The Role Played by Autophagy in FcεRI-Dependent Activation of Mast Cells

 ,
,  and
and 
Abstract
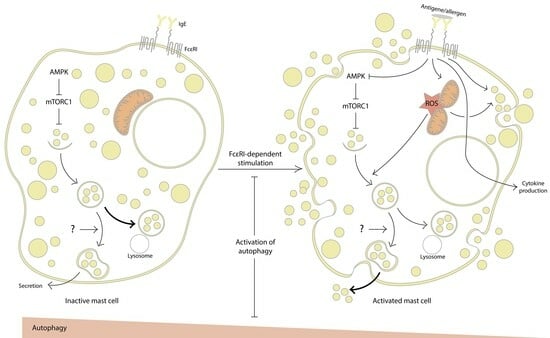
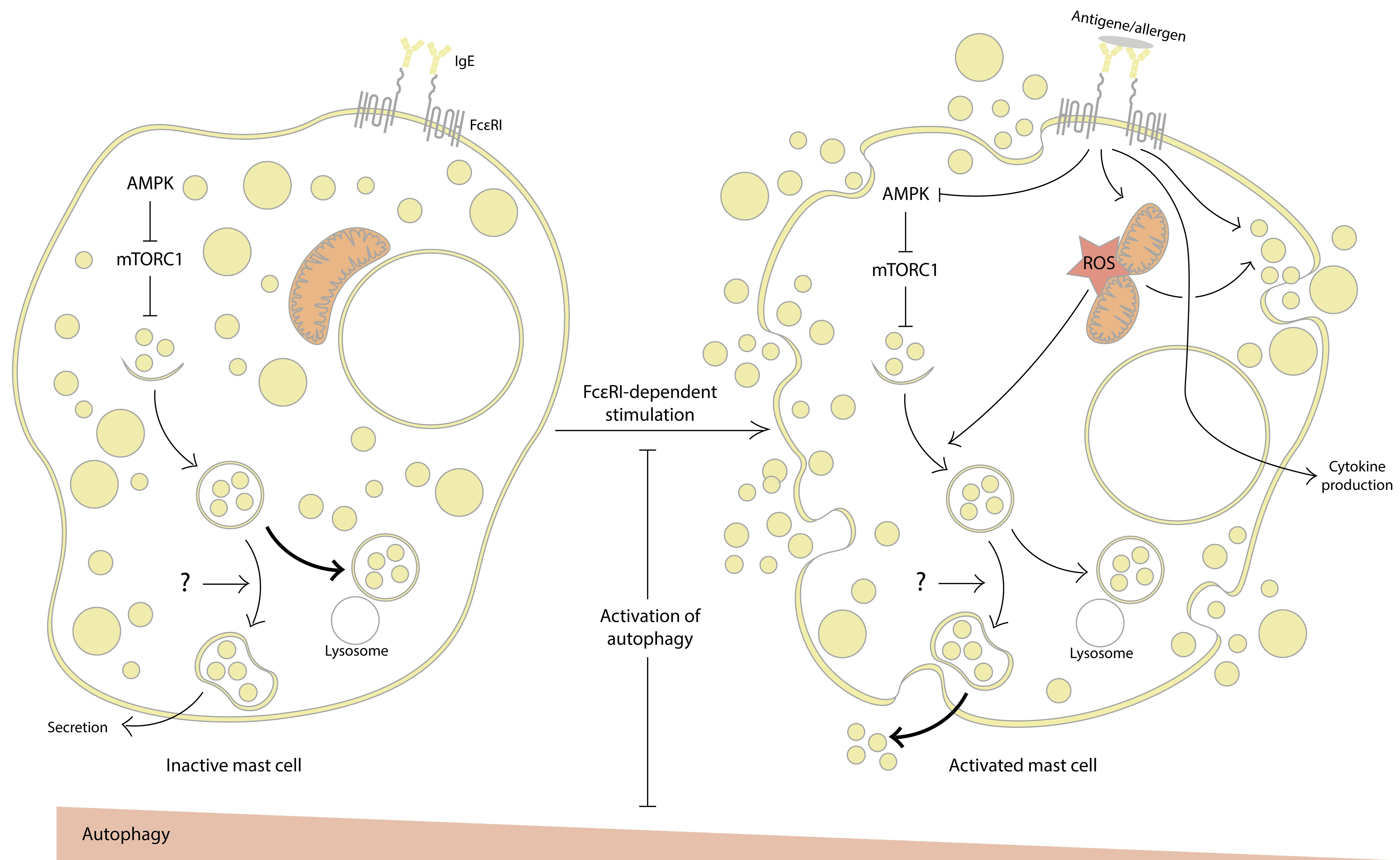
1. Introduction
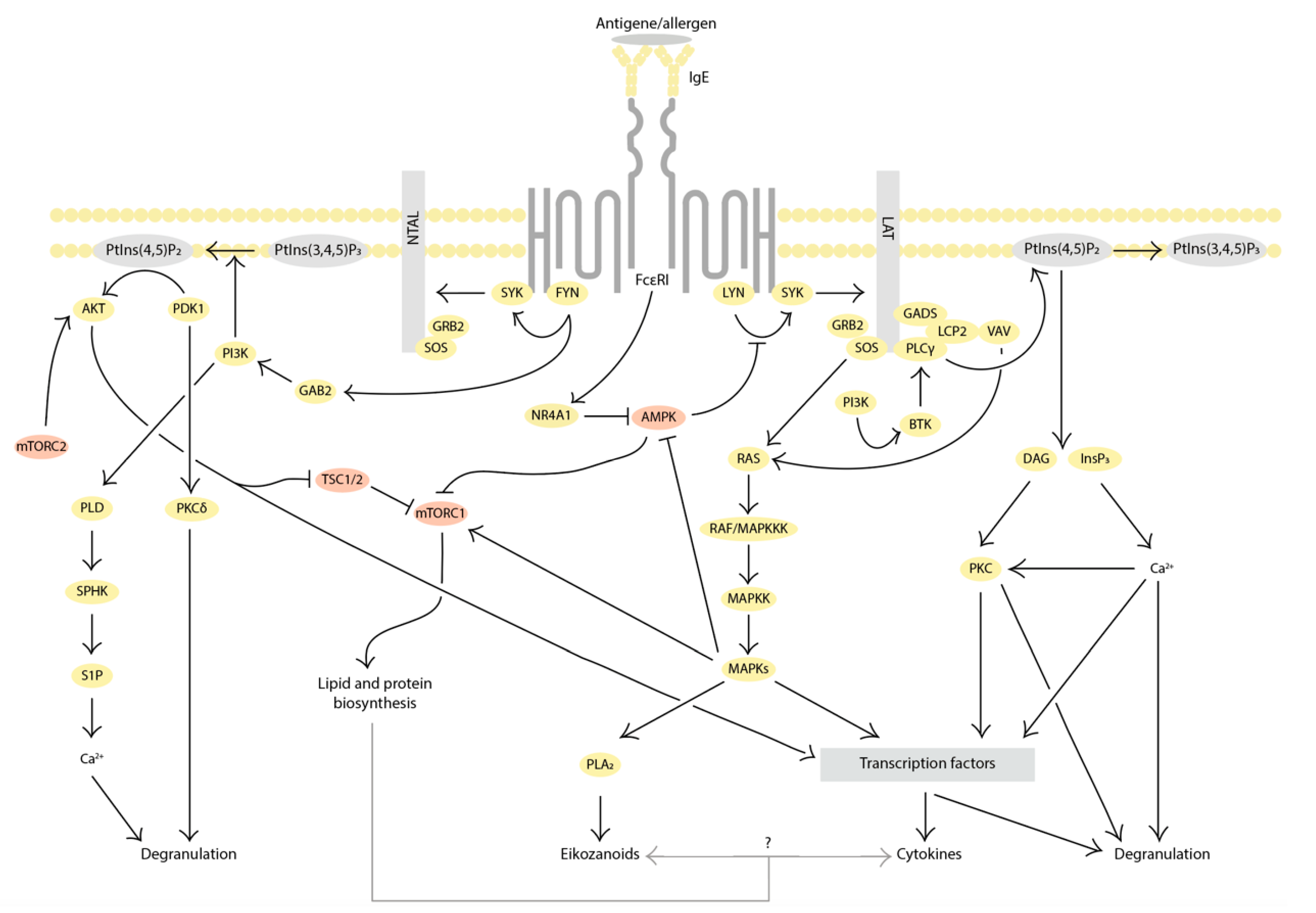
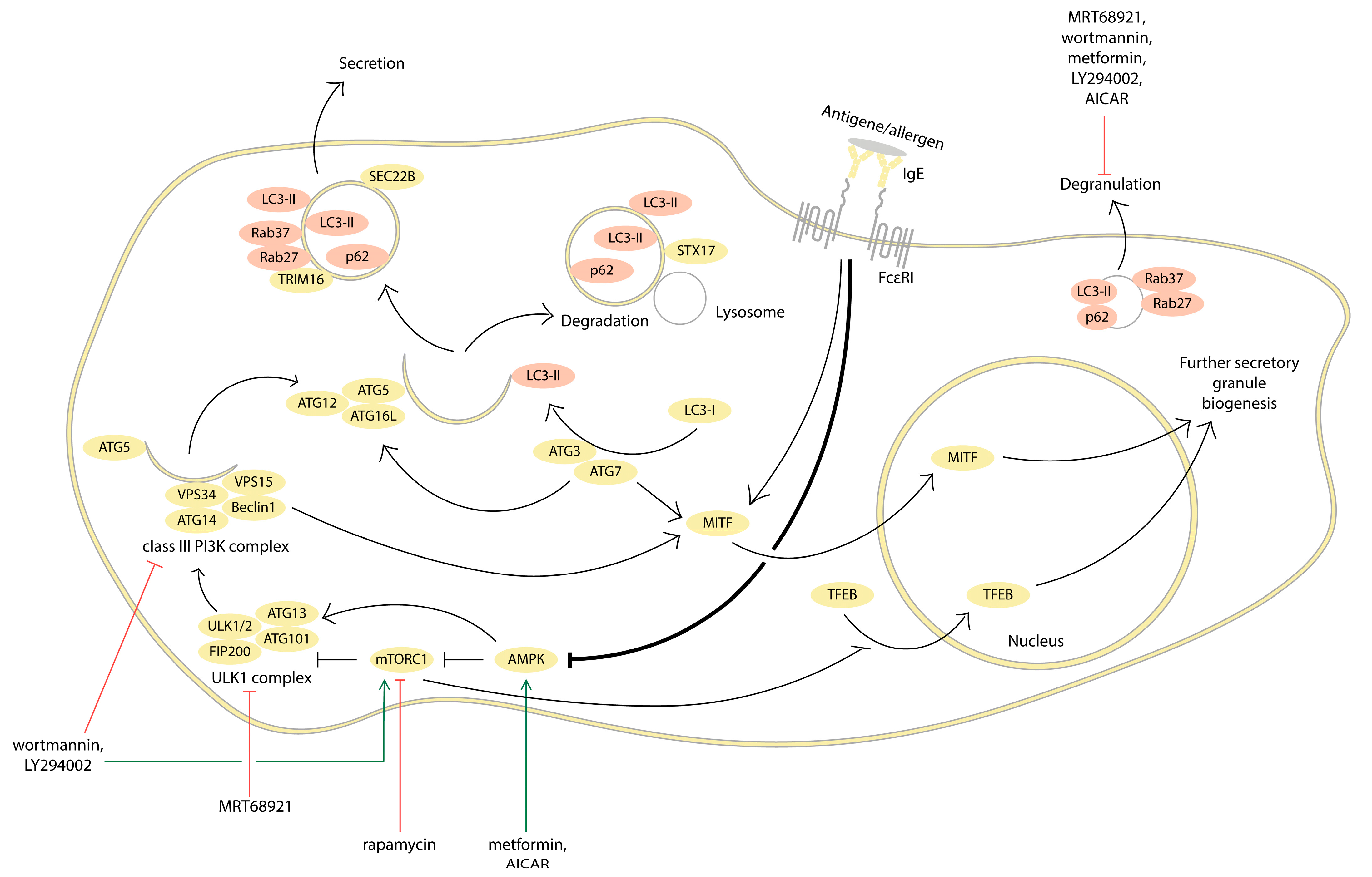
2. Evidence for the Involvement of Autophagy in MC Activation
2.1. Inhibition of Autophagy Promotes MC Activation
2.2. Autophagy Activation Interferes with MC Activation
3. Mitochondria at the Crossroads of Autophagy and MC Activation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- da Silva, E.Z.M.; Jamur, M.C.; Oliver, C. Mast Cell Function: A New Vision of an Old Cell. J. Histochem. Cytochem. 2014, 62, 698–738. [Google Scholar] [CrossRef] [PubMed]
- Sismanopoulos, N.; Delivanis, D.-A.; Alysandratos, K.-D.; Angelidou, A.; Therianou, A.; Kalogeromitros, D.; Theoharides, T.C. Mast Cells in Allergic and Inflammatory Diseases. Curr. Pharm. Des. 2012, 18, 2261–2277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kurashima, Y. Two Sides of the Coin: Mast Cells as a Key Regulator of Allergy and Acute/Chronic Inflammation. Cells 2021, 10, 1615. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, S.; Pejler, G. Mast Cell Secretory Granules: Armed for Battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- McLeod, J.J.A.; Baker, B.; Ryan, J.J. Mast Cell Production and Response to IL-4 and IL-13. Cytokine 2015, 75, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Kaplan, M.H. Transcription Factors in the Development and Pro-Allergic Function of Mast Cells. Front Allergy 2021, 2, 679121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Leung, P.S.C.; Gershwin, M.E.; Song, J. New Mechanistic Advances in FcεRI-Mast Cell-Mediated Allergic Signaling. Clin. Rev. Allergy Immunol. 