
Immunotherapy of Hematological Malignancies of Human B-Cell Origin with CD19 CAR T Lymphocytes


Abstract
1. Acute Lymphoblastic Lymphoma
2. Standard Treatment Options for Acute Lymphoblastic Leukemia (ALL)
3. Non-Hodgkin’s Lymphoma (NHL)
4. Traditional Treatments for Non-Hodgkin’s Lymphoma (NHL)
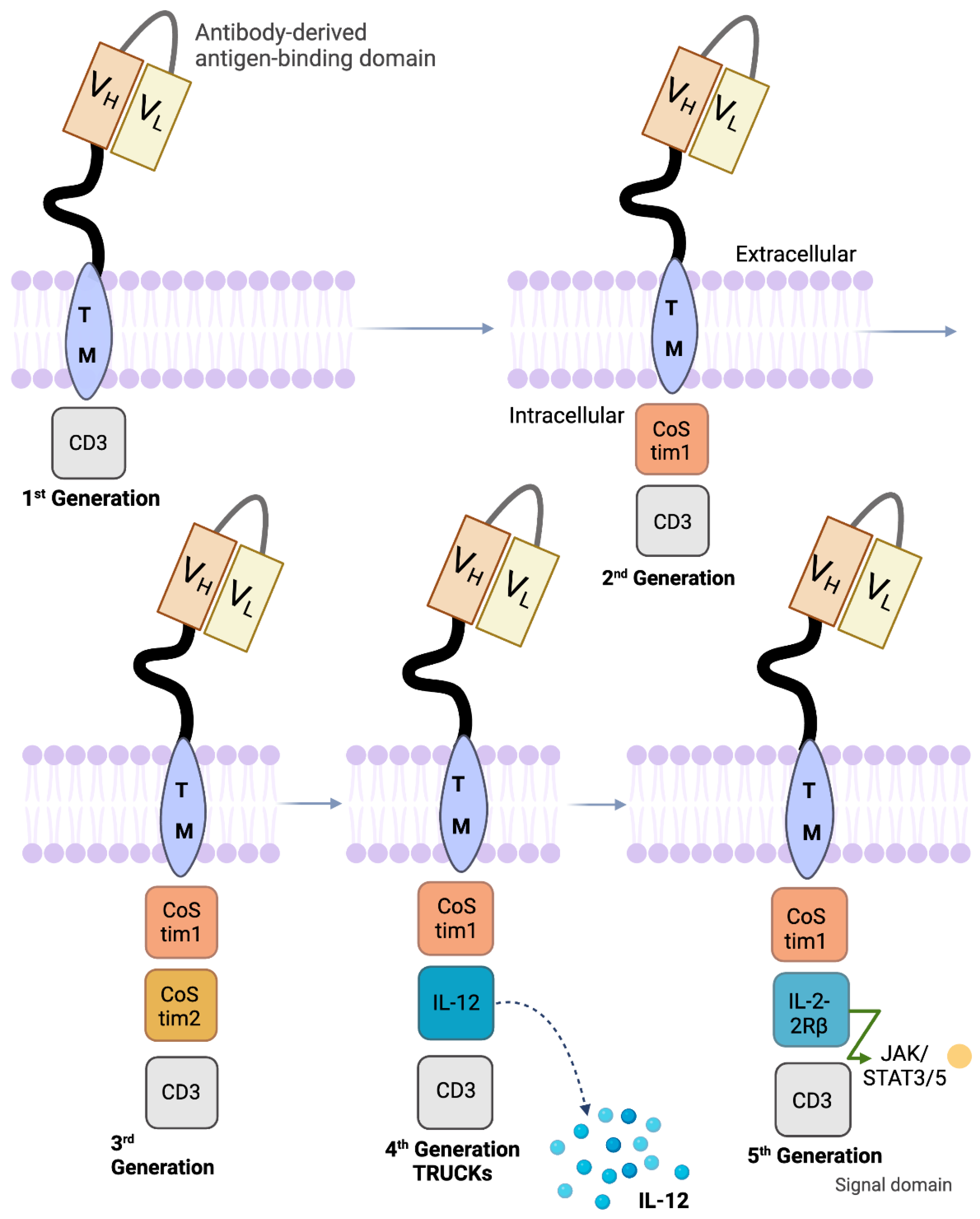
5. Design of Various CD19 CAR Constructs
6. Tisagenlecleucel and Axicabtagene Ciloleucel for the Treatment of Refractory Non-Hodgkin’s Lymphoma and Acute Lymphoblastic Leukemia
7. Summary of Clinical Trials
8. CD19 CAR-T Therapy in NHL
9. CD19 CAR-T Therapy in ALL
10. Clinical Trials of Tisagenlecleucel and Axicabtagene Ciloleucel for the Treatment of Refractory Non-Hodgkin’s Lymphoma and Acute Lymphoblastic Leukemia
11. Summary of Clinical Trials of Brexucabtagene Autoleucel (Tecartus) for the Treatment of ALL and NHL
12. Summary of Clinical Trials of Lisocabtagene Maraleucel (Breyanzi) for the Treatment of NHL
13. Application of CAR T Cell and CAR-NK Therapy in Solid Tumors
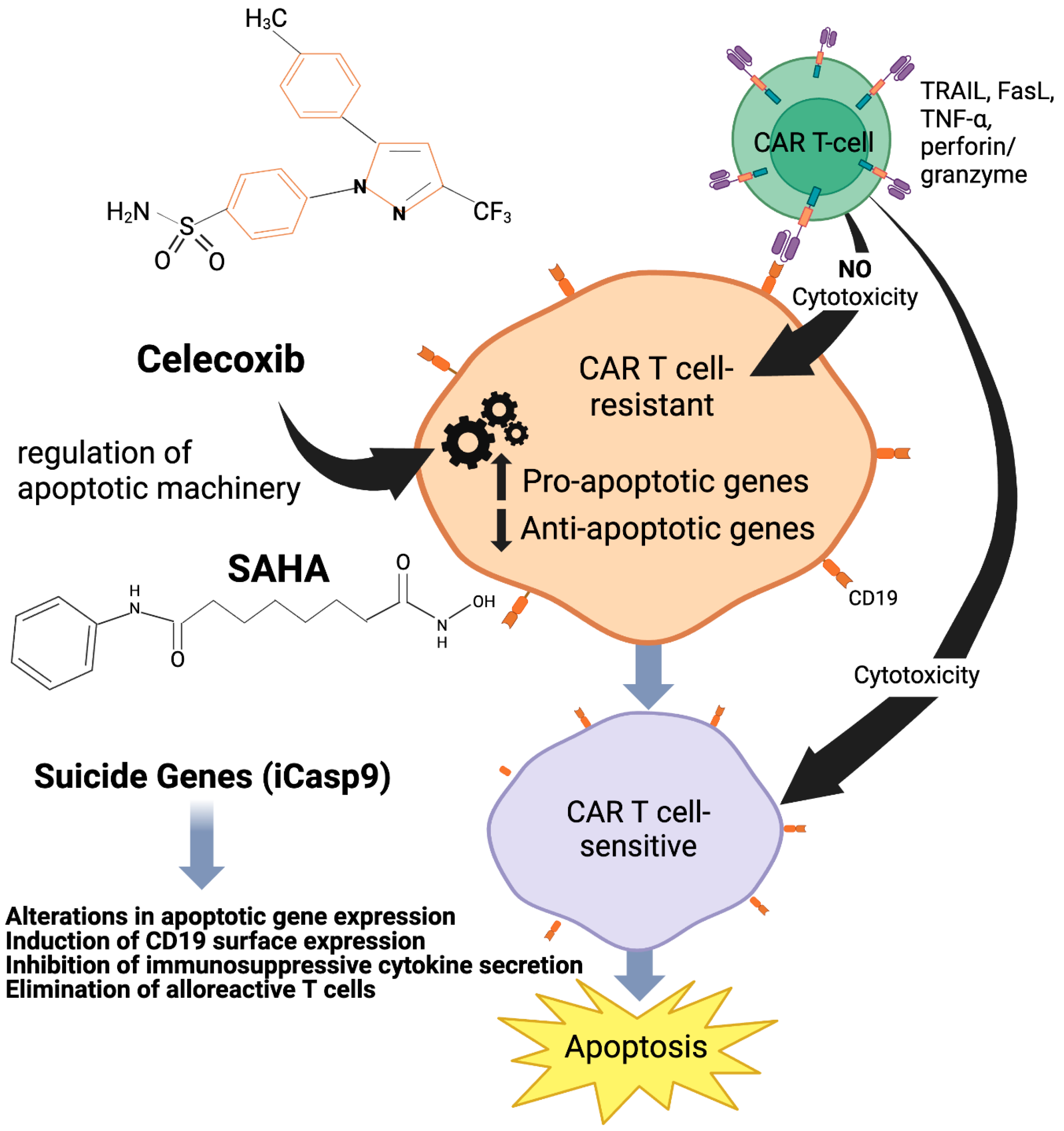
14. Incorporation of Suicide Gene to Increase Efficacy of CAR Therapy
15. FDA-Approved Small-Molecule Inhibitors to Increase the Efficacy of CAR T Cell Therapy
15.1. Celecoxib (Celebrex, COX2 Inhibitor)
15.2. Histone Deacetylase Inhibitors (HDACi) Vorinostat and Panobinostat
16. Regulation of Apoptotic Machinery by Cox-2 Inhibitor and HDACi
16.1. Celecoxib
16.2. Vorinostat (SAHA)
17. Conclusions/Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Mahmood, K.; Ubaid, M.; Rizvi, S.T. Multiple Osteolytic Lesions Causing Hypercalcemia: A Rare Presentation of Acute Lymphoblastic Leukemia. Case Rep. Med. 2017, 2017, 2347810. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Moriyama, T.; Relling, M.V.; Yang, J.J. Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood 2015, 125, 3988–3995. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Abbas, N.; Saba, T.; Rahman, S.I.U.; Mehmood, Z.; Kolivand, H. Classification of Acute Lymphoblastic Leukemia Using Deep Learning. Microsc. Res. Tech. 2018, 81, 1310–1317. [Google Scholar] [CrossRef]
- Mohapatra, S.; Patra, D.; Satpathy, S. An Ensemble Classifier System for Early Diagnosis of Acute Lymphoblastic Leukemia in Blood Microscopic Images. Neural Comput. Appl. 2014, 24, 1887–1904. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultra, C. Proposals for the Classification of the Acute Leukaemias French-American-British (FAB) Co-Operative Group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 Revision of the World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia: Rationale and Important Changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Cheung, Y.T.; Krull, K.R. Neurocognitive Outcomes in Long-term Survivors of Childhood Acute Lymphoblastic Leukemia Treated on Contemporary Treatment Protocols: A Systematic Review. Neurosci. Biobehav. Rev. 2015, 53, 108–120. [Google Scholar] [CrossRef]
- Wilejto, M.; Di Giuseppe, G.; Hitzler, J.; Gupta, S.; Alba, O. Treatment of Young Children with CNS-positive Acute Lymphoblastic Leukemia Without Cranial Radiotherapy. Pediatr. Blood Cancer 2015, 62, 1881–1885. [Google Scholar] [CrossRef]
