

Investigating Repeat Expansions in NIPA1, NOP56, and NOTCH2NLC Genes: A Closer Look at Amyotrophic Lateral Sclerosis Patients from Southern Italy




Abstract
1. Introduction
2. Materials and Methods
2.1. Population
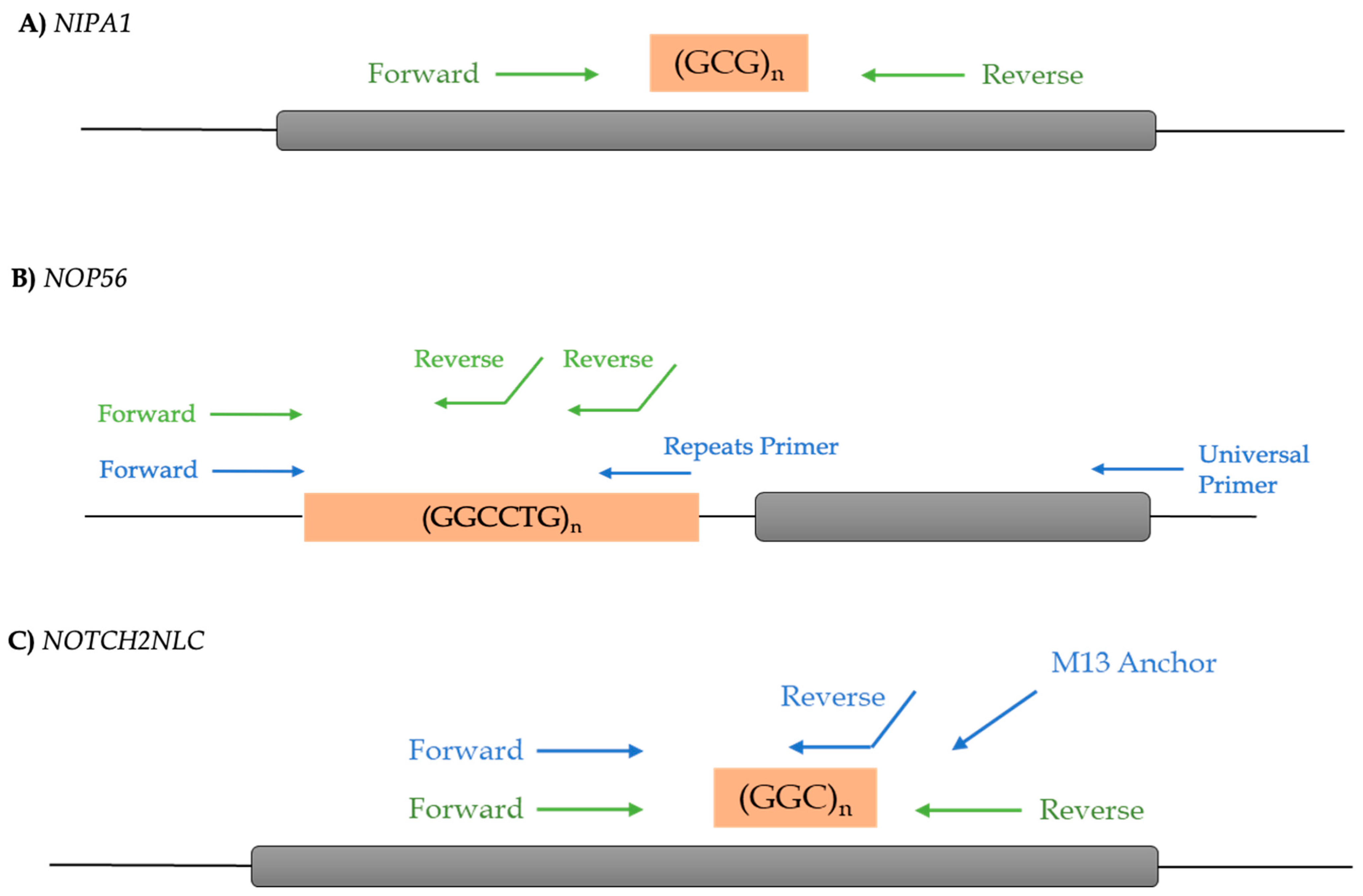
2.2. Genetic Analysis
2.3. Statistical Analysis
3. Results
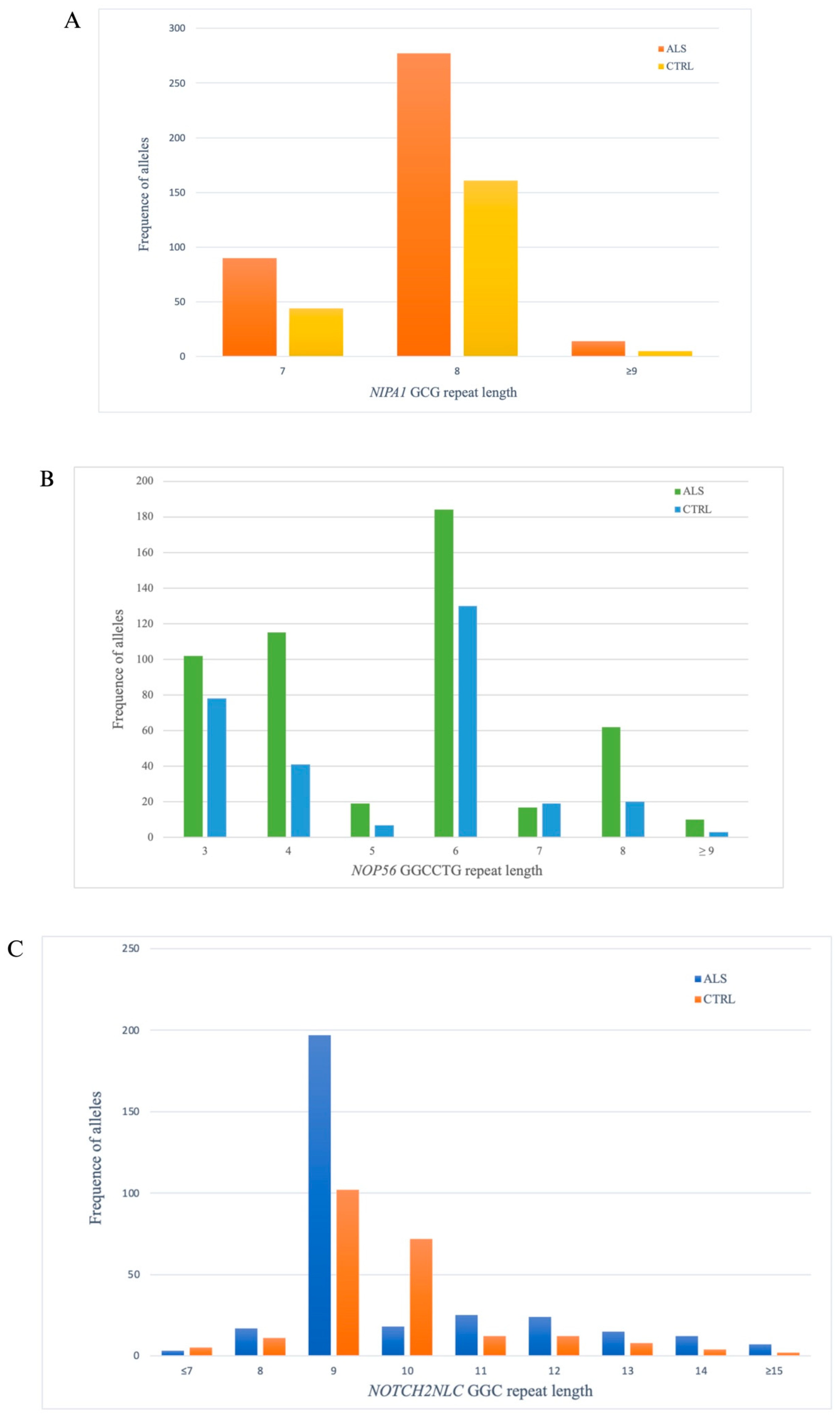
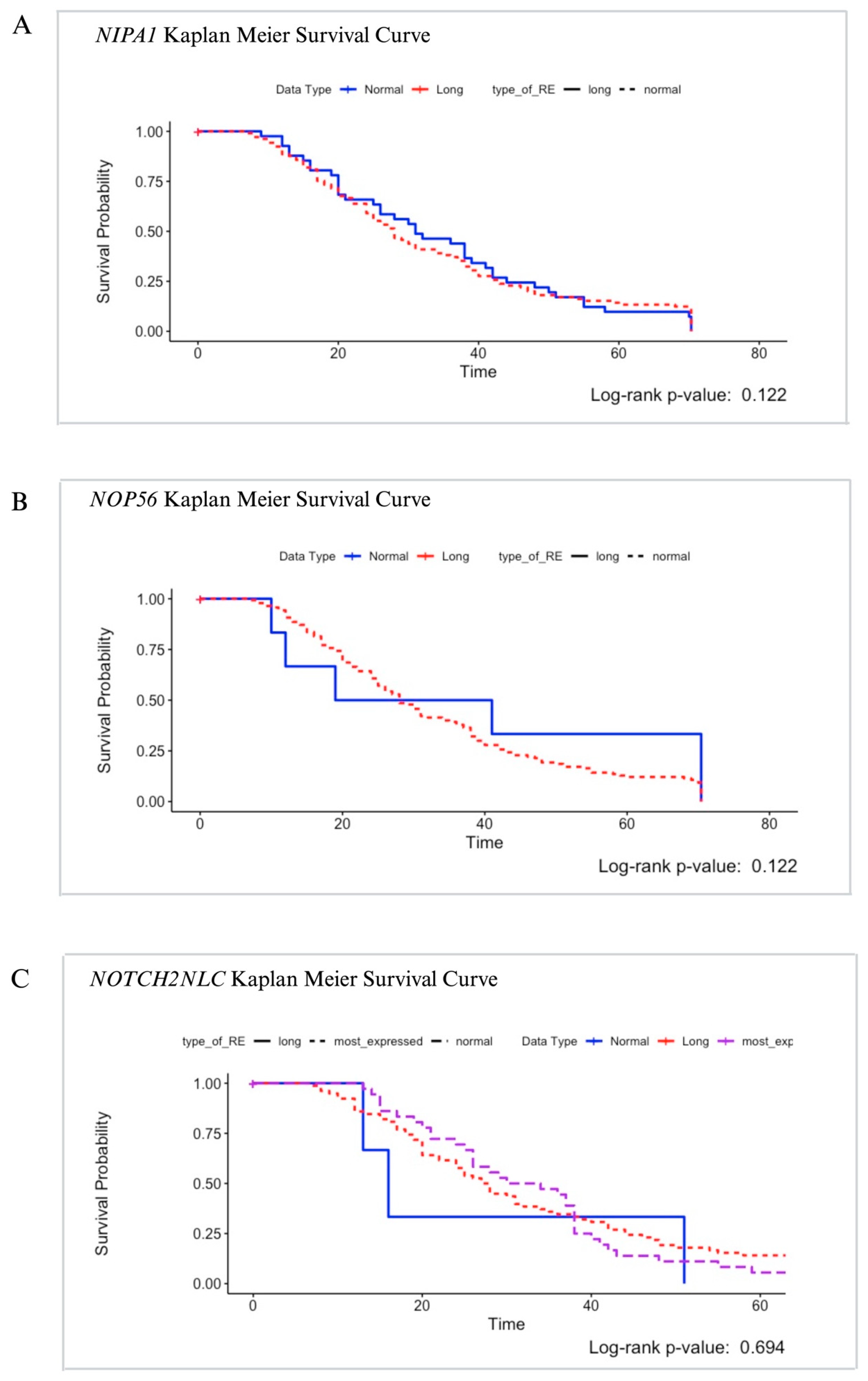
3.1. NIPA1
3.2. NOP56
3.3. NOTCH2NLC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chio, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar] [PubMed]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Perrone, B.; Conforti, F.L. Common mutations of interest in the diagnosis of amyotrophic lateral sclerosis: How common are common mutations in ALS genes? Expert. Rev. Mol. Diagn. 2020, 20, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Ji, A.L.; Zhang, X.; Chen, W.W.; Huang, W.J. Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J. Med. Genet. 2017, 54, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Pulst, S.M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski, T.J., Jr.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- Blauw, H.M.; Veldink, J.H.; van Es, M.A.; van Vught, P.W.; Saris, C.G.; van der Zwaag, B.; Franke, L.; Burbach, J.P.; Wokke, J.H.; Ophoff, R.A.; et al. Copy-number variation in sporadic amyotrophic lateral sclerosis: A genome-wide screen. Lancet Neurol. 2008, 7, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Henden, L.; Fearnley, L.G.; Grima, N.; McCann, E.P.; Dobson-Stone, C.; Fitzpatrick, L.; Friend, K.; Hobson, L.; Chan Moi Fat, S.; Rowe, D.B.; et al. Short tandem repeat expansions in sporadic amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Adv. 2023, 9, eade2044. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.L.; Spataro, R.; Sproviero, W.; Mazzei, R.; Cavalcanti, F.; Condino, F.; Simone, I.L.; Logroscino, G.; Patitucci, A.; Magariello, A.; et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 2012, 79, 2315–2320. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.F.; Pal, M.; Engelhardt, J.I.; Molnar, M.J.; Klivenyi, P.; Szell, M. Beyond C9orf72: Repeat expansions and copy number variations as risk factors of amyotrophic lateral sclerosis across various populations. BMC Med. Genomics 2024, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Blauw, H.M.; Al-Chalabi, A.; Andersen, P.M.; van Vught, P.W.; Diekstra, F.P.; van Es, M.A.; Saris, C.G.; Groen, E.J.; van Rheenen, W.; Koppers, M.; et al. A large genome scan for rare CNVs in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2010, 19, 4091–4099. [Google Scholar] [CrossRef] [PubMed]
- Blauw, H.M.; van Rheenen, W.; Koppers, M.; Van Damme, P.; Waibel, S.; Lemmens, R.; van Vught, P.W.; Meyer, T.; Schulte, C.; Gasser, T.; et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Tsai, P.C.; Guo, Y.C.; Hsiao, C.T.; Liu, G.T.; Liao, Y.C.; Soong, B.W. Spinocerebellar ataxia type 36 in the Han Chinese. Neurol. Genet. 2016, 2, e68. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zeng, J.; He, M.; Zeng, X.; Zhou, Y.; Liu, Z.; Xia, K.; Pan, Q.; Jiang, H.; Shen, L.; et al. Genetic and clinical analysis of spinocerebellar ataxia type 36 in Mainland China. Clin. Genet. 2016, 90, 141–148. [Google Scholar] [CrossRef]
- Miyazaki, K.; Yamashita, T.; Morimoto, N.; Sato, K.; Mimoto, T.; Kurata, T.; Ikeda, Y.; Abe, K. Early and selective reduction of NOP56 (Asidan) and RNA processing proteins in the motor neuron of ALS model mice. Neurol. Res. 2013, 35, 744–754. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Yuan, Y.; Liu, Z.; Hou, X.; Li, W.; Ni, J.; Huang, L.; Hu, Y.; Liu, P.; Hou, X.; Xue, J.; et al. Identification of GGC repeat expansion in the NOTCH2NLC gene in amyotrophic lateral sclerosis. Neurology 2020, 95, e3394–e3405. [Google Scholar] [CrossRef] [PubMed]