2022, 63, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast Cell Mediators: Their Differential Release and the Secretory Pathways Involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Kuehn, H.S.; Metcalfe, D.D.; Gilfillan, A.M. Activation and Function of the mTORC1 Pathway in Mast Cells1. J. Immunol. 2008, 180, 4586–4595. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shaw, R.J. AMPK Control of mTOR Signaling and Growth. In Structure, Function and Regulation of Tor Complexes from Yeasts to Mammals Part B; The enzymes; Elsevier: Amsterdam, The Netherlands, 2010; pp. 49–75. ISBN 9780123810052. [Google Scholar]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- New, J.; Thomas, S.M. Autophagy-Dependent Secretion: Mechanism, Factors Secreted, and Disease Implications. Autophagy 2019, 15, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The Mechanisms and Roles of Selective Autophagy in Mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 Ubiquitin-like Conjugation Systems in Macroautophagy. “Protein Modifications: Beyond the Usual Suspects” Review Series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-W.; Chu, M.-L.; Liu, H.-S. Autophagy and Metabolism. Kaohsiung J. Med. Sci. 2021, 37, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Solvik, T.A.; Nguyen, T.A.; Tony Lin, Y.-H.; Marsh, T.; Huang, E.J.; Wiita, A.P.; Debnath, J.; Leidal, A.M. Secretory Autophagy Maintains Proteostasis upon Lysosome Inhibition. J. Cell Biol. 2022, 221, e202110151. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.; McCaffery, J.M.; Curwin, A.J.; Duran, J.M.; Malhotra, V. Biogenesis of a Novel Compartment for Autophagosome-Mediated Unconventional Protein Secretion. J. Cell Biol. 2011, 195, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A Key Role for Autophagy and the Autophagy Gene Atg16l1 in Mouse and Human Intestinal Paneth Cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Torisu, T.; Torisu, K.; Lee, I.H.; Liu, J.; Malide, D.; Combs, C.A.; Wu, X.S.; Rovira, I.I.; Fergusson, M.M.; Weigert, R.; et al. Autophagy Regulates Endothelial Cell Processing, Maturation and Secretion of von Willebrand Factor. Nat. Med. 2013, 19, 1281–1287. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy Proteins Regulate the Secretory Component of Osteoclastic Bone Resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Ushio, H.; Ueno, T.; Kojima, Y.; Komatsu, M.; Tanaka, S.; Yamamoto, A.; Ichimura, Y.; Ezaki, J.; Nishida, K.; Komazawa-Sakon, S.; et al. Crucial Role for Autophagy in Degranulation of Mast Cells. J. Allergy Clin. Immunol. 2011, 127, 1267–1276.e6. [Google Scholar] [CrossRef]
- Kim, M.; Park, Y.; Kwon, Y.; Kim, Y.; Byun, J.; Jeong, M.S.; Kim, H.-U.; Jung, H.S.; Mun, J.Y.; Jeoung, D. MiR-135-5p-p62 Axis Regulates Autophagic Flux, Tumorigenic Potential, and Cellular Interactions Mediated by Extracellular Vesicles during Allergic Inflammation. Front. Immunol. 2019, 10, 738. [Google Scholar] [CrossRef]
- Huang, H.; Li, Y.; Liu, B. Transcriptional Regulation of Mast Cell and Basophil Lineage Commitment. Semin. Immunopathol. 2016, 38, 539–548. [Google Scholar] [CrossRef]
- Guo, Y.; Proaño-Pérez, E.; Muñoz-Cano, R.; Martin, M. Anaphylaxis: Focus on Transcription Factor Activity. Int. J. Mol. Sci. 2021, 22, 4935. [Google Scholar] [CrossRef]
- Lee, K.W.; Kim, M.; Lee, S.H.; Kim, K.D. The Function of Autophagy as a Regulator of Melanin Homeostasis. Cells 2022, 11, 2085. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-L.; Li, X.; Lu, Y.; Jin, Y.; Jeong, Y.-T.; Kim, Y.D.; Lee, I.-K.; Taketomi, Y.; Sato, H.; Cho, Y.S.; et al. AMP-Activated Protein Kinase Negatively Regulates FcεRI-Mediated Mast Cell Signaling and Anaphylaxis in Mice. J. Allergy Clin. Immunol. 2013, 132, 729–736.e12. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Li, X.; Deng, Y.; Timilshina, M.; Huang, B.; Kim, D.-Y.; Chang, J.