Mitochondrial Permeability Transition, Cell Death and Neurodegeneration

 ,
,  , ,
, ,  , and
, and 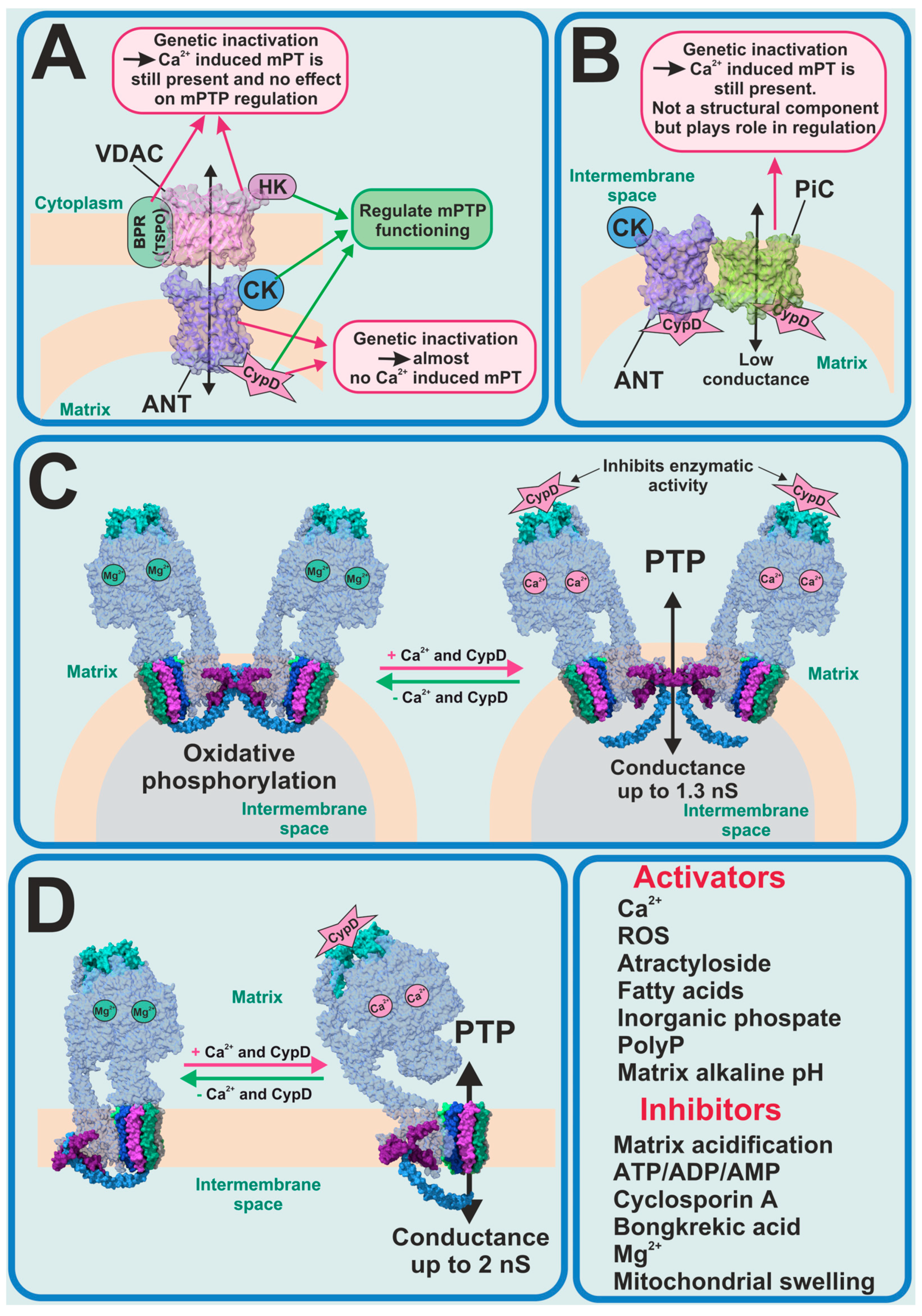{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
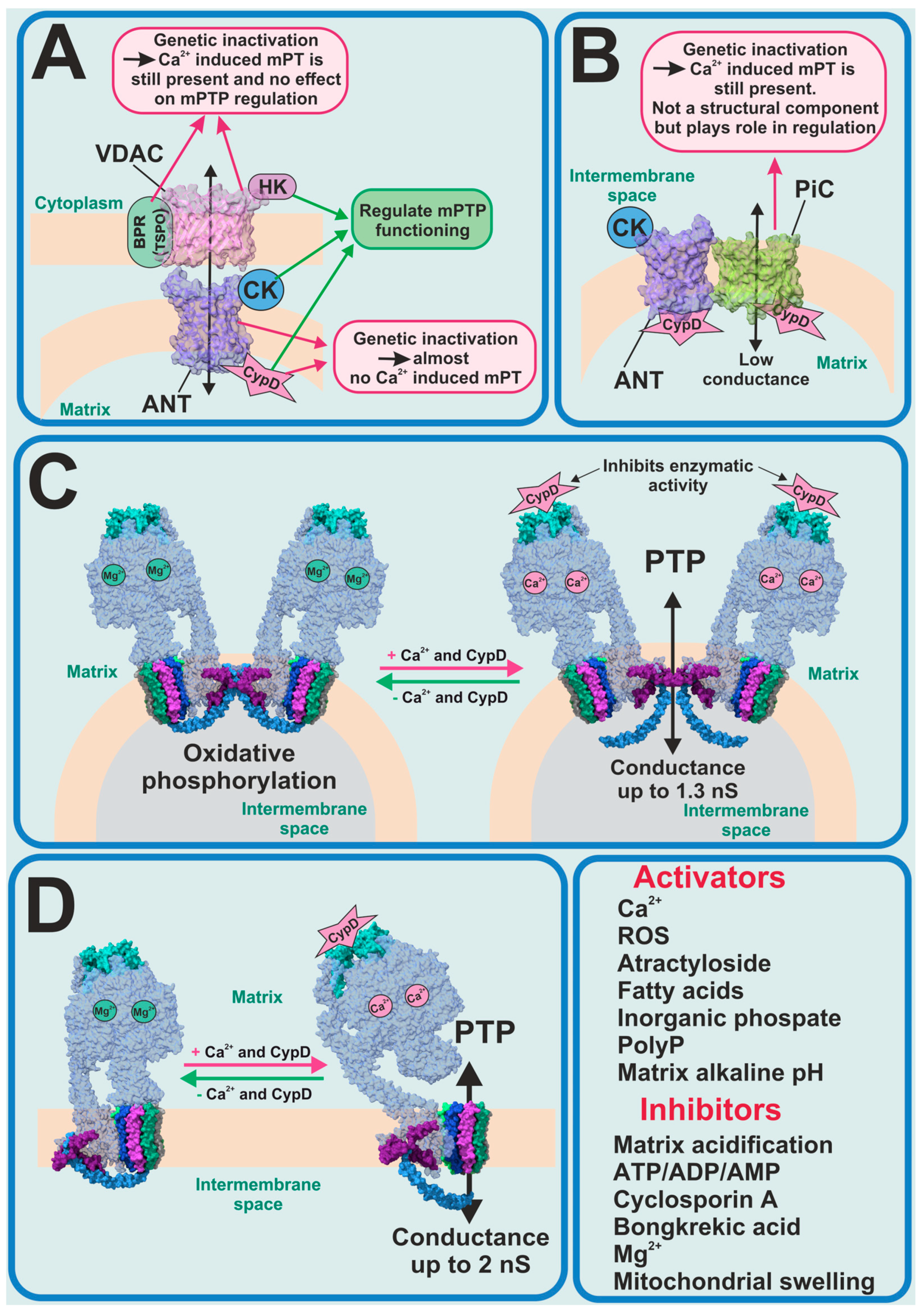
2. Permeability Transition
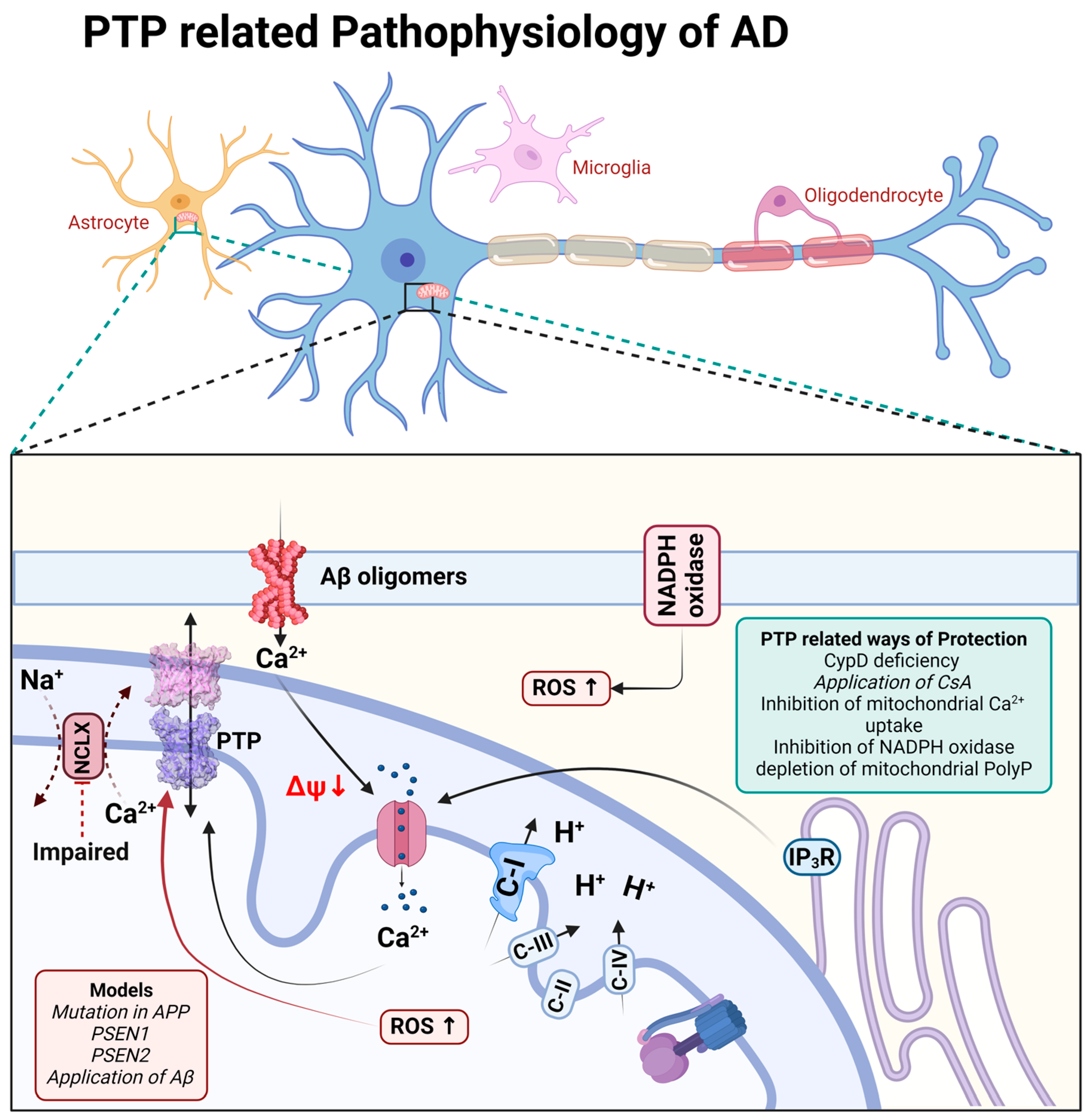
3. Alzheimer’s Disease
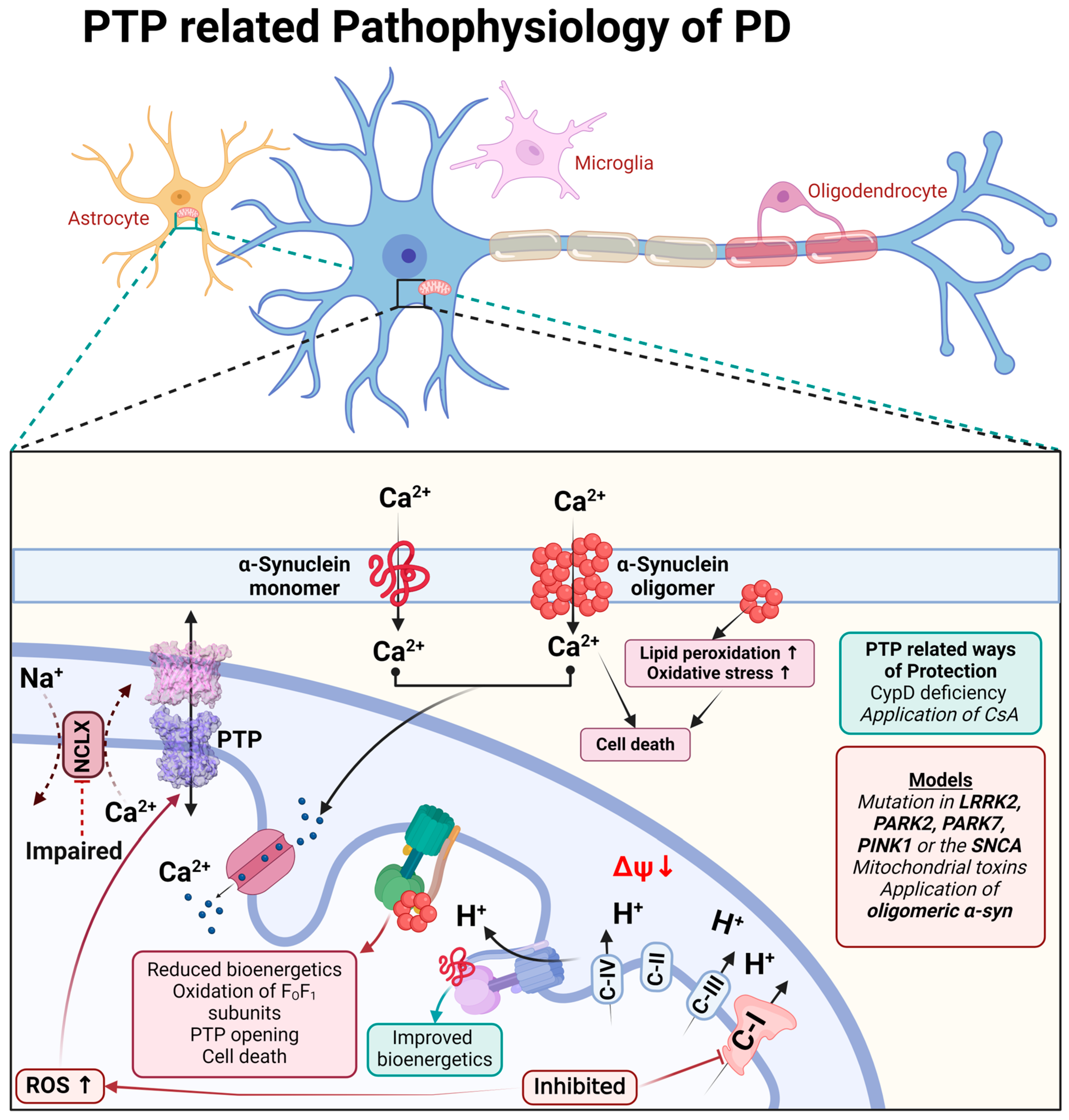
4. Parkinson’s Disease
5. Amyotrophic Lateral Sclerosis
6. Frontotemporal Dementia
7. Huntington’s Disease
8. Epilepsy, Multiple Sclerosis and mtDNA Mutations
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of alpha-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef]
- Rollo, J.; Crawford, J.; Hardy, J. A dynamical systems approach for multiscale synthesis of Alzheimer’s pathogenesis. Neuron 2023, 111, 2126–2139. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Dolgacheva, L.P. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem. Soc. Trans. 2017, 45, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Arber, C.; Bartolome, F.; de Vicente, M.; Preza, E.; Carro, E.; Houlden, H.; Gandhi, S.; Wray, S.; Abramov, A.Y. Mutations in valosin-containing protein (VCP) decrease ADP/ATP translocation across the mitochondrial membrane and impair energy metabolism in human neurons. J. Biol. Chem. 2017, 292, 8907–8917. [Google Scholar] [CrossRef]
- Ghosh, R.; Wood-Kaczmar, A.; Dobson, L.; Smith, E.J.; Sirinathsinghji, E.C.; Kriston-Vizi, J.; Hargreaves, I.P.; Heaton, R.; Herrmann, F.; Abramov, A.Y.; et al. Expression of mutant exon 1 huntingtin fragments in human neural stem cells and neurons causes inclusion formation and mitochondrial dysfunction. FASEB J. 2020, 34, 8139–8154. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Abeti, R.; Abramov, A.Y. Mitochondrial Ca2+ in neurodegenerative disorders. Pharmacol. Res. 2015, 99, 377–381. [Google Scholar] [CrossRef]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.V.; Shilov, E.S.; Mufazalov, I.A.; Gogvadze, V.; Nedospasov, S.A.; Zhivotovsky, B. Cytochrome c: The Achilles’ heel in apoptosis. Cell Mol. Life Sci. 2012, 69, 1787–1797. [Google Scholar] [CrossRef]
- Azzi, A.; Azzone, G.F. Swelling and shrinkage phenomena in liver mitochondria. II. Low amplitude swelling-shrinkage cycles. Biochim. Biophys. Acta 1965, 105, 265–278. [Google Scholar] [CrossRef]
- Azzi, A.; Azzone, G.F. Swelling and shrinkage phenomena in liver mitochondria. I. Large amplitude swelling induced by inorganic phosphate and by ATP. Biochim. Biophys. Acta 1965, 105, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Azzone, G.F.; Azzi, A. Volume changes in liver mitochondria. Proc. Natl. Acad. Sci. USA 1965, 53, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Brenner-Holzach, O.; Raaflaub, J. Correlation between the swelling of isolated mitochondria and decomposition of intramitochondrial adenosine nucleotides (ATP, ADP, AMP, CoA). Helv. Physiol. Pharmacol. Acta 1954, 12, 242–252. [Google Scholar]
- Chappell, J.B.; Crofts, A.R. Calcium Ion Accumulation and Volume Changes of Isolated Liver Mitochondria. Calcium Ion-Induced Swelling. Biochem. J. 1965, 95, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Fluharty, A.L.; Sanadi, D.R. On the Mechanism of Oxidative Phosphorylation. IV. Mitochondrial Swelling Caused by Arsenite in Combination with 2,3-Dimercaptopropanol and by Cadmium Ion*. Biochemistry 1962, 1, 276–281. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch. Biochem. Biophys. 1979, 195, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Broekemeier, K.M.; Dempsey, M.E.; Pfeiffer, D.R. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J. Biol. Chem. 1989, 264, 7826–7830. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Hayano, T.; Suzuki, M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 1989, 337, 473–475. [Google Scholar] [CrossRef]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Petronilli, V.; Szabo, I.; Zoratti, M. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett. 1989, 259, 137–143. [Google Scholar] [CrossRef]
