
Antagonizing Activin A/p15INK4b Signaling as Therapeutic Strategy for Liver Disease

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Activin A Antagonists
2.3. Isolation and Purification of Rat Hepatocytes
2.4. Rat Hepatic Stellate Cells (HSCs)
2.5. Cell Culture Experiments
2.5.1. Rat Hepatocytes
2.5.2. Rat HSCs
2.5.3. Human Huh-7 Cells
2.6. MTT Assay
2.7. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
2.8. PH, NUCC-555 Treatment, and Bromodeoxyuridine (BrdU) Administration in Rats
2.9. Thioacetamide (TAA)-Induced Liver Fibrosis and NUCC-555 Administration in Rats
2.10. Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis and NUCC-555 Administration in Mice
2.11. IHC/Histochemistry/Immunocytochemistry
2.11.1. Nuclear staining for BrdU
2.11.2. Collagen Staining
2.11.3. Immunohistochemical/Immunocytochemical Detection of α-SMA and Vimentin
2.12. Serological Analyses
2.13. Microscopy and Imaging
2.14. Statistical Data Analysis
3. Results
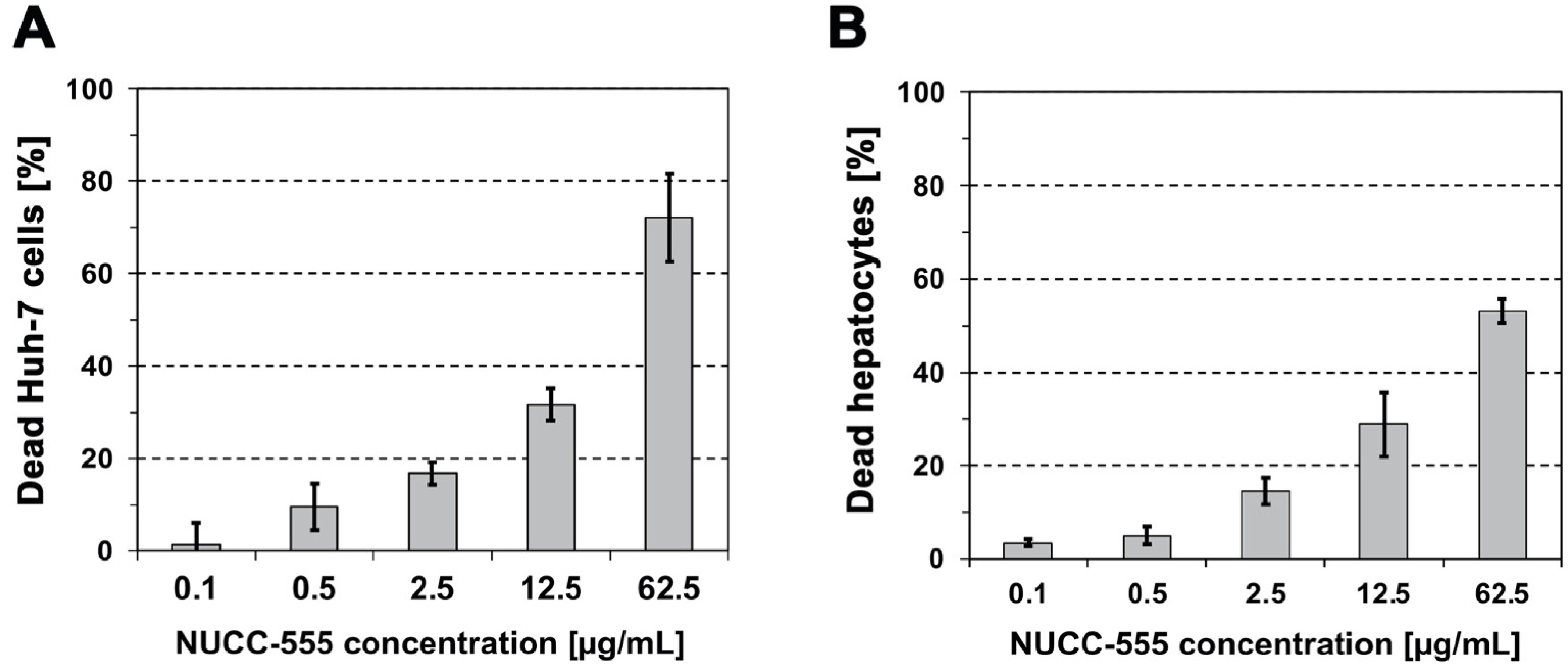
3.1. Cytotoxicity of NUCC-474/555 in Human Huh-7 Cells and Rat Hepatocytes
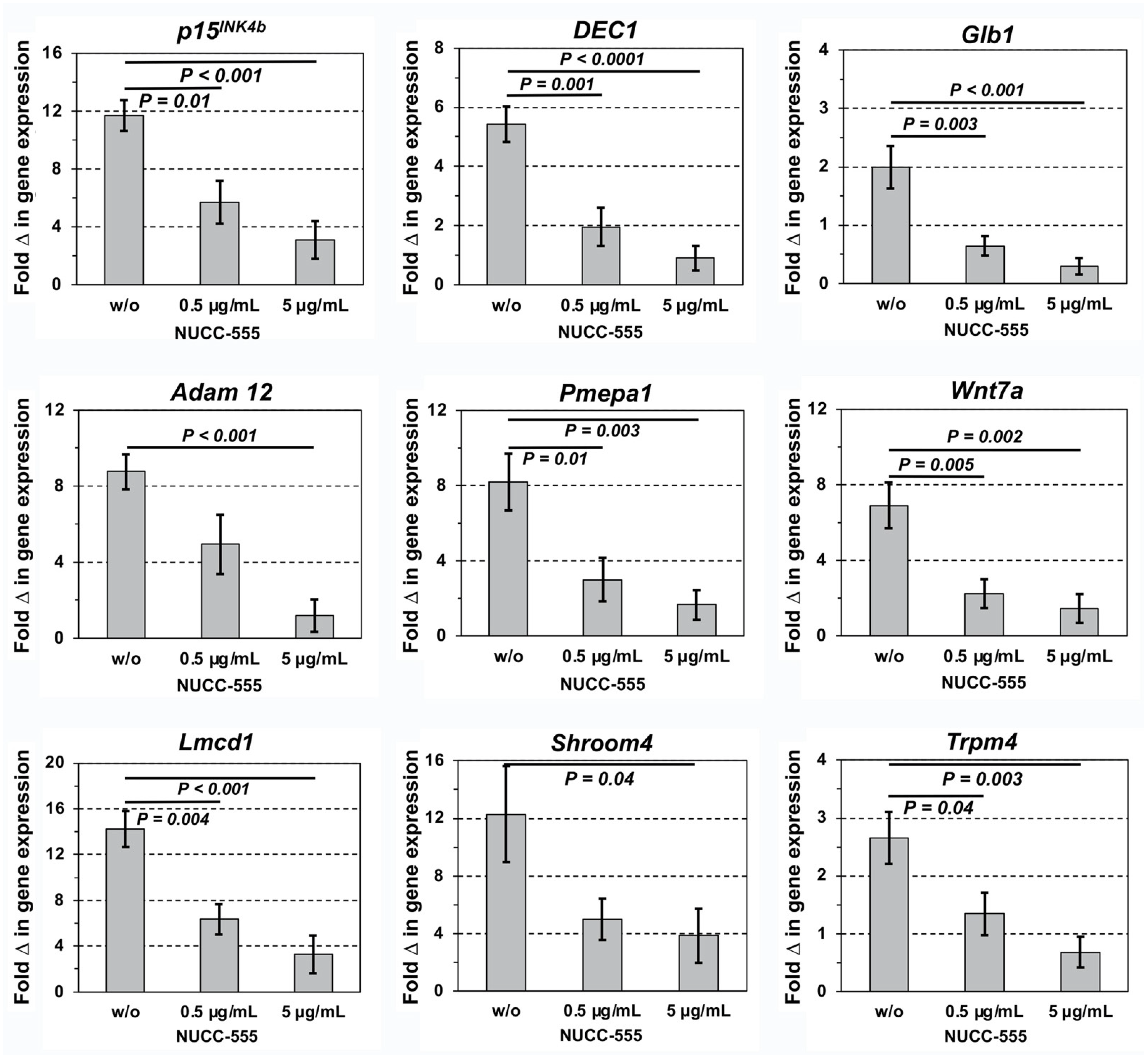
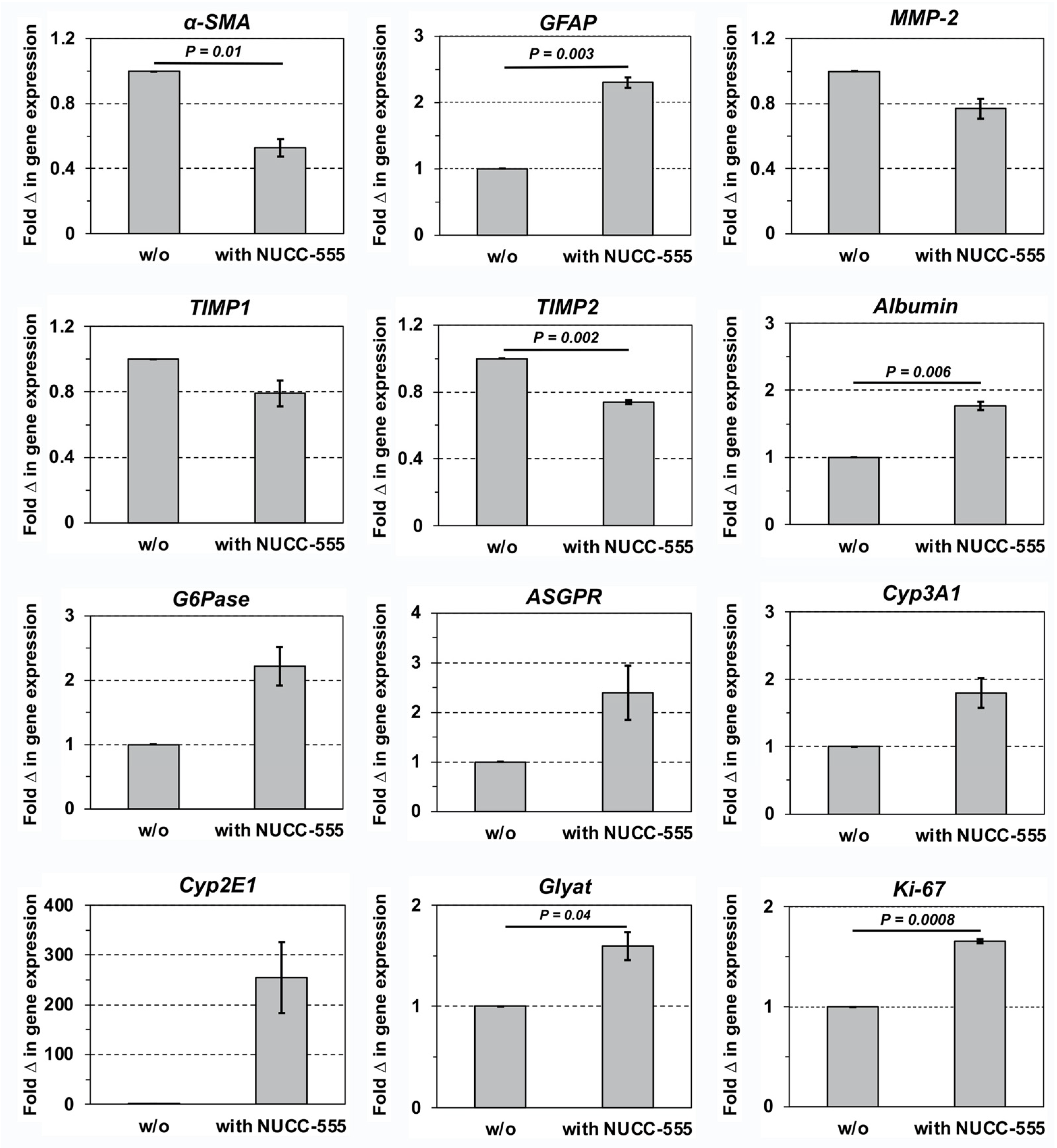
3.2. Blockage of Activin A-Induced Overexpression of Selected Genes by NUCC-555
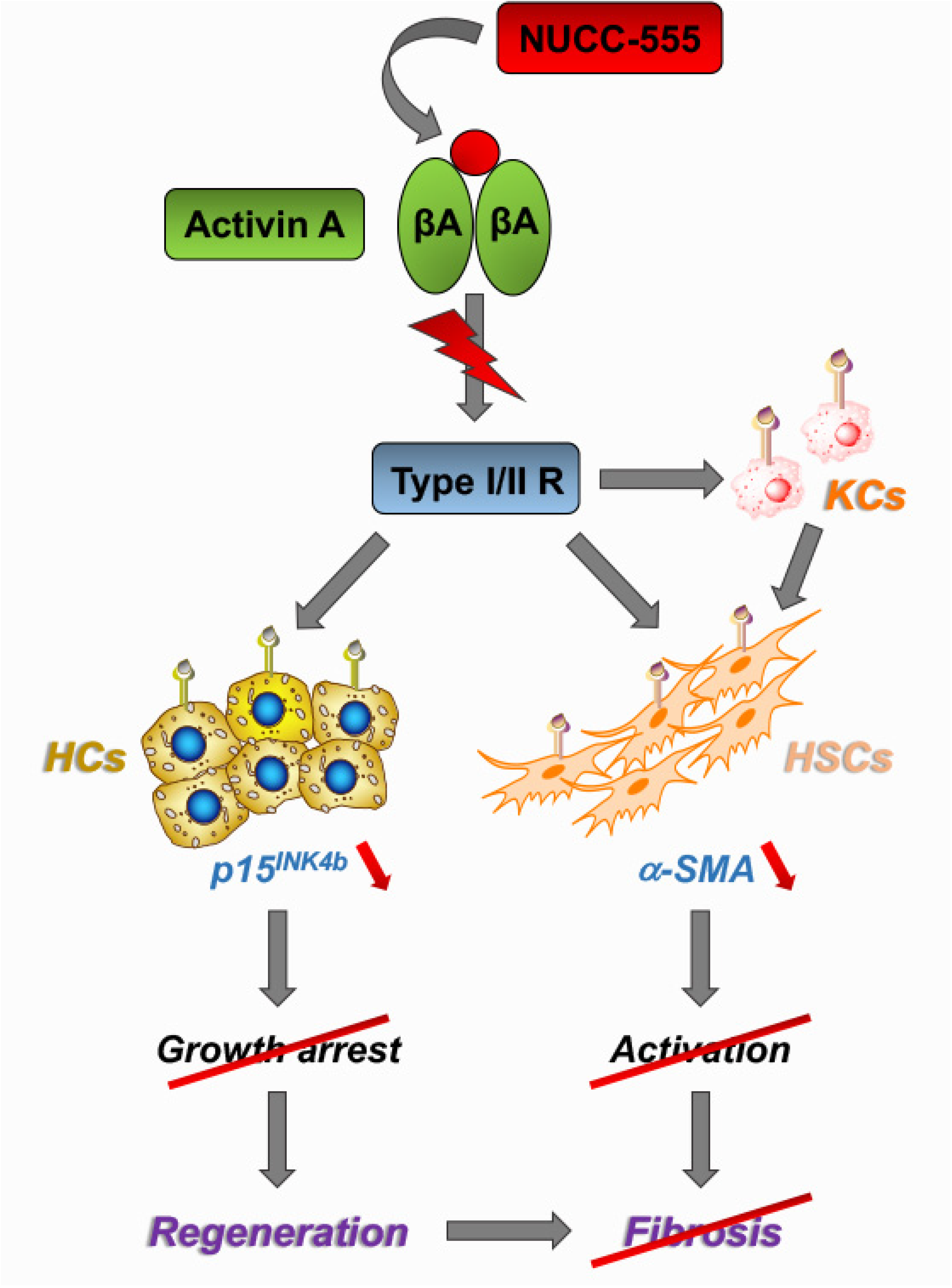
3.3. Role of Activin A Signaling in Rat HSCs
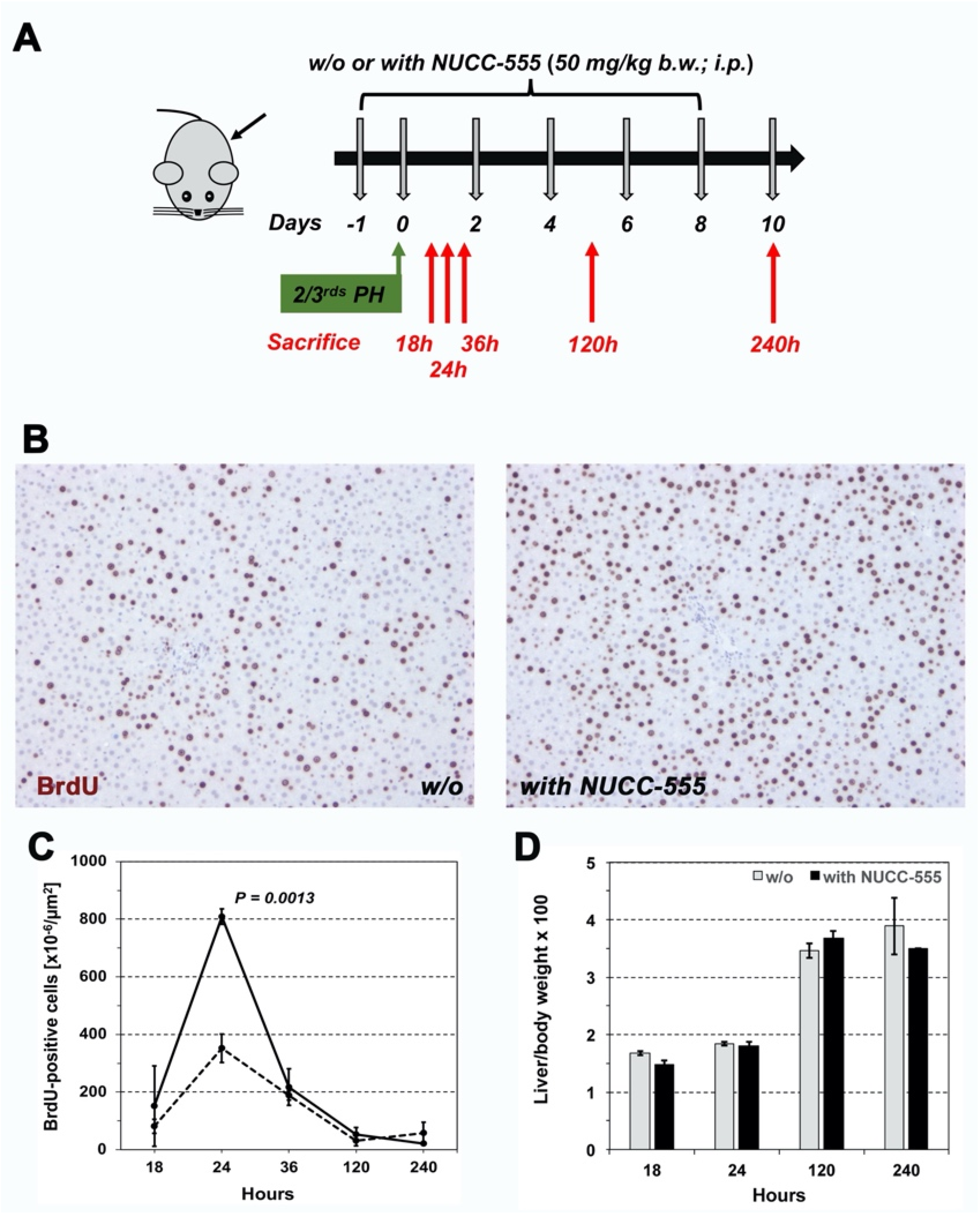
3.4. Accelerated Rat Liver Regeneration through NUCC-555 Administration
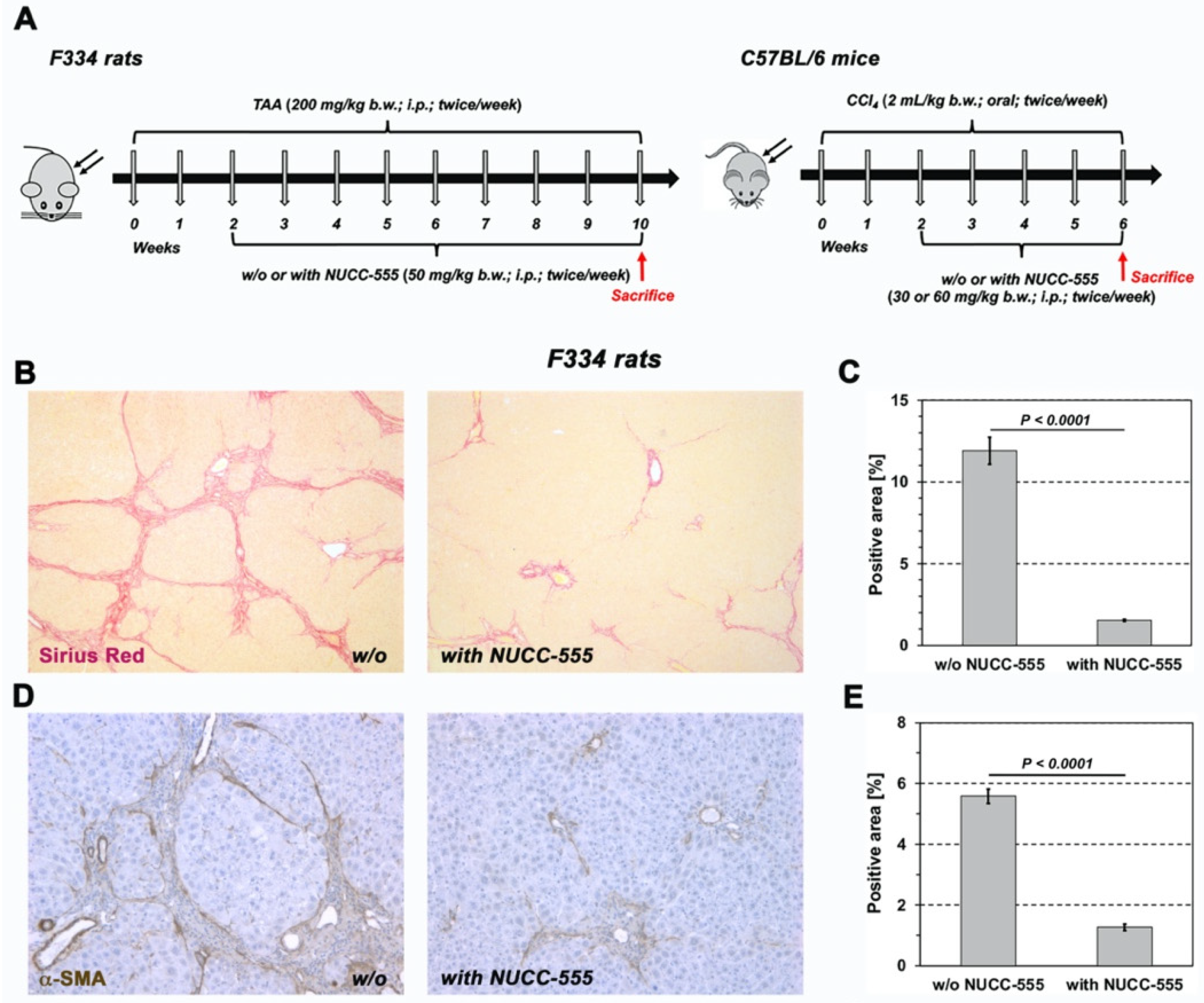
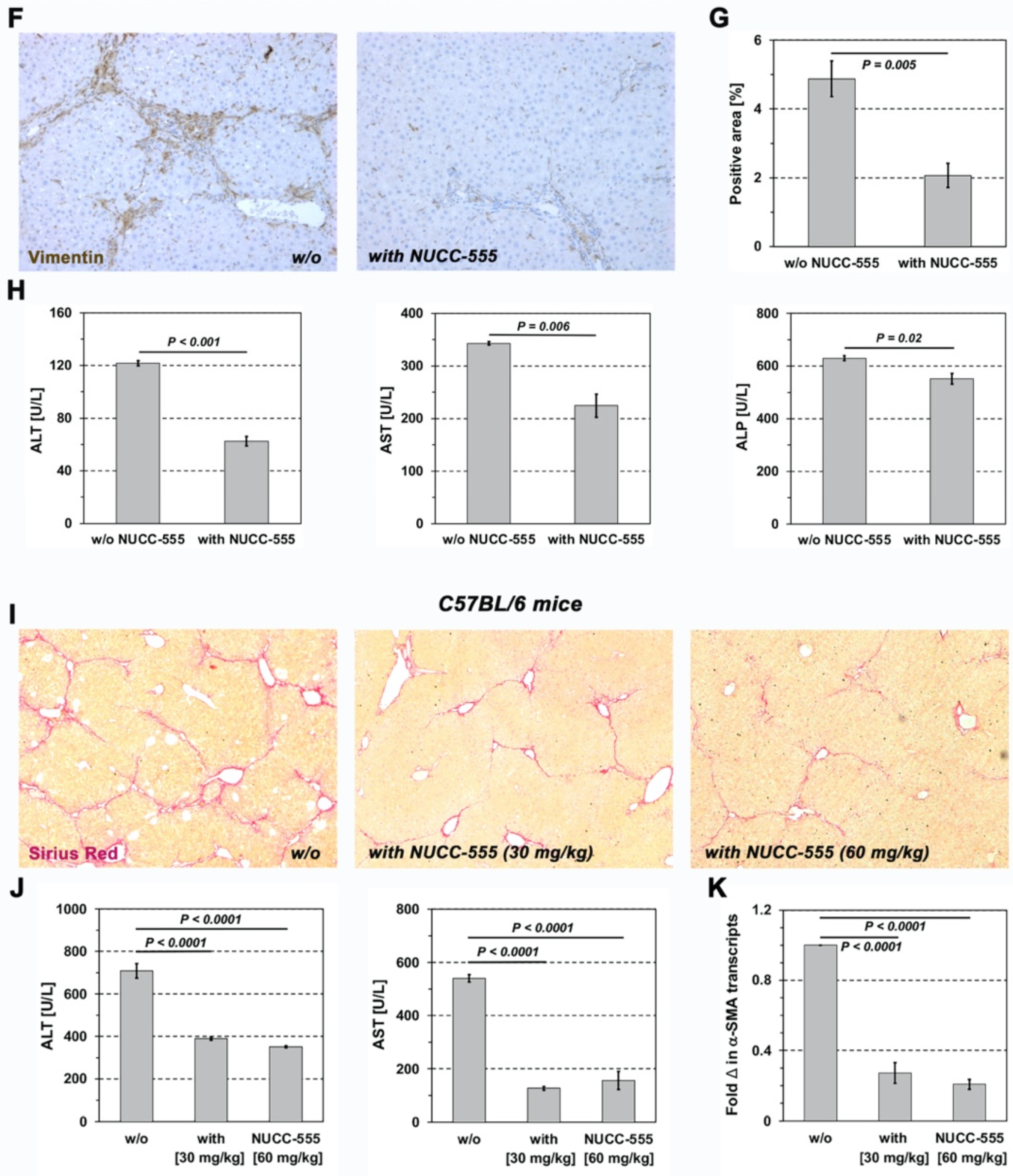