- Manini, A.; Gagliardi, D.; Meneri, M.; Antognozzi, S.; Del Bo, R.; Comi, G.P.; Corti, S.; Ronchi, D. NOTCH2NLC GGC repeats are not expanded in Italian amyotrophic lateral sclerosis patients. Sci. Rep. 2023, 13, 3187. [Google Scholar] [CrossRef]
- Sone, J.; Mori, K.; Inagaki, T.; Katsumata, R.; Takagi, S.; Yokoi, S.; Araki, K.; Kato, T.; Nakamura, T.; Koike, H.; et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain 2016, 139, 3170–3186. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wang, J.L.; Huang, W.; Zeng, S.; Jiao, B.; Liu, Z.; Chen, Z.; Li, Y.; Wang, Y.; Min, H.X.; et al. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet. 2019, 105, 166–176. [Google Scholar] [CrossRef]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Hannaford, A.; Pavey, N.; van den Bos, M.; Geevasinga, N.; Menon, P.; Shefner, J.M.; Kiernan, M.C.; Vucic, S. Diagnostic Utility of Gold Coast Criteria in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2021, 89, 979–986. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, C.; Sprovieri, T.; Morello, G.; Perrone, B.; Spampinato, A.G.; Simone, I.L.; Trojsi, F.; Monsurro, M.R.; Spataro, R.; La Bella, V.; et al. Genetic investigation of amyotrophic lateral sclerosis patients in south Italy: A two-decade analysis. Neurobiol. Aging 2021, 99, 99.e7–99.e14. [Google Scholar] [CrossRef]
- Garcia-Murias, M.; Quintans, B.; Arias, M.; Seixas, A.I.; Cacheiro, P.; Tarrio, R.; Pardo, J.; Millan, M.J.; Arias-Rivas, S.; Blanco-Arias, P.; et al. C’osta da Morte’ ataxia is spinocerebellar ataxia 36: Clinical and genetic characterization. Brain 2012, 135, 1423–1435. [Google Scholar] [CrossRef]