-H.; Ichinose, H.; Baek, S.-H.; Murakami, M.; et al. The Orphan Nuclear Receptor NR4A1 Promotes FcεRI-Stimulated Mast Cell Activation and Anaphylaxis by Counteracting the Inhibitory LKB1/AMPK Axis. Allergy 2019, 74, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, L.; Jiang, J.; Li, L.; Li, J.; Xu, C.; Jin, S.; Zhu, L.; Yan, G. Pterostilbene Inhibits FcεRI Signaling through Activation of the LKB1/AMPK Pathway in Allergic Response. J. Agric. Food Chem. 2020, 68, 3456–3465. [Google Scholar] [CrossRef]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-Monitoring and Autophagy-Deficient Mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef]
- Whitmarsh-Everiss, T.; Laraia, L. Small Molecule Probes for Targeting Autophagy. Nat. Chem. Biol. 2021, 17, 653–664. [Google Scholar] [CrossRef]
- Takayama, G.; Ohtani, M.; Minowa, A.; Matsuda, S.; Koyasu, S. Class I PI3K-Mediated Akt and ERK Signals Play a Critical Role in FcεRI-Induced Degranulation in Mast Cells. Int. Immunol. 2013, 25, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Rådinger, M.; Gilfillan, A.M. The Multiple Roles of Phosphoinositide 3-Kinase in Mast Cell Biology. Trends Immunol. 2008, 29, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.-U. Regulation of the Innate Immune System by Autophagy: Monocytes, Macrophages, Dendritic Cells and Antigen Presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Goishi, K.; Mizuno, K.; Nakanishi, H.; Sasaki, T. Involvement of Rab27 in Antigen-Induced Histamine Release from Rat Basophilic Leukemia 2H3 Cells. Biochem. Biophys. Res. Commun. 2004, 324, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Tolmachova, T.; Ushakov, D.S.; Romao, M.; Abrink, M.; Ferenczi, M.A.; Raposo, G.; Seabra, M.C. Rab27b Regulates Mast Cell Granule Dynamics and Secretion. Traffic 2007, 8, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Feng, W.; Wu, F.; Gao, Y.; Sun, Q.; Hu, N.; Lu, W.; Zhou, J. MiR-135-5p Inhibits TGF-β-Induced Epithelial-Mesenchymal Transition and Metastasis by Targeting SMAD3 in Breast Cancer. J. Cancer 2020, 11, 6402–6412. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Zhu, L.; Ding, L.; Yuan, J.; Du, L.; Pan, M.; Xue, F.; Xiao, H. MiR-135-5p Promotes Osteoblast Differentiation by Targeting HIF1AN in MC3T3-E1 Cells. Cell. Mol. Biol. Lett. 2019, 24, 51. [Google Scholar] [CrossRef] [PubMed]
- Rakhmanova, V.; Jin, M.; Shin, J. Inhibition of Mast Cell Function and Proliferation by mTOR Activator MHY1485. Immune Netw. 2018, 18, e18. [Google Scholar] [CrossRef] [PubMed]
- Rakhmanova, V.; Park, S.; Lee, S.; Kim, Y.H.; Shin, J. 3-Benzyl-5-((2-Nitrophenoxy) Methyl)-Dihydrofuran-2(3H)-One Suppresses FcεRI-Mediated Mast Cell Degranulation via the Inhibition of mTORC2-Akt Signaling. Biochem. Biophys. Res. Commun. 2020, 521, 72–76. [Google Scholar] [CrossRef]
- Shin, J.; Pan, H.; Zhong, X.-P. Regulation of Mast Cell Survival and Function by Tuberous Sclerosis Complex 1. Blood 2012, 119, 3306–3314. [Google Scholar] [CrossRef]
- Iskarpatyoti, J.A.; Shi, J.; Abraham, M.A.; Rathore, A.P.S.; Miao, Y.; Abraham, S.N. Mast Cell Regranulation Requires a Metabolic Switch Involving mTORC1 and a Glucose-6-Phosphate Transporter. Cell Rep. 2022, 40, 111346. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Shimabukuro-Demoto, S.; Tsutsui, H.; Toyama-Sorimachi, N. Type I Interferon Limits Mast Cell-Mediated Anaphylaxis by Controlling Secretory Granule Homeostasis. PLoS Biol. 2019, 17, e3000530. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Tsutsui, H.; Shimabukuro-Demoto, S.; Yoshida-Sugitani, R.; Karyu, H.; Furuyama-Tanaka, K.; Ohshima, D.; Kato, N.; Okamura, T.; Toyama-Sorimachi, N. Lysosome Biogenesis Regulated by the Amino-Acid Transporter SLC15A4 Is Critical for Functional Integrity of Mast Cells. Int. Immunol. 