- Carraro, M.; Bernardi, P. The mitochondrial permeability transition pore in Ca2+ homeostasis. Cell Calcium 2023, 111, 102719. [Google Scholar] [CrossRef]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef] [PubMed]
- Beutner, G.; Ruck, A.; Riede, B.; Brdiczka, D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta 1998, 1368, 7–18. [Google Scholar] [CrossRef]
- Ruck, A.; Dolder, M.; Wallimann, T.; Brdiczka, D. Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS Lett. 1998, 426, 97–101. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323. [Google Scholar] [CrossRef] [PubMed]
- Herick, K.; Kramer, R.; Luhring, H. Patch clamp investigation into the phosphate carrier from Saccharomyces cerevisiae mitochondria. Biochim. Biophys. Acta 1997, 1321, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Aguilar, M.; Douglas, D.L.; Gibson, A.K.; Domeier, T.L.; Molkentin, J.D.; Baines, C.P. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell. Cardiol. 2014, 72, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Fraley, C.; Diao, C.T.; Winkfein, R.; Colicos, M.A.; Duchen, M.R.; French, R.J.; Pavlov, E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 18091–18096. [Google Scholar] [CrossRef]
- Baev, A.Y.; Negoda, A.; Abramov, A.Y. Modulation of mitochondrial ion transport by inorganic polyphosphate—essential role in mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Baev, A.Y.; Berezhnov, A.V.; Abramov, A.Y. Role of inorganic polyphosphate in mammalian cells: From signal transduction and mitochondrial metabolism to cell death. Biochem. Soc. Trans. 2016, 44, 40–45. [Google Scholar] [CrossRef]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef]
- Mironova, G.D.; Gritsenko, E.; Gateau-Roesch, O.; Levrat, C.; Agafonov, A.; Belosludtsev, K.; Prigent, A.F.; Muntean, D.; Dubois, M.; Ovize, M. Formation of palmitic acid/Ca2+ complexes in the mitochondrial membrane: A possible role in the cyclosporin-insensitive permeability transition. J. Bioenerg. Biomembr. 2004, 36, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.; Saris, N.E.; Andersson, L.C.; Belosludtseva, N.; Agafonov, A.; Sharma, A.; Moshkov, D.A.; Mironova, G.D. On the mechanism of palmitic acid-induced apoptosis: The role of a pore induced by palmitic acid and Ca2+ in mitochondria. J. Bioenerg. Biomembr. 2006, 38, 113–120. [Google Scholar] [CrossRef]
- Peterson, A.A.; Rangwala, A.M.; Thakur, M.K.; Ward, P.S.; Hung, C.; Outhwaite, I.R.; Chan, A.; Usanov, D.L.; Mootha, V.K.; Seeliger, M.A.; et al. Discovery and molecular basis of subtype-selective cyclophilin inhibitors. Nat. Chem. Biol. 2022, 18, 1184. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Akhter, F.; Roy, A.; Chen, D.; Turner, B.; Wang, Y.; Clemente, N.; Wang, C.; Swerdlow, R.H.; Battaile, K.P.; et al. New cyclophilin D inhibitor rescues mitochondrial and cognitive function in Alzheimer’s disease. Brain 2023, 26, awad432. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, T.; Lai, X.; Xie, H.; Tang, H.; Wu, S.; Li, Y. Rational design peptide inhibitors of Cyclophilin D as a potential treatment for acute pancreatitis. Medicine 2023, 102, e36188. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Ionov, M.; Pavlov, E.; Duchen, M.R. Membrane cholesterol content plays a key role in the neurotoxicity of β-amyloid: Implications for Alzheimer’s disease. Aging Cell 2011, 10, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Rego, A.C.; Oliveira, C. Effect of amyloid beta-peptide on permeability transition pore: A comparative study. J. Neurosci. Res. 2002, 69, 257–267. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Shevtsova, E.P.; Kireeva, E.G.; Oxenkrug, G.F.; Sablin, S.O. Mitochondria as a Target for Neurotoxins and Neuroprotective Agents. Ann. N. Y. Acad. Sci. 2003, 993, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Abramov, A.Y.; Duchen, M.R. β-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 2011, 134, 1658–1672. [Google Scholar] [CrossRef]