- Kimura, F.; Fujimura, C.; Ishida, S.; Nakajima, H.; Furutama, D.; Uehara, H.; Shinoda, K.; Sugino, M.; Hanafusa, T. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006, 66, 265–267. [Google Scholar] [CrossRef]
- Kjaeldgaard, A.L.; Pilely, K.; Olsen, K.S.; Jessen, A.H.; Lauritsen, A.O.; Pedersen, S.W.; Svenstrup, K.; Karlsborg, M.; Thagesen, H.; Blaabjerg, M.; et al. Prediction of survival in amyotrophic lateral sclerosis: A nationwide, Danish cohort study. BMC Neurol. 2021, 21, 164. [Google Scholar] [CrossRef] [PubMed]
- Hannaford, A.; Byth, K.; Pavey, N.; Henderson, R.D.; Mathers, S.; Needham, M.; Schultz, D.; Menon, P.; Kiernan, M.C.; Vucic, S. Clinical and neurophysiological biomarkers of disease progression in amyotrophic lateral sclerosis. Muscle Nerve 2023, 67, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Leon, J. Pearson Winsorized: A robust coefficient for correlations with small samples. Rev. Chil. Pediatr. 2020, 91, 642–643. [Google Scholar] [CrossRef] [PubMed]
- Tazelaar, G.H.P.; Dekker, A.M.; van Vugt, J.; van der Spek, R.A.; Westeneng, H.J.; Kool, L.; Kenna, K.P.; van Rheenen, W.; Pulit, S.L.; McLaughlin, R.L.; et al. Association of NIPA1 repeat expansions with amyotrophic lateral sclerosis in a large international cohort. Neurobiol. Aging 2019, 74, 234.e9–234.e15. [Google Scholar] [CrossRef] [PubMed]
- Tazelaar, G.H.P.; Boeynaems, S.; De Decker, M.; van Vugt, J.; Kool, L.; Goedee, H.S.; McLaughlin, R.L.; Sproviero, W.; Iacoangeli, A.; Moisse, M.; et al. ATXN1 repeat expansions confer risk for amyotrophic lateral sclerosis and contribute to TDP-43 mislocalization. Brain Commun. 2020, 2, fcaa064. [Google Scholar] [CrossRef] [PubMed]
