, J.A.; Flores, C.; Gonzalez, S.; Cereijido, M.; Matter, K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 1996, 134, 1031–1049. [Google Scholar] [CrossRef]
- Benais-Pont, G.; Punn, A.; Flores-Maldonado, C.; Eckert, J.; Raposo, G.; Fleming, T.P.; Cereijido, M.; Balda, M.S.; Matter, K. Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J. Cell Biol. 2003, 160, 729–740. [Google Scholar] [CrossRef]
- Kreis, T.E. Microtubules containing detyrosinated tubulin are less dynamic. EMBO J. 1987, 6, 2597–2606. [Google Scholar] [CrossRef]
- Nie, M.; Balda, M.S.; Matter, K. Stress- and Rho-activated ZO-1–associated nucleic acid binding protein binding to p21 mRNA mediates stabilization, translation, and cell survival. Proc. Natl. Acad. Sci. USA 2012, 109, 10897–10902. [Google Scholar] [CrossRef]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A generalist algorithm for cellular segmentation. Nat. Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Legland, D.; Arganda-Carreras, I.; Andrey, P. MorphoLibJ: Integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 2016, 32, 3532–3534. [Google Scholar] [CrossRef]
- Haase, R.; Royer, L.A.; Steinbach, P.; Schmidt, D.; Dibrov, A.; Schmidt, U.; Weigert, M.; Maghelli, N.; Tomancak, P.; Jug, F.; et al. CLIJ: GPU-accelerated image processing for everyone. Nat. Methods 2020, 17, 5–6. [Google Scholar] [CrossRef]
- Huang, X.; Li, X.; Shen, H.; Zhao, Y.; Zhou, Z.; Wang, Y.; Yao, J.; Xue, K.; Wu, D.; Qiu, Y. Transcriptional repression of beige fat innervation via a YAP/TAZ-S100B axis. Nat. Commun. 2023, 14, 7102. [Google Scholar] [CrossRef]
- Aravamudhan, A.; Haak, A.J.; Choi, K.M.; Meridew, J.A.; Caporarello, N.; Jones, D.L.; Tan, Q.; Ligresti, G.; Tschumperlin, D.J. TBK1 regulates YAP/TAZ and fibrogenic fibroblast activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L852–L863. [Google Scholar] [CrossRef]
- Aijaz, S.; D’Atri, F.; Citi, S.; Balda, M.S.; Matter, K. Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev. Cell 2005, 8, 777–786. [Google Scholar] [CrossRef]
- Chiang, H.-S.; Zhao, Y.; Song, J.-H.; Liu, S.; Wang, N.; Terhorst, C.; Sharpe, A.H.; Basavappa, M.; Jeffrey, K.L.; Reinecker, H.-C. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat. Immunol. 2014, 15, 63–71. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef]
- Goult, B.T.; Brown, N.H.; Schwartz, M.A. Talin in mechanotransduction and mechanomemory at a glance. J. Cell Sci. 2021, 134, jcs258749. [Google Scholar] [CrossRef]
- Nie, M.; Aijaz, S.; Leefa Chong San, I.V.; Balda, M.S.; Matter, K. The Y-box factor ZONAB/DbpA associates with GEF-H1/Lfc and mediates Rho-stimulated transcription. EMBO Rep. 2009, 10, 1125–1131. [Google Scholar] [CrossRef]
- Jesaitis, L.A.; Goodenough, D.A. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J. Cell Biol. 1994, 124, 949–961. [Google Scholar] [CrossRef]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef]
- Wittchen, E.S.; Haskins, J.; Stevenson, B.R. Exogenous expression of the amino-terminal half of the tight junction protein ZO-3 perturbs junctional complex assembly. J. Cell Biol. 2000, 151, 825–836. [Google Scholar] [CrossRef]
- Oka, T.; Remue, E.; Meerschaert, K.; Vanloo, B.; Boucherie, C.; Gfeller, D.; Bader, G.D.; Sidhu, S.S.; Vandekerckhove, J.; Gettemans, J.; et al. Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem. Biochem. J. 2010, 432, 461–472. [Google Scholar] [CrossRef]