- Shevtsova, E.F.; Angelova, P.R.; Stelmashchuk, O.A.; Esteras, N.; Vasil’eva, N.A.; Maltsev, A.V.; Shevtsov, P.N.; Shaposhnikov, A.V.; Fisenko, V.P.; Bachurin, S.O.; et al. Pharmacological sequestration of mitochondrial calcium uptake protects against dementia and β-amyloid neurotoxicity. Sci. Rep. 2022, 12, 12766. [Google Scholar] [CrossRef]
- Keller, J.N.; Guo, Q.; Holtsberg, F.W.; Bruce-Keller, A.J.; Mattson, M.P. Increased Sensitivity to Mitochondrial Toxin-Induced Apoptosis in Neural Cells Expressing Mutant Presenilin-1 Is Linked to Perturbed Calcium Homeostasis and Enhanced Oxyradical Production. J. Neurosci. 1998, 18, 4439–4450. [Google Scholar] [CrossRef]
- Toglia, P.; Ullah, G. The gain-of-function enhancement of IP3-receptor channel gating by familial Alzheimer’s disease-linked presenilin mutants increases the open probability of mitochondrial permeability transition pore. Cell Calcium 2016, 60, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; A Sosunov, A.; McKhann, M.G.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef]
- Gauba, E.; Chen, H.; Guo, L.; Du, H. Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 138–147. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef]
- Twyning, M.J.; Tufi, R.; Gleeson, T.P.; Kolodziej, K.M.; Campesan, S.; Terriente-Felix, A.; Collins, L.; De Lazzari, F.; Giorgini, F.; Whitworth, A.J. Partial loss of MCU mitigates pathology in vivo across a diverse range of neurodegenerative disease models. Cell Rep. 2024, 43, 113681. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Sharipov, R.R.; Boyarkin, D.P.; Belosludtseva, N.V.; Dubinin, M.V.; Krasilnikova, I.A.; Bakaeva, Z.V.; Zgodova, A.E.; Pinelis, V.G.; Surin, A.M. The effect of DS16570511, a new inhibitor of mitochondrial calcium uniporter, on calcium homeostasis, metabolism, and functional state of cultured cortical neurons and isolated brain mitochondria. BBA-Gen Subj. 2021, 1865, 129847. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Vinogradova, D.; Neganova, M.E.; Serkova, T.P.; Sokolov, V.V.; Bachurin, S.O.; Shevtsova, E.F.; Abramov, A.Y. Pharmacological Sequestration of Mitochondrial Calcium Uptake Protects Neurons against Glutamate Excitotoxicity. Mol. Neurobiol. 2019, 56, 2244–2255. [Google Scholar] [CrossRef]
- Griffiths, K.K.; Wang, A.; Jonas, E.A.; Levy, R.J. Sulfide quinone oxidoreductase contributes to voltage sensing of the mitochondrial permeability transition pore. FASEB J. 2024, 38, e23494. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Sherer, T.B.; Betarbet, R.; Panov, A.V. Complex I and Parkinson’s Disease. IUBMB Life 2001, 52, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Angelova, P.R. Cellular mechanisms of complex I-associated pathology. Biochem. Soc. Trans. 2019, 47, 1963–1969. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid—different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Seaton, T.A.; Cooper, J.M.; Schapira, A.H.V. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res. 1998, 809, 12–17. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Parks, J.K.; Parker, W.D.; Bennett, J.P. The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 1999, 1453, 49–62. [Google Scholar] [CrossRef]