- Dekker, A.M.; Seelen, M.; van Doormaal, P.T.; van Rheenen, W.; Bothof, R.J.; van Riessen, T.; Brands, W.J.; van der Kooi, A.J.; de Visser, M.; Voermans, N.C.; et al. Large-scale screening in sporadic amyotrophic lateral sclerosis identifies genetic modifiers in C9orf72 repeat carriers. Neurobiol. Aging 2016, 39, 220.e9–220.e15. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Brunetti, M.; Di Pierro, A.; Barberis, M.; Croce, R.; Bersano, E.; De Marchi, F.; Zuccala, M.; Barizzone, N.; Calvo, A.; et al. Analysis of the GCG repeat length in NIPA1 gene in C9orf72-mediated ALS in a large Italian ALS cohort. Neurol. Sci. 2019, 40, 2537–2540. [Google Scholar] [CrossRef] [PubMed]
- van Blitterswijk, M.; Mullen, B.; Heckman, M.G.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; Murray, M.E.; Hsiung, G.Y.; Stewart, H.; Karydas, A.M.; et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 2014, 35, 2421.e13–2421.e17. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Abe, K.; Matsuura, T.; Ikeda, Y.; Hitomi, T.; Akechi, Y.; Habu, T.; Liu, W.; Okuda, H.; Koizumi, A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 2011, 89, 121–130. [Google Scholar] [CrossRef]
- Nel, M.; Mavundla, T.; Gultig, K.; Botha, G.; Mulder, N.; Benatar, M.; Wuu, J.; Cooley, A.; Myers, J.; Rampersaud, E.; et al. Repeats expansions in ATXN2, NOP56, NIPA1 and ATXN1 are not associated with ALS in Africans. IBRO Neurosci. Rep. 2021, 10, 130–135. [Google Scholar] [CrossRef]
- Maharjan, N.; Saxena, S. It Takes Two to Tango: DPRs in ALS and SCA36. Neuron 2020, 107, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Doi, H.; Fukai, R.; Fujita, A.; Mitsuhashi, S.; Hashiguchi, S.; Kishida, H.; Ueda, N.; Morihara, K.; Ogasawara, A.; et al. GGC Repeat Expansion of NOTCH2NLC in Adult Patients with Leukoencephalopathy. Ann. Neurol. 