- Pui, C.; Campana, D.; Deqing, P.; Bowman, P.; Sandlund, J.; Kaste, S.; Ribeiro, R.; Rubnitz, J.; Raimondi, S.; Onciu, M.; et al. Treating Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Porkka, K.; Perttu, K.; Jundán, T.; Rimpiläinen, J.; Mustjoki, S.; Smykla, R.; Wild, R.; Luo, R.; Arnan, M.; Brethon, B.; et al. Dasatinib Crosses the Blood-brain Barrier and is an Efficient Therapy for Central Nervous System Philadelphia Chromosome–positive Leukemia. Blood 2008, 112, 1005–1012. [Google Scholar] [CrossRef]
- Juliusson, G.; Karlsson, K.; Lazarevic, V.L.; Wahlin, A.; Brune, M.; Antunovic, P.; Derolf, Å.; Hägglund, H.; Karbach, H.; Lehmann, S.; et al. Hematopoietic Stem Cell Transplantation Rates and Long-term Survival in Acute Myeloid and Lymphoblastic Leukemia: Real-world Population-based Data from the Swedish Acute Leukemia Registry 1997–2006. Cancer 2011, 117, 4238–4246. [Google Scholar] [CrossRef] [PubMed]
- Olaya-Vargas, A.; Pérez-García, M.; Ramírez-Uribe, N.; Del Campo-Martinez, M.A.; Lopez-Hernández, G.; Hernández-García, M.; Amador-Zarco, J.; Garcia-Vega, G.; Melchor-Vidal, Y.; Zapata-Tarres, M.Z.; et al. Low Dose Total Body Irradiation (600 cGy) as a Conditioning Regimen in Allogenic Hematopoietic Stem Cell Transplant in Children with Acute Lymphoblastic Leukemia. J. Cancer Ther. 2016, 7, 586–592. [Google Scholar] [CrossRef][Green Version]
- Park, J.; Choi, E.K.; Kim, J.H.; Lee, S.W.; Song, S.Y.; Yoon, S.M.; Kim, Y.S.; Kim, S.S.; Park, J.H.; Park, J.; et al. Effects of Total Body Irradiation-Based Conditioning on Allogeneic Stem Cell Transplantation for Pediatric Acute Leukemia: A Single-Institution Study. Radiat. Oncol. J. 2014, 32, 198–207. [Google Scholar] [CrossRef][Green Version]
- Fisher, S.G.; Fisher, R.I. The epidemiology of non-Hodgkin’s lymphoma. Oncogene 2004, 23, 6524–6534. [Google Scholar] [CrossRef]
- Bleyer, A.; Budd, T.; Montello, M. Adolescents and Young Adults with Cancer: The Scope of the Problem and Criticality of Clinical Trials. Cancer 2006, 107, 1645–1655. [Google Scholar] [CrossRef]
- Thieblemont, C.; Grossoeuvre, A.; Houot, R.; Broussais-Guillaumont, F.; Salles, G.; Traullé, G.; Espinouse, D.; Coiffier, B. Non-Hodgkin’s Lymphoma in Very Elderly Patients Over 80 Years. A Descriptive Analysis of Clinical Presentation and Outcome. Ann. Oncol. 2008, 19, 774–779. [Google Scholar] [CrossRef]
- Chihara, D.; Nastoupil, L.J.; Williams, J.N.; Lee, P.; Koff, J.L.; Flowers, C.R. New Insights into the Epidemiology of Non-Hodgkin Lymphoma and Implications for Therapy. Expert Rev. Anticancer Ther. 2015, 15, 531–544. [Google Scholar] [CrossRef]
- Pulte, D.; Gondos, A.; Brenner, H. Ongoing Improvement in Outcomes for Patients Diagnosed as Having Non-Hodgkin Lymphoma From the 1990s to the Early 21st Century. Arch. Intern. Med. 2008, 168, 469–476. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, Y.; Zheng, T.; Zheng, T.; Ma, S. Risk Factors of Non-Hodgkin Lymphoma. Expert Opin. Med. Diagn. 2011, 5, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Weledji, E.P.; Orock, G.E. Surgery for Non-Hodgkin’s Lymphoma. Oncol. Rev. 2015, 9, 274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Groan, T.M. New Classification of Low-Grade Lymphoma. Ann. Oncol. 1996, 7, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Ribrag, V.; Caballero, D.; Fermé, C.; Zucca, E.; Arranz, R.; Briones, J.; Gisselbrecht, C.; Salles, G.; Gianni, A.M.; Gomez, H.; et al. Multicenter Phase II Study Of Plitidepsin In Patients With Relapsed/Refractory Non-Hodgkin’s Lymphoma. Haematologica 2013, 98, 357–363. [Google Scholar] [CrossRef]