3.5. Effect of NUCC-555 in Rodent Fibrosis Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kang, L.I.; Mars, W.M.; Michalopoulos, G.K. Signals and cells involved in regulating liver regeneration. Cells 2012, 1, 1261–1292. [Google Scholar] [CrossRef] [PubMed]
- Gilgenkrantz, H.; Collin de l’Hortet, A. Understanding Liver Regeneration: From Mechanisms to Regenerative Medicine. Am. J. Pathol. 2018, 188, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Pajalunga, D.; Mazzola, A.; Salzano, A.M.; Biferi, M.G.; De Luca, G.; Crescenzi, M. Critical requirement for cell cycle inhibitors in sustaining nonproliferative states. J. Cell Biol. 2007, 176, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, D.L.; Sanchez, H. Liver regeneration and aging: A current perspective. Curr. Gerontol. Geriatr. Res. 2011, 2011, 526379. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef]
- Rodgarkia-Dara, C.; Vejda, S.; Erlach, N.; Losert, A.; Bursch, W.; Berger, W.; Schulte-Hermann, R.; Grusch, M. The activin axis in liver biology and disease. Mutat. Res. 2006, 613, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Wang, Q.; Lin, S.L.; Chang, C.D.; Chuang, J.; Ying, S.Y. Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Exp. Biol. Med. Maywood 2006, 231, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Haridoss, S.; Yovchev, M.I.; Schweizer, H.; Megherhi, S.; Beecher, M.; Locker, J.; Oertel, M. Activin A is a prominent autocrine regulator of hepatocyte growth arrest. Hepatol. Commun. 2017, 1, 852–870. [Google Scholar] [CrossRef]