- Kiener, T.K.; Selptsova-Friedrich, I.; Hunziker, W. Tjp3/zo-3 is critical for epidermal barrier function in zebrafish embryos. Dev. Biol. 2008, 316, 36–49. [Google Scholar] [CrossRef]
- Schwayer, C.; Shamipour, S.; Pranjic-Ferscha, K.; Schauer, A.; Balda, M.; Tada, M.; Matter, K.; Heisenberg, C.-P. Mechanosensation of Tight Junctions Depends on ZO-1 Phase Separation and Flow. Cell 2019, 179, 937–952.e18. [Google Scholar] [CrossRef]
- Adachi, M.; Inoko, A.; Hata, M.; Furuse, K.; Umeda, K.; Itoh, M.; Tsukita, S. Normal establishment of epithelial tight junctions in mice and cultured cells lacking expression of ZO-3, a tight-junction MAGUK protein. Mol. Cell Biol. 2006, 26, 9003–9015. [Google Scholar] [CrossRef]
- Gao, Z.; Gao, Z.; Zhang, H.; Hou, S.; Zhou, Y.; Liu, X. Targeting STING: From antiviral immunity to treat osteoporosis. Front. Immunol. 2022, 13, 1095577. [Google Scholar] [CrossRef]
- Miranda, A.; Shirley, C.A.; Jenkins, R.W. Emerging roles of TBK1 in cancer immunobiology. Trends Cancer 2024. [Google Scholar] [CrossRef]
- Gurfinkel, Y.; Polain, N.; Sonar, K.; Nice, P.; Mancera, R.L.; Rea, S.L. Functional and structural consequences of TBK1 missense variants in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurobiol. Dis. 2022, 174, 105859. [Google Scholar] [CrossRef]
- Tsapara, A.; Luthert, P.; Greenwood, J.; Hill, C.S.; Matter, K.; Balda, M.S. The RhoA activator GEF-H1/Lfc is a transforming growth factor-beta target gene and effector that regulates alpha-smooth muscle actin expression and cell migration. Mol. Biol. Cell 2010, 21, 860–870. [Google Scholar] [CrossRef]
- Yu, H.; He, J.; Su, G.; Wang, Y.; Fang, F.; Yang, W.; Gu, K.; Fu, N.; Wang, Y.; Shen, Y.; et al. Fluid shear stress activates YAP to promote epithelial-mesenchymal transition in hepatocellular carcinoma. Mol. Oncol. 2021, 15, 3164–3183. [Google Scholar] [CrossRef]
- Nakao, Y.; Nakagawa, S.; Yamashita, Y.I.; Umezaki, N.; Okamoto, Y.; Ogata, Y.; Yasuda-Yoshihara, N.; Itoyama, R.; Yusa, T.; Yamashita, K.; et al. High ARHGEF2 (GEF-H1) Expression is Associated with Poor Prognosis Via Cell Cycle Regulation in Patients with Pancreatic Cancer. Ann. Surg. Oncol. 2021, 28, 4733–4743. [Google Scholar] [CrossRef]
- Kent, O.A.; Sandi, M.J.; Rottapel, R. Co-dependency between KRAS addiction and ARHGEF2 promotes an adaptive escape from MAPK pathway inhibition. Small GTPases 2019, 10, 441–448. [Google Scholar] [CrossRef]
- Joo, E.; Olson, M.F. Regulation and functions of the RhoA regulatory guanine nucleotide exchange factor GEF-H1. Small GTPases 2021, 12, 358–371. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haas, A.J.; Karakus, M.; Zihni, C.; Balda, M.S.; Matter, K. ZO-1 Regulates Hippo-Independent YAP Activity and Cell Proliferation via a GEF-H1- and TBK1-Regulated Signalling Network. Cells 2024, 13, 640. https://doi.org/10.3390/cells13070640
Haas AJ, Karakus M, Zihni C, Balda MS, Matter K. ZO-1 Regulates Hippo-Independent YAP Activity and Cell Proliferation via a GEF-H1- and TBK1-Regulated Signalling Network. Cells. 2024; 13(7):640. https://doi.org/10.3390/cells13070640
Chicago/Turabian StyleHaas, Alexis J., Mert Karakus, Ceniz Zihni, Maria S. Balda, and Karl Matter. 2024. "ZO-1 Regulates Hippo-Independent YAP Activity and Cell Proliferation via a GEF-H1- and TBK1-Regulated Signalling Network" Cells 13, no. 7: 640. https://doi.org/10.3390/cells13070640