- Fragkandrea, I.; Nixon, J.A.; Panagopoulou, P. Signs and Symptoms of Childhood Cancer: A Guide for Early Recognition. Am. Fam. Physician 2013, 88, 185–192. [Google Scholar]
- Armitage, J.O. Staging Non-Hodgkin Lymphoma. CA Cancer J. Clin. 2009, 55, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Keating, M.J.; Freireich, E.J. Toward the Potential Cure of Leukemias in the Next Decade. Cancer 2018, 125, 4301–4313. [Google Scholar] [CrossRef]
- Sonneveld, P.; de Ridder, M.; van der Lelie, H.; Nieuwenhuis, K.; Schouten, H.; Mulder, A.; van Reijswoud, I.; Hop, W.; Lowenberg, B. Comparison of Doxorubicin and Mitoxantrone in the Treatment of Elderly Patients with Advanced Diffuse Non-Hodgkin’s Lymphoma Using CHOP versus CNOP Chemotherapy. J. Clin. Oncol. 1995, 13, 2530–2539. [Google Scholar] [CrossRef]
- Fisher, R.I.; Gaynor, E.R.; Dahlberg, S.; Oken, M.M.; Grogan, T.M.; Mize, E.M.; Glick, J.H.; Coltman, C.A., Jr.; Miller, T.P. Comparison of a Standard Regimen (CHOP) with Three Intensive Chemotherapy Regimens for Advanced Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 1993, 328, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Grillo-Lopez, A.J. Rituximab (Rituxan/MabThera): The First Decade (1993–2003). Expert Rev. Anticancer Ther. 2003, 3, 767–779. [Google Scholar] [CrossRef]
- Pfreundschuh, M.; Trumper, L.; Osterborg, A.; Pettengell, R.; Trneny, M.; Imrie, K.; Ma, D.; Gill, D.; Walewski, J.; Zinzani, P.; et al. CHOP-like Chemotherapy Plus Rituximab versus CHOP-like Chemotherapy Alone in Young Patients with Good-prognosis Diffuse Large-B-cell Lymphoma: A Randomised Controlled Trial by the MabThera International Trial (MInT) group. Lancet Oncol. 2006, 7, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Lepage, E.; Brière, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP Chemotherapy Plus Rituximab Compared with CHOP Alone in Elderly Patients with Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 235–342. [Google Scholar] [CrossRef]
- Möricke, A.; Zimmermann, M.; Valsecchi, M.G.; Stanulla, M.; Biondi, A.; Mann, G.; Locatelli, F.; Cazzaniga, G.; Niggli, F.; Aricò, M.; et al. Dexamethasone vs Prednisone in Induction Treatment of Pediatric ALL: Results of the Randomized Trial AIEOP-BFM ALL 2000. Blood 2016, 127, 2101–2112. [Google Scholar] [CrossRef]
- Atra, A.; Gerrard, M.; Hobson, R.; Imeson, J.D.; Hann, I.M.; Pinkerton, C.R. Outcome of Relapsed or Refractory Childhood B-Cell Acute Lymphoblastic Leukaemia and B-Cell Non-Hodgkin’s Lymphoma Treated with the UKCCSG 9003/9002 Protocols. Br. J. Haematol. 2001, 112, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Liu, X.; Jiang, S.; June, C.H.; Grupp, S.A.; Zhao, Y. Regimen-Specific Effects of RNA-Modified Chimeric Antigen Receptor T Cells in Mice with Advanced Leukemia. Hum. Gene Ther. 2013, 24, 717–727. [Google Scholar] [CrossRef]
- Subklewe, M.; von Bergwelt-Baildon, M.; Humpe, A. Chimeric antigen receptor T cells: A race to revolutionize cancer therapy. Transfus. Med. Hemother. 2019, 46, 15–24. [Google Scholar] [CrossRef]