- Yasuda, H.; Mine, T.; Shibata, H.; Eto, Y.; Hasegawa, Y.; Takeuchi, T.; Asano, S.; Kojima, I. Activin A: An autocrine inhibitor of initiation of DNA synthesis in rat hepatocytes. J. Clin. Investig. 1993, 92, 1491–1496. [Google Scholar] [CrossRef]
- Schwall, R.H.; Robbins, K.; Jardieu, P.; Chang, L.; Lai, C.; Terrell, T.G. Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology 1993, 18, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Zhang, Y.Q.; Kogure, K.; Hasegawa, Y.; Takagi, H.; Mori, M.; Kojima, I. Transforming growth factor beta and activin tonically inhibit DNA synthesis in the rat liver. Hepatology 2001, 34, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Nakatani, M.; Hitachi, K.; Uezumi, A.; Sunada, Y.; Ageta, H.; Inokuchi, K. Activin signaling as an emerging target for therapeutic interventions. Cell Commun. Signal 2009, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Attisano, L.; Lee-Hoeflich, S.T. The Smads. Genome Biol. 2001, 2, 3010.1–3010.8. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Menthena, A.; Koehler, C.; Sandhu, J.S.; Yovchev, M.I.; Hurston, E.; Shafritz, D.A.; Oertel, M. Activin A, p15INK4b signaling, and cell competition promote stem/progenitor cell repopulation of livers in aging rats. Gastroenterology 2011, 140, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Patella, S.; Phillips, D.J.; de Kretser, D.M.; Evans, L.W.; Groome, N.P.; Sievert, W. Characterization of serum activin-A and follistatin and their relation to virological and histological determinants in chronic viral hepatitis. J. Hepatol. 