- Thomas, B.; Banerjee, R.; Starkova, N.N.; Zhang, S.F.; Calingasan, N.Y.; Yang, L.; Wille, E.; Lorenzo, B.J.; Ho, D.J.; Beal, M.F.; et al. Mitochondrial Permeability Transition Pore Component Cyclophilin D Distinguishes Nigrostriatal Dopaminergic Death Paradigms in the MPTP Mouse Model of Parkinson’s Disease. Antioxid. Redox Signal. 2012, 16, 855–868. [Google Scholar] [CrossRef]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Kostic, M.; Horne, A.; Gandhi, S.; Sekler, I.; Abramov, A.Y. LRRK2 deficiency induced mitochondrial Ca2+ efflux inhibition can be rescued by Na+/Ca2+/Li+ exchanger upregulation. Cell Death Dis. 2019, 10, 265. [Google Scholar] [CrossRef]
- Kostic, M.; Ludtmann, M.H.R.; Bading, H.; Hershfinkel, M.; Steer, E.; Chu, C.T.; Abramov, A.Y.; Sekler, I. PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cell Rep. 2015, 13, 376–386. [Google Scholar] [CrossRef]
- Gandhi, S.; Vaarmann, A.; Yao, Z.; Duchen, M.R.; Wood, N.W.; Abramov, A.Y. Dopamine induced neurodegeneration in a PINK1 model of Parkinson’s disease. PLoS ONE 2012, 7, e375642012. [Google Scholar] [CrossRef]
- Kim, E.Y.; Kang, K.-h.; Koh, H. Cyclophilin 1 (Cyp1) mutation ameliorates oxidative stress-induced defects in a Drosophila DJ-1 null mutant. Biochem. Biophys. Res. Commun. 2018, 505, 823–829. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Cook, C.; Petrucelli, L. Genetic Convergence Brings Clarity to the Enigmatic Red Line in ALS. Neuron 2019, 101, 1057–1069. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Matveeva, L.A.; Belosludtsev, K.N. Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 6833. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 1068–1082. [Google Scholar] [CrossRef]
- Parone, P.A.; Cruz, S.D.; Han, J.S.; McAlonis-Downes, M.; Vetto, A.P.; Lee, S.K.; Tseng, E.; Cleveland, D.W. Enhancing Mitochondrial Calcium Buffering Capacity Reduces Aggregation of Misfolded SOD1 and Motor Neuron Cell Death without Extending Survival in Mouse Models of Inherited Amyotrophic Lateral Sclerosis. J. Neurosci. 2013, 33, 4657–4671. [Google Scholar] [CrossRef]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial Alterations in the Spinal Cord of Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 186–197. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective Mitochondrial Dynamics Is an Early Event in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e618. [Google Scholar] [CrossRef]
- Kostic, V.; Jackson-Lewis, V.; de Bilbao, F.; Dubois-Dauphin, M.; Przedborski, S. Bcl-2: Prolonging Life in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. Science 1997, 277, 559–563. [Google Scholar] [CrossRef]
- Pasinelli, P.; Borchelt, D.R.; Houseweart, M.K.; Cleveland, D.W.; Brown, R.H. Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc. Natl. Acad. Sci. USA 1998, 95, 15763–15768. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic Lateral Sclerosis-Associated SOD1 Mutant Proteins Bind and Aggregate with Bcl-2 in Spinal Cord Mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Vukosavic, S.; Dubois-Dauphin, M.; Romero, N.; Przedborski, S. Bax and Bcl-2 Interaction in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 1999, 73, 2460–2468. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Fisher, J.; Pasinelli, P.; Brown, R.H.; Korsmeyer, S.; Glimcher, L. The proapoptotic BCL-2 family member BIM mediates motoneuron loss in a model of amyotrophic lateral sclerosis. Cell Death Differ. 2007, 14, 1386–1389. [Google Scholar] [CrossRef]
- Soo, K.Y.; Atkin, J.D.; Farg, M.; Walker, A.K.; Horne, M.K.; Nagley, P. Bim Links ER Stress and Apoptosis in Cells Expressing Mutant SOD1 Associated with Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e35413. [Google Scholar] [CrossRef]