2019, 86, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.Y.; Xu, Q.; Tian, Y.; Hu, Z.M.; Qin, L.X.; Yang, J.X.; Huang, W.; Xue, J.; Li, J.C.; Zeng, S.; et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain 2020, 143, 222–233. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| South Italian ALS Cohort (n = 302) | |
|---|---|
| Age at onset, years, mean (± SD) | 61.6 (± 12.6) |
| Male, n (%) | 163 (54%) |
| Sporadic ALS, n (%) | 275 (91%) |
| Familiar ALS, n (%) | 27 (9%) |
| C9orf72-ALS, n (%) | 16 (5.3%) |
| FUS-ALS, n (%) | 2 (0.7%) |
| SOD1-ALS, n (%) | 1 (0.3%) |
| TARDBP-ALS | 6 (2%) |
| Family history of NDs, n (%) | |
| Yes | 68 (22.5%) |
| No | 234 (77.5%) |
| Region of symptom onset, n (%) | |
| Spinal | 209 (69.2%) |
| Bulbar | 61 (20.2%) |
| Repeat Alleles | ALS | Controls | p-Value | Odd Ratio |
|---|---|---|---|---|
| NIPA1 | n (%) | n (%) | ||
| Short (7 repeats) | 101 (17.1%) | 48 (14.4%) | 0.39 | 0.1 |
| Normal (8 repeats) | 476 (80.7%) | 279 (85.5%) | ||
| Long (≥9 repeats) | 13 (2.2%) | 5 (1.5%) | ||
| NOP56 | ||||
| 8 repeats | 66 (11.1%) | 20 (5.98%) | 0.40 | 0.93 |
| ≥9 repeats | 10 (1.7%) | 3 (0.9%) | ||
| NOTCH2NLC | ||||
| 9 repeats | 335 (56.8%) | 158 (47.3%) | 0.42 | 0.8 |
| ≥15 repeats | 7 (1.2%) | 2 (0.6%) |
| NIPA1 | ||||||
| GCG repeat allele | Cohort | Age of onset (mean *) | p-value (Paired t-test) | Gender | p-value (Fisher’s exact test) | OR (Batista Pike) |
| Short (7) | ALS | 61.74 (13.9) | 0.2544 | 43 M 47 F | 0.7137 | 0.835 |
| CTRLs | 63.07 (16.8) | 23 M 21 F | ||||
| Normal (8) | ALS | 61.45 (12.1) | 149 M 128 F | 0.4251 | 1.192 | |
| CTRLs | 63.07 (16.8) | 78 M 79 F | ||||
| Long (≥9) | ALS | 58.8 (11.7) | 9 M 4 F | 0.2545 | 3.375 | |
| CTRLs | 57.9 (15.1) | 2 M 3 F | ||||
| NOP56 | ||||||
| GGCCTG repeat allele | Cohort | Age of onset (mean *) | p-value (Paired t-test) | Gender | p-value (Fisher’s exact test) | OR (Batista Pike) |