2001, 34, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Norris, S.; Evans, L.W.; Langley, P.G.; Hughes, R.D. Transforming growth factor-beta 1, activin and follistatin in patients with hepatocellular carcinoma and patients with alcoholic cirrhosis. Scand. J. Gastroenterol. 2002, 37, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.D.; Evans, L.W. Activin A and follistatin in acute liver failure. Eur. J. Gastroenterol. Hepatol. 2003, 15, 127–131. [Google Scholar] [CrossRef]
- Deli, A.; Kreidl, E.; Santifaller, S.; Trotter, B.; Seir, K.; Berger, W.; Schulte-Hermann, R.; Rodgarkia-Dara, C.; Grusch, M. Activins and activin antagonists in hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 1699–1709. [Google Scholar] [CrossRef]
- Yndestad, A.; Haukeland, J.W.; Dahl, T.B.; Bjøro, K.; Gladhaug, I.P.; Berge, C.; Damås, J.K.; Haaland, T.; Løberg, E.M.; Linnestad, P.; et al. A complex role of activin A in non-alcoholic fatty liver disease. Am. J. Gastroenterol. 2009, 104, 2196–2205. [Google Scholar] [CrossRef] [PubMed]
- Voumvouraki, A.; Notas, G.; Koulentaki, M.; Georgiadou, M.; Klironomos, S.; Kouroumalis, E. Increased serum activin-A differentiates alcoholic from cirrhosis of other aetiologies. Eur. J. Clin. Investig. 2012, 42, 815–822. [Google Scholar] [CrossRef]
- Kiagiadaki, F.; Kampa, M.; Voumvouraki, A.; Castanas, E.; Kouroumalis, E.; Notas, G. Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mishra, R.K.; Schiltz, G.E.; Makanji, Y.; Scheidt, K.A.; Mazar APWoodruff, T.K. Virtual high-throughput screening to identify novel activin antagonists. J. Med. Chem. 2015, 58, 5637–5648. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.A.; Gray, P.C.; Blount, A.L.; MacConell, L.A.; Wiater, E.; Bilezikjian, L.M.; Vale, W. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 2000, 404, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.B.; Lerch, T.F.; Cook, R.W.; Woodruff, T.K.; Jardetzky, T.S. The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 2005, 9, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Fainsod, A.; Deissler, K.; Yelin, R.; Marom, K.; Epstein, M.; Pillemer, G.; Steinbeisser, H.; Blum, M. The dorsalizing and neural inducing gene follistatin is an antagonist of BMP-4. Mech. Dev. 1997, 63, 39–50. [Google Scholar] [CrossRef]
- Amthor, H.; Christ, B.; Rashid-Doubell, F.; Kemp, C.F.; Lang, E.; Patel, K. Follistatin regulates bone morphogenetic protein-7 (BMP-7) activity to stimulate embryonic muscle growth. Dev. Biol. 2002, 243, 115–127. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef]
- DaCosta Byfield, S.; Major, C.; Laping, N.J.; Roberts, A.B. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2004, 65, 744–752. [Google Scholar] [CrossRef]
- Block, G.D.; Locker, J.; Bowen, W.C.; Petersen, B.E.; Katyal, S.; Strom, S.C.; Riley, T.; Howard, T.A.; Michalopoulos, G.K. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined (HGM) medium. J. Cell Biol. 1996, 132, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gehart, H.; Artegiani, B.; Lopez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-term expansion of functional mouse and human hepatocytes as 3D organoids. Cell 2018, 175, 1591–1606. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of junctional adhesion molecule A promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 2016, 151, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Oertel, M.; Shafritz, D.A. Liver repopulation by fetal liver stem/progenitor cells. In Methods in Bioengineering: Cell Transplantation, Soto-Gutierrez, A., Navarro-Alvarez, N., Fox, I.J., Eds.; Artech House: London, UK, 2011; Chapter 13; pp. 201–217. [Google Scholar]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.J.; Zhang, X.; Wheatley, A.M.; Mellor, S.L.; Cranfield, M.; Risbridger, G.P.; Groome, N.P.; Fleming, J.S. betaA- and betaC-activin, follistatin, activin receptor mRNA and betaC-activin peptide expression during rat liver regeneration. J. Mol. Endocrinol. 2005, 34, 505–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zhang, J.; Yan, B.; Chen, X. DEC1, a basic helix-loop-helix transcription factor and a novel target gene of the p53 family, mediates p53-dependent premature senescence. J. Biol. Chem. 2008, 283, 2896–2905. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Damaschke, N.; Yang, B.; Truong, M.; Guenther, C.; McCormick, J.; Huang, W.; Jarrard, D. Overexpression of the novel senescence marker β-galactosidase (GLB1) in prostate cancer predicts reduced PSA recurrence. PLoS ONE 2015, 10, e0124366. [Google Scholar] [CrossRef] [PubMed]
- Atfi, A.; Dumont, E.; Colland, F.; Bonnier, D.; L’helgoualc’h, A.; Prunier, C.; Ferrand, N.; Clément, B.; Wewer, U.M.; Théret, N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-beta signaling through interaction with the type II receptor. J. Cell Biol. 2007, 178, 201–208. [Google Scholar] [CrossRef]