- Glockshuber, R.; Malia, M.; Pfitzinger, I.; Plückthun, A. A Comparison of Strategies to Stabilize Immunoglobulin Fv-Fragments. Biochemistry 1990, 29, 1362–1367. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.V.; Sadelain, M. Antigen-Dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Jain, M.D.; Bachmeier, C.A.; Phuoc, V.H.; Chavez, J.C. Axicabtagene Ciloleucel (KTE-C19), an Anti-CD19 CAR T Therapy for the Treatment of Relapsed/Refractory Aggressive B-Cell Non-Hodgkin’s Lymphoma. Ther. Clin. Risk Manag. 2018, 14, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 Costimulation by a Chimeric Antigen Receptor Abrogates CD28 and IL-2 Induced IL-10 Secretion by Redirected CD4+ T cells. Oncoimmunology 2012, 1, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of Large, Established Tumor Xenografts with Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/bcl-XL Activation and CD8+ T Cell-Mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Zhang, L.; Feldman, S.A.; Zheng, Z.; Chinnasamy, N.; Xu, H.; Nahvi, A.V.; Dudley, M.E.; Rosenberg, S.A.; Morgan, R.A. Evaluation of γ-Retroviral Vectors that Mediate the Inducible Expression of IL-12 for Clinical Application. J. Immunother. 2012, 35, 430–439. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, L.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- Chow, V.A.; Shadman, M.; Gopal, A.K. Translating Anti-CD19 CAR T-cell Therapy into Clinical Practice for Relapsed/refractory Diffuse Large B-cell Lymphoma. Blood 2018, 132, 777–781. [Google Scholar] [CrossRef]
- Maloney, D.G. CAR-T cells. Clin. Lymphoma Myeloma Leuk. 2019, S100–S101. [Google Scholar] [CrossRef]
- Hopfinger, G.; Worel, N. CAR T-Cell Therapy in Diffuse Large B-Cell Lymphoma. memo-Mag. Eur. Med. Oncol. 2020, 13, 32–35. [Google Scholar] [CrossRef]
- Bouchkouj, N.; Kasamon, Y.L.; de Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA Approval Summary: Axicabtagene Bouchkouj Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N.V.; Nazimuddin, F.; et al. Sustained Remissions with CD19-Specific Chimeric Antigen Receptor (CAR)-Modified T Cells in Children with Relapsed/Refractory ALL. J. Clin. Oncol. 2016, 34, 3011. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Bouchkouj, N.; Lin, X.; Wang, X.; Przepiorka, D.; Xu, Z.; Purohit-Sheth, T.; Theoret, M. FDA Approval Summary: Brexucabtagene Autoleucel for Treatment of Adults with Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Oncologist 2022, 27, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.; Jacobson, C.; Hill, B.; Timmerman, J.; Holmes, H.; Jaglowski, S.; Flinn, I.; et al. Three-Year Follow-Up of KTE-X19 in Patients with Relapsed/Refractory Mantle Cell Lymphoma, Including High-Risk Subgroups, in the ZUMA-2 Study. J. Clin. Oncol. 2023, 41, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Ghobadi, A.; Oluwole, O.; Logan, A.; Boissel, N.; Cassaday, R.; Leguay, T.; Bishop, M.; Topp, M.; Tzachanis, D.; et al. Two-Year Follow-Up of KTE-X19 in Patients with Relapsed or Refractory Adult B-Cell Acute Lymphoblastic Leukemia in ZUMA-3 and Its Contextualization with SCHOLAR-3, an External Historical Control Study. J. Hematol. Oncol. 2022, 15, 170. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Solomon, S.; Arnason, J.; Johnston, P.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene Maraleucel as Second-Line Therapy for Large B-Cell Lymphoma: Primary Analysis of the Phase 3 TRANSFORM Study. Blood 2023, 141, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.; Jacobson, C.; Hill, B.; Timmerman, J.; Holmes, H.; Jaglowski, S.; Flinn, I.; et al. KXE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.J.; Gödel, P.; Rüger, M.A.; Onur, Ö.A.; Shimabukuro-Vornhagen, A.; Kochanek, M.; Böll, B. In the Eye of the Storm: Immune-mediated Toxicities Associated with CAR-T Cell Therapy. Hemasphere 2019, 3, e191. [Google Scholar] [CrossRef]