- Izumikawa, K.; Nobe, Y.; Yoshikawa, H.; Ishikawa, H.; Miura, Y.; Nakayama, H.; Nonaka, T.; Hasegawa, M.; Egawa, N.; Inoue, H.; et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci. Rep. 2017, 7, 7709. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Barrett, J.N.; Garcia-Chacon, L.; David, G.; Barrett, E.F. Repetitive nerve stimulation transiently opens the mitochondrial permeability transition pore in motor nerve terminals of symptomatic mutant SOD1 mice. Neurobiol. Dis. 2011, 42, 381–390. [Google Scholar] [CrossRef]
- Klivenyi, P.; Ferrante, R.J.; Matthews, R.T.; Bogdanov, M.B.; Klein, A.M.; Andreassen, O.A.; Mueller, G.; Wermer, M.; Kaddurah-Daouk, R.; Beal, M.F. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat. Med. 1999, 5, 347–350. [Google Scholar] [CrossRef]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef]
- Bordet, T.; Berna, P.; Abitbol, J.L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345–368. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Sileikyte, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabo, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef]
- Leroy, M.; Bertoux, M.; Skrobala, E.; Mode, E.; Adnet-Bonte, C.; Le Ber, I.; Bombois, S.; Cassagnaud, P.; Chen, Y.H.; Deramecourt, V.; et al. Characteristics and progression of patients with frontotemporal dementia in a regional memory clinic network. Alzheimers Res. Ther. 2021, 13, 19. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138, 54–70. [Google Scholar] [CrossRef]
- Antonioni, A.; Raho, E.M.; Lopriore, P.; Pace, A.P.; Latino, R.R.; Assogna, M.; Mancuso, M.; Gragnaniello, D.; Granieri, E.; Pugliatti, M.; et al. Frontotemporal Dementia, Where Do We Stand? A Narrative Review. Int. J. Mol. Sci. 2023, 24, 11732. [Google Scholar] [CrossRef]
- Chu, M.; Liu, L.; Nan, H.T.; Jiang, D.M.; Wang, Y.H.; Rosa-Neto, P.; Piao, Y.S.; Wu, L.Y. Extremely Early-Onset Frontotemporal Dementia: A Case Report and Literature Review. J. Alzheimers Dis. 2022, 90, 1139–1151. [Google Scholar] [CrossRef]
- Onyike, C.U.; Diehl-Schmid, J. The epidemiology of frontotemporal dementia. Int. Rev. Psychiatr. 2013, 25, 130–137. [Google Scholar] [CrossRef]
- Anoar, S.; Woodling, N.S.; Niccoli, T. Mitochondria Dysfunction in Frontotemporal Dementia/Amyotrophic Lateral Sclerosis: Lessons From Models. Front. Neurosci. 2021, 15, 6076. [Google Scholar] [CrossRef]
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.; Hautbergue, G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis. Front Neurosci. 2021, 15, 3624. [Google Scholar] [CrossRef]
- Wagner, M.; Lorenz, G.; Volk, A.E.; Brunet, T.; Edbauer, D.; Berutti, R.; Zhao, C.; Anderl-Straub, S.; Bertram, L.; Danek, A.; et al. Clinico-genetic findings in 509 frontotemporal dementia patients. Mol. Psychiatr. 2021, 26, 5824–5832. [Google Scholar] [CrossRef]
- Rademakers, R.; Neumann, M.; Mackenzie, I.R. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 2012, 8, 423–434. [Google Scholar] [CrossRef]
- Jara, C.; Aranguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef]
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Esteras, N.; Kopach, O.; Maiolino, M.; Lariccia, V.; Amoroso, S.; Qamar, S.; Wray, S.; Rusakov, D.A.; Jaganjac, M.; Abramov, A.Y. Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimers Dement. 2022, 18, 318–338. [Google Scholar] [CrossRef]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.A.; Siskova, Z.; Mandelkow, E.; Mandelkow, E.M. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Sahara, N.; Kanaan, N.M.; Tsukita, K.; Kondo, T.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Kawakami, K.; Hotta, A.; et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 2016, 6, 904. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; D’Souza, I.; Crudder, C.H.; Onodera, H.; Itoyama, Y.; Poorkaj, P.; Bird, T.D.; Schellenberg, G.D. Pro-apoptotic effects of tau mutations in chromosome 17 frontotemporal dementia and parkinsonism. Neuroreport 2000, 11, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Wang, Y.; Yao, P.J.; Fu, W.M.; Mattson, M.P.; Itoyama, Y.; Onodera, H.; D’Souza, I.; Poorkaj, P.H.; Bird, T.D.; et al. Alteration in calcium channel properties is responsible for the neurotoxic action of a familial frontotemporal dementia tau mutation. J. Neurochem. 2003, 87, 427–436. [Google Scholar] [CrossRef]