- Le Pabic, H.; Bonnier, D.; Wewer, U.M.; Coutand, A.; Musso, O.; Baffet, G.; Clément, B.; Théret, N. ADAM12 in human liver cancers: TGF-beta-regulated expression in stellate cells is associated with matrix remodeling. Hepatology 2003, 37, 1056–1066. [Google Scholar] [CrossRef]
- Fournier, P.G.; Juárez, P.; Jiang, G.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.; Mohammad, K.S.; et al. The TGF-β Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef]
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007, 132, 1937–1946. [Google Scholar] [CrossRef]
- Kogure, K.; Omata, W.; Kanzaki, M.; Zhang, Y.Q.; Yasuda, H.; Mine, T.; Kojima, I. A single intraportal administration of follistatin accelerates liver regeneration in partially hepatectomized rats. Gastroenterology 1995, 108, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Bell, A.; Brooks, J.M.; Setoyama, K.; Melis, M.; Han, B.; Fukumitsu, K.; Handa, K.; Tian, J.; Kaestner, K.H.; et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J. Clin. Investig. 2015, 125, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Yovchev, M.I.; Xue, Y.; Shafritz, D.A.; Locker, J.; Oertel, M. Repopulation of the fibrotic/cirrhotic rat liver by transplanted hepatic stem/progenitor cells and mature hepatocytes. Hepatology 2014, 59, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; De Bleser, P.J.; Xu, G.; Van Den Berg, K.; Wisse, E.; Geerts, A. Comparison of glial fibrillary acidic protein and desmin staining in normal and CCl4-induced fibrotic rat livers. Hepatology 1996, 23, 1538–1545. [Google Scholar] [CrossRef]