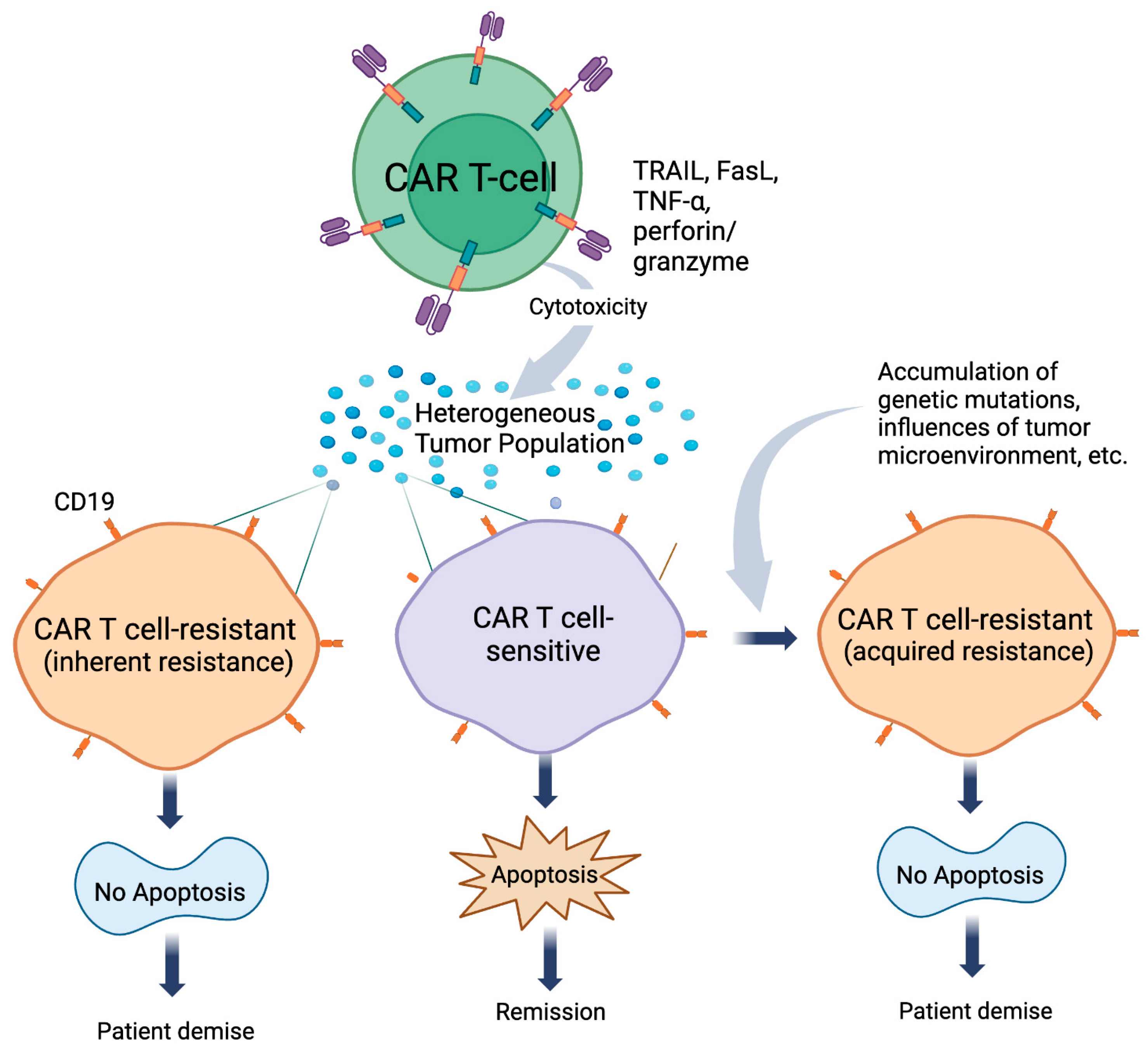
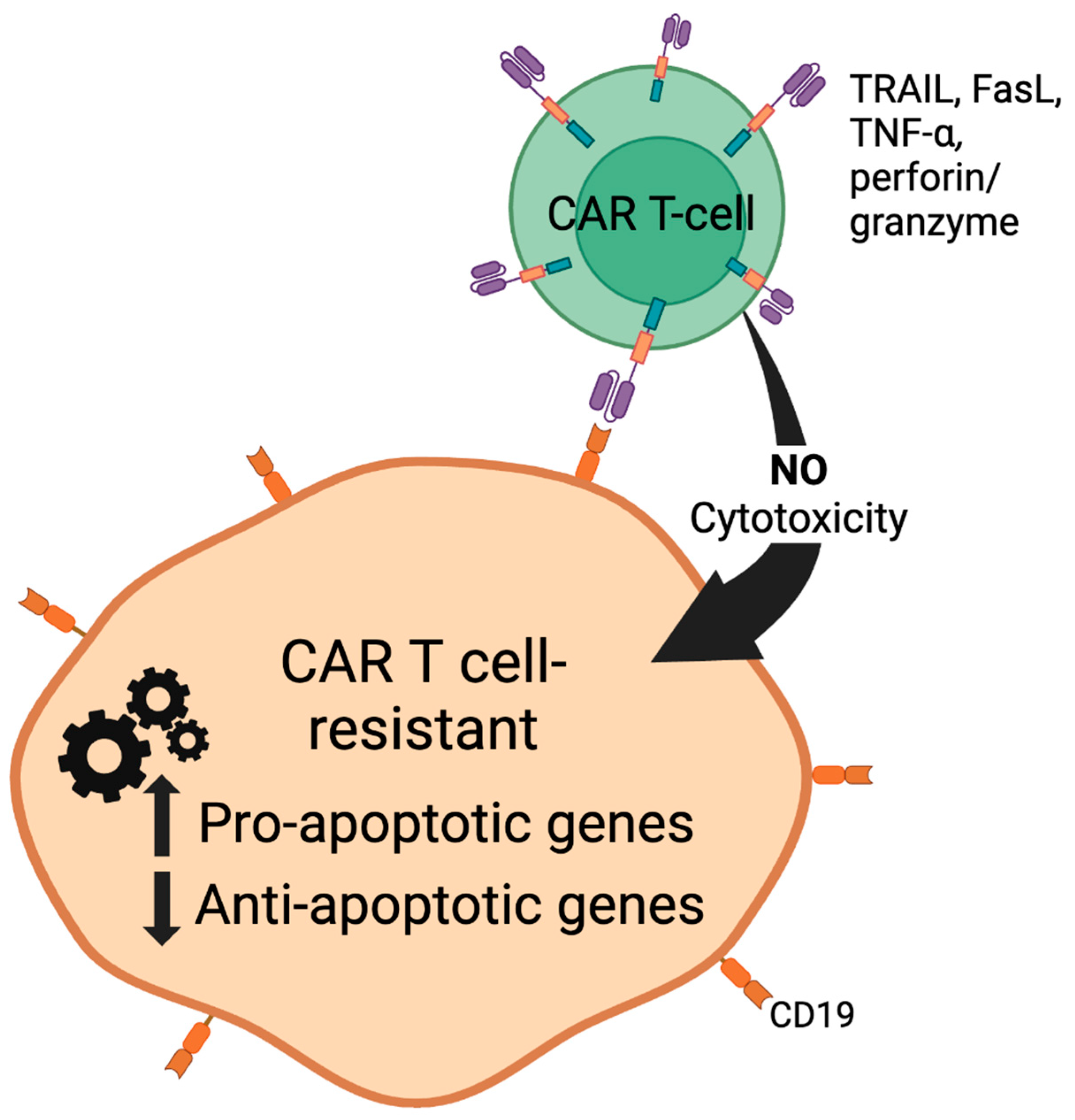
- Qin, Y.; Xu, G. Enhancing CAR T-cell therapies against solid tumors: Mechanisms and reversion of resistance. Front. Immunol. 2022, 13, 1053120. [Google Scholar] [CrossRef]
- Bagley, S.J.; Logun, M.; Fraietta, J.A.; Wang, X.; Desai, A.S.; Bagley, L.J.; Nabavizadeh, A.; Jarocha, D.; Martins, R.; Maloney, E.; et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: Phase 1 trial interim results. Nat. Med. 2024. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, Y.; He, Z.; Li, L.; Liu, S.; Jiang, M. Breakthrough of solid tumor treatment: CAR-NK immunotherapy. Cell Death Discov. 2024, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.; O’Dwyer, M. Realizing Innate Potential: CAR-NK Cell Therapies for Acute Myeloid Leukemia. Cancers 2021, 13, 1568. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wu, L.; Yin, L.; Shi, H.; Gu, Y.; Xing, N. Combined treatment with anti-PSMA CAR NK-92 cell and anti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin. Transl. Med. 2022, 12, e901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, C.; He, M.; Xing, W.; Hou, R.; Zhang, H. Co-expression of IL-21-Enhanced NKG2D CAR-NK cell therapy for lung cancer. BMC Cancer 2024, 24, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, M.; Song, B.; Miao, W.; Zhan, R.; Yang, S.; Han, Z.; Cai, H.; Xu, X.; Zhao, Y.; et al. Decoding the multidimensional signatures of resident and expanded natural killer cells generated from perinatal blood. Am. J. Cancer Res. 2022, 12, 2132–2145. [Google Scholar] [PubMed]
- Jazirehi, A.R.; Dinh, T.N.M. Approaches to Improve Clinical Efficacy of CD19-Redirected Chimeric Antigen Receptor (CD19 CAR) T Cell Immunotherapy of Non-Hodgkin’s Lymphoma. Cancer Ther. Oncol. Int. J. 2017, 6, 555682. [Google Scholar] [CrossRef]
- Onea, A.S.; Jazirehi, A.R. CD19 Chimeric Antigen Receptor (CD19 CAR)-Redirected Adoptive T-Cell Immunotherapy for the Treatment of Relapsed or Refractory B-Cell Non-Hodgkin’s Lymphomas. Am. J. Cancer Res. 2016, 6, 403–424. [Google Scholar] [PubMed]
- Zhang, J.; Kale, V.; Chen, M. Gene-directed Enzyme Prodrug Therapy. AAPS J. 2015, 17, 102–110. [Google Scholar] [CrossRef]
- Springer, C.J.; Niculescu-Duvaz, I. Prodrug-activating Systems in Suicide Gene Therapy. J. Clin. Investig. 2000, 105, 1161–1167. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Manganini, M.; Borleri, G.; Bonamino, M.; Imberti, L.; Biondi, A.; Golay, J.; Rambaldi, A.; Introna, M. Characterization of CD20-Transduced T lymphocytes as an Alternative Suicide Gene Therapy Approach for the Treatment of Graft-Versus-Host Disease. Hum. Gene Ther. 2004, 15, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J. COX1 and COX2 Tissue Expression: Implications and Predictions. J. Rheumatol. Suppl. 1997, 49, 15–19. [Google Scholar] [PubMed]