| 8 REs | ALS | 65.08 (10.1) | 0.6837 | 32 M 30 F | 0.2118 | 1.981 |
| CTRLs | 63.13 (16.7) | 7 M 13 F | ||||
| ≥9 REs | ALS | 61.27 (13.4) | 4 M 6 F | 0.5594 | 0.3333 | |
| CTRLs | 62.86 (16.3) | 2 M 1 F | ||||
| NOTCH2NLC | ||||||
| GGC repeat allele | Cohort | Age of onset (mean *) | p-value (Paired t-test) | Gender | p-value (Fisher’s exact test) | OR (Batista Pike) |
| 9 REs | ALS | 61.66 (12.7) | 0.3634 | 95 M 101 F | 0.4084 | 0.8 |
| CTRLs | 62.9 (16.6) | 61 M 52 F | ||||
| ≥15 REs | ALS | 61.05 (13.9) | 4 M 3 F | 0.5 | 0 | |
| CTRLs | 72.8 (12.5) | 2 M 0 F | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruffo, P.; De Amicis, F.; La Bella, V.; Conforti, F.L. Investigating Repeat Expansions in NIPA1, NOP56, and NOTCH2NLC Genes: A Closer Look at Amyotrophic Lateral Sclerosis Patients from Southern Italy. Cells 2024, 13, 677. https://doi.org/10.3390/cells13080677
Ruffo P, De Amicis F, La Bella V, Conforti FL. Investigating Repeat Expansions in NIPA1, NOP56, and NOTCH2NLC Genes: A Closer Look at Amyotrophic Lateral Sclerosis Patients from Southern Italy. Cells. 2024; 13(8):677. https://doi.org/10.3390/cells13080677
Chicago/Turabian StyleRuffo, Paola, Francesca De Amicis, Vincenzo La Bella, and Francesca Luisa Conforti. 2024. "Investigating Repeat Expansions in NIPA1, NOP56, and NOTCH2NLC Genes: A Closer Look at Amyotrophic Lateral Sclerosis Patients from Southern Italy" Cells 13, no. 8: 677. https://doi.org/10.3390/cells13080677
APA StyleRuffo, P., De Amicis, F., La Bella, V., & Conforti, F. L. (2024). Investigating Repeat Expansions in NIPA1, NOP56, and NOTCH2NLC Genes: A Closer Look at Amyotrophic Lateral Sclerosis Patients from Southern Italy. Cells, 13(8), 677. https://doi.org/10.3390/cells13080677