- Adante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. BBA-Bioenergetics 2008, 1777, 1289–1300. [Google Scholar] [CrossRef]
- Camilleri, A.; Ghio, S.; Caruana, M.; Weckbecker, D.; Schmidt, F.; Kamp, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Giese, A.; et al. Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. BBA-Biomembranes 2020, 1862, 64. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A Mitochondria-Associated Oxidative Stress Perspective on Huntington’s Disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Oliveros, J.C.; Naranjo, J.R.; Carafoli, E. Neuronal Ca(2+) dyshomeostasis in Huntington disease. Prion 2013, 7, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.S.; Johnson, G.V.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.S.; Slow, E.; Lupu, V.; Stavrovskaya, I.G.; Sugimori, M.; Llinas, R.; Kristal, B.S.; Hayden, M.R.; Bezprozvanny, I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2602–2607. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Nguyen, H.P.; Trumbeckaite, S.; Vielhaber, S.; Seppet, E.; Zierz, S.; Landwehrmeyer, B.; Riess, O.; von Horsten, S.; et al. Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats. J. Biol. Chem. 2008, 283, 30715–30724. [Google Scholar] [CrossRef] [PubMed]
- Milakovic, T.; Quintanilla, R.A.; Johnson, G.V. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: Functional consequences. J. Biol. Chem. 2006, 281, 34785–34795. [Google Scholar] [CrossRef] [PubMed]
- Gizatullina, Z.Z.; Lindenberg, K.S.; Harjes, P.; Chen, Y.; Kosinski, C.M.; Landwehrmeyer, B.G.; Ludolph, A.C.; Striggow, F.; Zierz, S.; Gellerich, F.N. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann. Neurol. 2006, 59, 407–411. [Google Scholar] [CrossRef]
- Fernandes, H.B.; Baimbridge, K.G.; Church, J.; Hayden, M.R.; Raymond, L.A. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 13614–13623. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Jin, Y.N.; von Bernhardi, R.; Johnson, G.V. Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol. Neurodegener. 2013, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Neuroprotective effect of cyclosporine and FK506 against 3-nitropropionic acid induced cognitive dysfunction and glutathione redox in rat: Possible role of nitric oxide. Neurosci. Res. 2009, 63, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Amico, E.; Martinello, K.; Draghi, F.; Cavalletti, E.; Fucile, S.; Maglione, V.; Pardo, A.D. L14 Inhibition of mitochondrial permeability transition pore (MPTP), by GNX-4728, is beneficial in an in-vitro huntington’s disease model. J. Neurol. Neurosurg. Psychiatry 2016, 87, A94–A952016. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Jekabsons, M.B.; Chen, S.; Lin, A.; Rego, A.C.; Goncalves, J.; Ellerby, L.M.; Nicholls, D.G. Mitochondrial dysfunction in Huntington’s disease: The bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 2007, 101, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; LaFrance, R.; Purl, K.J.; Brustovetsky, T.; Keene, C.D.; Low, W.C.; Dubinsky, J.M. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington’s Disease. J. Neurochem. 2005, 93, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- De Mario, A.; Scarlatti, C.; Costiniti, V.; Primerano, S.; Lopreiato, R.; Cali, T.; Brini, M.; Giacomello, M.; Carafoli, E. Calcium Handling by Endoplasmic Reticulum and Mitochondria in a Cell Model of Huntington’s Disease. PLoS Curr. 2016, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Pellman, J.J.; Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Ca(2+) handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington’s disease. J. Neurochem. 2015, 134, 652–667. [Google Scholar] [CrossRef]