- McCormack, P.L. Celecoxib: A Review of its Use for Symptomatic Relief in the Treatment of Osteoarthritis, Rheumatoid Arthritis and Ankylosing Spondylitis. Drugs 2011, 71, 2457. [Google Scholar] [CrossRef] [PubMed]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Daniels, S.; Robbins, J.; West, C.R.; Nemeth, M.N. Celecoxib in the Treatment of Primary Dysmenorrhea: Results From Two Randomized, Double-Blind, Active- and Placebo-Controlled, Crossover Studies. Clin. Ther. 2009, 31, 1192–1208. [Google Scholar] [CrossRef] [PubMed]
- Bubna, A.K. Vorinostat—An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef] [PubMed]
- Lehrmann, H.; Pritchard, L.L.; Harel-Bellan, A. Histone Acetyltransferase and Deacetylases in the Control of Cell Proliferation and Differentiation. Adv. Cancer Res. 2002, 86, 41–65. [Google Scholar] [CrossRef] [PubMed]
- Gallouet, A.S.; Travert, M.; Bresson-Bepoldin, L.; Guilloton, F.; Pangault, C.; Caulet-Maugendre, S.; Lamy, T.; Tarte, K.; Guillaudeux, T. COX2-independent Effects of Celecoxib Sensitize Lymphoma B Cells to TRAIL-Mediated Apoptosis. Clin. Cancer Res. 2014, 20, 2663–2673. [Google Scholar] [CrossRef]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The Histone Deacetylase Inhibitor Suberic Bishydroxymate Regulates the Expression of Multiple Apoptotic Mediators and Induces Mitochondria—Dependent Apoptosis of Melanoma Cells. Mol. Cancer Ther. 2004, 3, 425–435. [Google Scholar] [CrossRef]
- Dean, J.L.E.; Brook, M.; Clark, A.R.; Saklatvala, J. p38 Mitogen-activated Protein Kinase Regulates Cyclooxygenase 2 mRNA Stability and Transcription in Lipopolysacaride-treated Human Monocytes. J. Biol. Chem. 1999, 274, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Handrick, R.; Elsaesser, S.J.; Rudner, J.; Henke, G.; Ganswindt, U.; Belka, C.; Jendrossek, V. Improtance of Bak for Celecoxib-induced Apoptosis. Biochem. Pharmacol. 2008, 76, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone Deacetylase Inhibitor Activation of p21WAFI Involves Changes in Promoter-associated Proteins, Including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary of Trial | Drug/ Treatment | Overall Response Rate (ORR) | Objective Response Rate (ORR) | Complete Remission (CR) | Disease Free Progression (DFP) | Toxicites (Most Common) | References |
|---|---|---|---|---|---|---|---|
| n = 30 Children/ Adults r/r ALL | CTL019 (Kymriah) | OS = 78% | 90% | Sustained remissions at 6-month event -free survival = 67% Predicted probability of persistence with CTL019 = 68% Probability of relapse-free B-cell aplasia = 73% | Severe: CRS = 27% Any grade: CRS = 100% NT = 43% B-cell aplasia In patients with a response = 100% | [61] | |
| n = 28 DLBCL or r/r FL | CTL019 (Kymriah) | 64% | FL = 71% DLBCL = 43% | Sustained remissions of patients with response at 28.6 month (median) follow-up: FL = 89% DLBCL = 86% | Severe: CRS = 18% NT = 11% Any grade: CRS = 57% NT = 39% | [62] | |
| n = 93 adults r/r DLBCL (JULIET) | Tisagenlecleucel (Kymriah) | 52% | Complete response 40% | 12-month post first response: Relapse-free survival = 65% CR relapse-free survival = 79% | Severe: CRS = 22% Neurologic events = 12% Cytopenias (>28 days) = 32% Any grade: CRS = 58% Neurologic events = 21% Cytopenias = 44% | [58] | |