- Fields, S.Z.; Parshad, S.; Anne, M.; Raftopoulos, H.; Alexander, M.J.; Sherman, M.L.; Laadem, A.; Sung, V.; Terpos, E. Activin receptor antagonists for cancer-related anemia and bone disease. Expert. Opin. Investig. Drugs 2013, 22, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Activin receptor ligand traps in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 351–357. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekala, S.; Rai, R.; Reed, S.L.; Bowen, B.; Michalopoulos, G.K.; Locker, J.; Raeman, R.; Oertel, M. Antagonizing Activin A/p15INK4b Signaling as Therapeutic Strategy for Liver Disease. Cells 2024, 13, 649. https://doi.org/10.3390/cells13070649
Mekala S, Rai R, Reed SL, Bowen B, Michalopoulos GK, Locker J, Raeman R, Oertel M. Antagonizing Activin A/p15INK4b Signaling as Therapeutic Strategy for Liver Disease. Cells. 2024; 13(7):649. https://doi.org/10.3390/cells13070649
Chicago/Turabian StyleMekala, Sowmya, Ravi Rai, Samantha Loretta Reed, Bill Bowen, George K. Michalopoulos, Joseph Locker, Reben Raeman, and Michael Oertel. 2024. "Antagonizing Activin A/p15INK4b Signaling as Therapeutic Strategy for Liver Disease" Cells 13, no. 7: 649. https://doi.org/10.3390/cells13070649
APA StyleMekala, S., Rai, R., Reed, S. L., Bowen, B., Michalopoulos, G. K., Locker, J., Raeman, R., & Oertel, M. (2024). Antagonizing Activin A/p15INK4b Signaling as Therapeutic Strategy for Liver Disease. Cells, 13(7), 649. https://doi.org/10.3390/cells13070649