- Brustovetsky, N. Mutant Huntingtin and Elusive Defects in Oxidative Metabolism and Mitochondrial Calcium Handling. Mol. Neurobiol. 2016, 53, 2944–2953. [Google Scholar] [CrossRef]
- Lopes, C.; Ferreira, I.L.; Maranga, C.; Beatriz, M.; Mota, S.I.; Sereno, J.; Castelhano, J.; Abrunhosa, A.; Oliveira, F.; De Rosa, M.; et al. Mitochondrial and redox modifications in early stages of Huntington’s disease. Redox Biol. 2022, 56, 102424. [Google Scholar] [CrossRef]
- Tashakori-Miyanroudi, M.; Souresrafil, A.; Hashemi, P.; Jafar Ehsanzadeh, S.; Farrahizadeh, M.; Behroozi, Z. Prevalence of depression, anxiety, and psychological distress in patients with epilepsy during COVID-19: A systematic review. Epilepsy Behav. 2021, 125, 108410. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, S.; Madireddy, S. Therapeutic Strategies to Ameliorate Neuronal Damage in Epilepsy by Regulating Oxidative Stress, Mitochondrial Dysfunction, and Neuroinflammation. Brain Sci. 2023, 13, 784. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef]
- Kovac, S.; Domijan, A.M.; Walker, M.C.; Abramov, A.Y. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J. Cell Sci. 2012, 125, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Debska-Vielhaber, G.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. The Mechanism of Neuroprotection by Topiramate in an Animal Model of Epilepsy. Epilepsy 2004, 45, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Kovács, R.; Kardos, J.; Heinemann, U.; Kann, O. Mitochondrial Calcium Ion and Membrane Potential Transients Follow the Pattern of Epileptiform Discharges in Hippocampal Slice Cultures. J. Neurosci. 2005, 25, 4260–4269. [Google Scholar] [CrossRef]
- Neganova, M.E.; Blik, V.A.; Klochkov, S.G.; Chepurnova, N.E.; Shevtsova, E.F. Investigation of the Antioxidant Characteristics of a New Tryptamine Derivative of Securinine and its Influence on Seizure Activity in the Brain in Experimental Epilepsy. Neurochem. J. 2011, 5, 208–214. [Google Scholar] [CrossRef]
- Barrientos, S.A.; Martinez, N.W.; Yoo, S.; Jara, J.S.; Zamorano, S.; Hetz, C.; Twiss, J.L.; Alvarez, J.; Court, F.A. Axonal Degeneration Is Mediated by the Mitochondrial Permeability Transition Pore. J. Neuroscience. 2011, 31, 966–978. [Google Scholar] [CrossRef]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Rahder, M.; Stem, K.; et al. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar] [CrossRef]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboidl, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66 adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Su, K.G.; Savino, C.; Marracci, G.; Chaudhary, P.; Yu, X.L.; Morris, B.; Galipeau, D.; Giorgio, M.; Forte, M.; Bourdette, D. Genetic inactivation of the p66 isoform of ShcA is neuroprotective in a murine model of multiple sclerosis. Eur. J. Neurosci. 2012, 35, 562–571. [Google Scholar] [CrossRef]
- Warne, J.; Pryce, G.; Hill, J.M.; Shi, X.; Lennerås, F.; Puentes, F.; Kip, M.; Hilditch, L.; Walker, P.; Simone, M.I.; et al. Selective Inhibition of the Mitochondrial Permeability Transition Pore Protects against Neurodegeneration in Experimental Multiple Sclerosis. J. Biol. Chem. 2016, 291, 4356–4373. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. BBA-Bioenergetics 2015, 1847, 1401–1411. [Google Scholar] [CrossRef]
- Bazzani, V.; Redin, M.E.; McHale, J.; Perrone, L.; Vascotto, C. Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing. Int. J. Mol. Sci. 2022, 23, 1391. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Cha, M.-Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef]