| n = 101 r/r DLBCL, PMBCL or (transformed) FL (ZUMA-II) | Axicabtagene Ciloleucel (Yescarta) | 82% | 54% | Sustained remissions with a 15.4-month (median) event-free survival = 42% CR at 15.4 month (median) = 40% OS at 18-months = 52% | Severe: Thrombocytopenia = 38% Neutropenia = 78% Anemia = 43% CRS = 13% Neurologic events = 28% | [59,60] | |
| n = 20 adults r/r CLL or ALL | CAR T-19/ CTL019 (Kymriah) | CLL = 42.9% ALL = 83.3% | CLL = 21.4% ALL = 83.4% | Severe: CRS (CLL) = 42.86% CRS (ALL) = 83.33% | [56] | ||
| n = 7 Refractory DLBCL | KTE-C19 (Yescarta) | 71% | N/A | 57% | CR at 12+ months 0.43 | Severe: CRS = 14% NT = 57% Any grade: CRS = 85% NT = 44% | [57] |
| n = 53 adults Relapsed B-cell ALL | Autologous 19 − 28z + CAR T-cells | N/A | N/A | 83% | 29-month (median) Follow-up: Event-free survival = 6.1 months (median) OS = 12.9 months (median) | Severe: CRS = 26% NT = 42% Any grade: CRS = 85% NT = 44% | [63] |
| n = 59 Children/adults r/r ALL | CTL019 (Kymriah) | OS at 12 month (median) Follow-up = 79% | 93% (1-month Post Infusion) | N/P | 12-month (median) Follow-up: CR = 58% | Severe: CRS = 27% B-cell aplasia (continuing CR patients) = 71% Any grade: CRS = 88% | [64] |
| n = 75 Pediatric/young Adult r/r B-cell ALL (ELIANA) | Tisagenlecleucel (Kymriah) | 81% (3 mo. ≥) | PR = 21% | 3-month overall remission rate = 81% CR = 60% Incomplete hematologic recovery + CR = 21% | 6-month follow-up: Patients with relapse-free survival = 80% OS = 90% 12-month follow-up: patients with relapse-free survival = 59% OS = 76% | B-cell aplasia in Patients with a response = 100% Severe: CRS = 46% Neurologic event = 13% Infection = 24% Any grade: CRS = 77% Neurologic event = 40% Infection = 43% | [57] |
| Summary of Trial | Infusion Amount (Per kg of Body Weight) | ORR% | CR% | PR% | DFP% | Toxicities ≥ 3 | Mortalities Post Infusion | Reference |
|---|---|---|---|---|---|---|---|---|
| n = 75 (ELIANA) | 1.0 × 108 (median) | 81% (3 mo. ≥) | 60 | 21 | 59 (12 mo.) | CRS: 46 NT:13 FN:35 TLS:4 | 3 (CRS and other AE), 13 (DP + subsequent therapies, etc), 1 (unknown) | [57] |
| n = 30 | 0.76 × 106 To 20.6 × 106 | 78 | 90 | NP | 67 (8 mo) | CRS: 27 | 7 (DP) | [61] |
| n = 59 | 1 × 107 to 1 × 108 | 79 (12 mo.) | NP | 93 (at 1 mo.) | 58 (12 mo.) | CRS: 27 | NP | [64] |
| n = 63 | >50 kg infusion amt = 0.1 − 2.5 × 108 or ≤50 kg infusion amt = 0.2 − 5 × 106 (single doses) | 83 | 63 | NP | Not reached at 4.8 mo. (median) follow-up | CRS: 49 NT: 18 (safety population n = 68) | 0 | [54] |
| n = 6 (ELIANA subgroup) | >50 kg infusion amt = 0.1 − 2.5 × 108 or ≤50 kg infusion amt = 0.2 − 5.0 × 106 (single doses) | 66.7 (3 mo.) | 42.8 | NP | 100 (median time of 6 mo.) | CRS: 5 NT: 0 | 2 (DP, 71, and 352 days post infusion) | [65] |
| Summary of trial | Infusion amount (per kg of body weight) | ORR% | CR% | PR% | DFP% | Toxicities ≥3 | Mortalities post infusion | Reference |
| n = 93 r/r DLBCL, tFL or HGBL-DH/TH (JULIET) | 3.0 × 108 (median) | 52 | 40 | 12 | 65 (12 mo.) | CRS:22 NT:12 FN:15 TLS:1 | 3 (DP, 30 days post infusion) | [58] |
| n = 28 DBCL or r/r FL | 3.1 × 106 to 8.9 × 106 | 64 | FL: 71 DLBCL: 43 Combined: 57 | 18 (at 3 mo.) | FL:89 DLBCL: 86 (28.6 mo) | CRS:18 NT: 11 | 1 (NT) | [62] |
| n = 9 r/r DLBCL (JULIET subgroup) | 2.0 × 108 (median) | 77.8 | 55.6 | 22.2 | 100 (25 − 550 + day range) | CRS: 2 NT: 1 | 2 (DP, 30 days post infusion | [66] |
| Summary of Trial | Infusion Amount (Per kg of Body Weight) | ORR% | CR% | PR% | DFP% | Toxicities ≥3 | Mortalities Post Infusion | Reference |
|---|---|---|---|---|---|---|---|---|
| n = 108 r/r DLBCL, tFL or PMBCL (ZUMA-I & II) | 2.0 × 106 | 82 (12 mo.) | 58 | 29 | 41 (15 mo.) | CRS: 13 NT:28 | 2 (CRS), 42 (DP + subsequent therapies, etc.) | [59,60] |
| n = 53 r/r DLBCL or FL | 1 × 106 or 3 × 106 | - | 83 | - | - | CRS: 26 NT:42 | 1 | [63] |
| n = 22 r/r DLBCL, FL, or mantle cell lymphoma | 1 × 106, 2 × 106 or 6 × 106 | 73 | 55 | 18 | 63.3 (12 mo.) | NT: 55 | 0 | [67] |
| Summary of Trials | Drug/ Treatment | Overall Response | Objective Response | Complete Remission | Disease Free Progression | Toxicities (Most Common) | Reference | |
|---|---|---|---|---|---|---|---|---|
| n = 54 n = 78 r/r B-ALL (all leukapheresed patients) r/r B-cell precursor (ALL) (ZUMA 3, Phase II) | Brexucabtagene autoleucel KTE-X19 (Tecartus) | 83.60% (16.4% of people showed no response) | 71% | 52% | Median duration of remission (DOR) = 14.6 mo. Median relapse-free survival (RFS) = 11.6 mo. Median overall survival (OS) = 25.4 mo | Total = 79% Severe: CRS = 26% NT = 35% Any grade: CRS = 92% NT = 87% | [62,65] | |
| n = 68 r/r mantle cell lymphoma (NHL) (ZUMA 2, Phase II) | Brexucabtagene autoleucel KTE-X19 (Tecartus) | 91% | 91% | 68% | Median OS = 46.6 mo. | Grade 1 or 2: CRS = 76% NT = 32% Severe: CRS = 15% NT = 31% | [66,67] | |
| n = 92 LBCL (NHL) (TRANSFORM, Phase III) | Lisocabtagene maraleucel (Breyanzi) | 87% | 74% | Not reached (NR) | Grade 3: CRS: 1% NT: 4% No grade 4 or 5 events Prolonged cytopenia = 43% | [63] | ||
| Summary | Infusion amount (per kg of body weight) | ORR% | CR% | PR% | DFP% | Toxicities ≥3 | Mortalities post infusion | Reference |
| n = 54 n = 78 r/r B-ALL (ZUMA 3, Phase II) | 1 × 106 | 83.6 | 52 | 15 | 39 (complete remission) | 97% Febrile neutropenia: 35% Infections: 30% CRS: 26% NT: 35% | Fatal adverse reactions = 5% (cerebral edema and infections) | [62,65] |
| Summary | Infusion amount (per kg of body weight) | ORR% | CR% | PR% | DFP% | Toxicities ≥3 | Mortalities post infusion | Reference |
| n = 68 r/r mantle cell lymphoma (NHL) (ZUMA 2, Phase II) | 2 × 106 | 91 | 68 | 24 | 24.9— to not estimable | 99% Cytopenias: 94% CRS: 91% NT: 63% | No death from CRS No death from NT 16 deaths total (24%): Death from progressive disease = 14 patients (21%) Grade 5 AE = 2 patients (3%) | [66,67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khvorost, D.; Kendall, B.; Jazirehi, A.R. Immunotherapy of Hematological Malignancies of Human B-Cell Origin with CD19 CAR T Lymphocytes. Cells 2024, 13, 662. https://doi.org/10.3390/cells13080662
Khvorost D, Kendall B, Jazirehi AR. Immunotherapy of Hematological Malignancies of Human B-Cell Origin with CD19 CAR T Lymphocytes. Cells. 2024; 13(8):662. https://doi.org/10.3390/cells13080662
Chicago/Turabian StyleKhvorost, Darya, Brittany Kendall, and Ali R. Jazirehi. 2024. "Immunotherapy of Hematological Malignancies of Human B-Cell Origin with CD19 CAR T Lymphocytes" Cells 13, no. 8: 662. https://doi.org/10.3390/cells13080662
APA StyleKhvorost, D., Kendall, B., & Jazirehi, A. R. (2024). Immunotherapy of Hematological Malignancies of Human B-Cell Origin with CD19 CAR T Lymphocytes. Cells, 13(8), 662. https://doi.org/10.3390/cells13080662