- Turnbull, H.E.; Lax, N.Z.; Diodato, D.; Ansorge, O.; Turnbull, D.M. The mitochondrial brain: From mitochondrial genome to neurodegeneration. BBA-Mol. Basis Dis. 2010, 1802, 111–121. [Google Scholar] [CrossRef]
- Soong, N.W.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a Specific Somatic Mitochondrial-DNA Mutation in Adult Human Brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar] [CrossRef]
- Khotina, V.A.; Vinokurov, A.Y.; Ekta, M.B.; Sukhorukov, V.N.; Orekhov, A.N. Creation of Mitochondrial Disease Models Using Mitochondrial DNA Editing. Biomedicines 2023, 11, 532. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Smulders-Srinivasan, T.K.; Kirby, D.M.; Acin-Perez, R.; Enriquez, J.A.; Lightowlers, R.N.; Duchen, M.R.; Turnbull, D.M. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain 2010, 133, 797–807. [Google Scholar] [CrossRef]
- Siddiqui, A.; Rivera-Sánchez, S.; Castro, M.d.R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA damage Is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Chen, Q.; Wang, X.H.; Wang, Q.C.; Wang, Y.; Cheng, H.P.; Guo, C.X.; Sun, Q.M.; Chen, Q.; Tang, T.S. Dysregulation of Mitochondrial Calcium Signaling and Superoxide Flashes Cause Mitochondrial Genomic DNA Damage in Huntington Disease. J. Biol. Chem. 2013, 288, 3070–3084. [Google Scholar] [CrossRef] [PubMed]
- Warita, H.; Hayashi, T.; Murakami, T.; Manabe, Y.; Abe, K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Mol. Brain Res. 2001, 89, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Zhang, D.K.; Zassenhaus, H.P. Mitochondrial DNA mutations, apoptosis, and the misfolded protein response. Rejuv. Res. 2005, 8, 216–226. [Google Scholar] [CrossRef]
- Uchino, H.; Hatakeyama, K.; Morota, S.; Tanoue, T.; Nishiyama, T.; Usui, D.; Taguchi, C.; Suzuki, M.; Hansson, M.J.; Elmér, E. Cyclophilin-D Inhibition in Neuroprotection: Dawn of a New Era of Mitochondrial Medicine. Brain Edema 2013, 15, 311–315. [Google Scholar]
- Pflugrad, H.; Schrader, A.K.; Tryc, A.B.; Ding, X.; Lanfermann, H.; Jäckel, E.; Schrem, H.; Beneke, J.; Barg-Hock, H.; Klempnauer, J.; et al. Longterm calcineurin inhibitor therapy and brain function in patients after liver transplantation. Liver Transplant. 2018, 24, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Duchen, M.R. Actions of ionomycin, 4-BrA23187 and a novel electrogenic Ca2+ ionophore on mitochondria in intact cells. Cell Calcium 2003, 33, 101–112. [Google Scholar] [CrossRef]
- Macho, A.; Blanco-Molina, M.; Spagliardi, P.; Appendino, G.; Bremner, P.; Heinrich, M.; Fiebich, B.L.; Muñoz, E. Calcium ionophoretic and apoptotic effects of ferutinin in the human Jurkat T-cell line. Biochem. Pharmacol. 2004, 68, 875–883. [Google Scholar] [CrossRef]
- Nazrullaev, S.S.; Khubaktova, Z.A.; Syrov, V.N.; Alieva, D.A.; Akhmetkhodzhaeva Kh, S. Experimental evaluation of the effect of a new medicinal form of tefestrol on reproduction in rats. Eksperimental’naia I Klin. Farmakol. 2006, 69, 35–39. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baev, A.Y.; Vinokurov, A.Y.; Potapova, E.V.; Dunaev, A.V.; Angelova, P.R.; Abramov, A.Y. Mitochondrial Permeability Transition, Cell Death and Neurodegeneration. Cells 2024, 13, 648. https://doi.org/10.3390/cells13070648
Baev AY, Vinokurov AY, Potapova EV, Dunaev AV, Angelova PR, Abramov AY. Mitochondrial Permeability Transition, Cell Death and Neurodegeneration. Cells. 2024; 13(7):648. https://doi.org/10.3390/cells13070648
Chicago/Turabian StyleBaev, Artyom Y., Andrey Y. Vinokurov, Elena V. Potapova, Andrey V. Dunaev, Plamena R. Angelova, and Andrey Y. Abramov. 2024. "Mitochondrial Permeability Transition, Cell Death and Neurodegeneration" Cells 13, no. 7: 648. https://doi.org/10.3390/cells13070648