2017, 29, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Ishimaru, K.; Kobayashi, A.; Yu, G.; Nakamura, Y.; Oh-Oka, K.; Suzuki-Inoue, K.; Kono, K.; Nakao, A. Resveratrol Inhibits IL-33-Mediated Mast Cell Activation by Targeting the MK2/3-PI3K/Akt Axis. Sci. Rep. 2019, 9, 18423. [Google Scholar] [CrossRef] [PubMed]
- Nian, J.-B.; Zeng, M.; Zheng, J.; Zeng, L.-Y.; Fu, Z.; Huang, Q.-J.; Wei, X. Epithelial Cells Expressed IL-33 to Promote Degranulation of Mast Cells through Inhibition on ST2/PI3K/mTOR-Mediated Autophagy in Allergic Rhinitis. Cell Cycle 2020, 19, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Lazki-Hagenbach, P.; Ali, H.; Sagi-Eisenberg, R. Authentic and Ectopically Expressed MRGPRX2 Elicit Similar Mechanisms to Stimulate Degranulation of Mast Cells. Cells 2021, 10, 376. [Google Scholar] [CrossRef]
- Wang, B.; Kundu, M. Canonical and Noncanonical Functions of ULK/Atg1. Curr. Opin. Cell Biol. 2017, 45, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Henkel, G.; Brown, M.A. PU.1 and GATA: Components of a Mast Cell-Specific Interleukin 4 Intronic Enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Rivera Vargas, T.; Cai, Z.; Shen, Y.; Dosset, M.; Benoit-Lizon, I.; Martin, T.; Roussey, A.; Flavell, R.A.; Ghiringhelli, F.; Apetoh, L. Selective Degradation of PU.1 during Autophagy Represses the Differentiation and Antitumour Activity of T9 Cells. Nat. Commun. 2017, 8, 559. [Google Scholar] [CrossRef]
- Chen, Y.; Pappu, B.P.; Zeng, H.; Xue, L.; Morris, S.W.; Lin, X.; Wen, R.; Wang, D. B Cell Lymphoma 10 Is Essential for FcεR-Mediated Degranulation and IL-6 Production in Mast Cells1. J. Immunol. 2007, 178, 49–57. [Google Scholar] [CrossRef]
- Paul, S.; Kashyap, A.K.; Jia, W.; He, Y.-W.; Schaefer, B.C. Selective Autophagy of the Adaptor Protein Bcl10 Modulates T Cell Receptor Activation of NF-κB. Immunity 2012, 36, 947–958. [Google Scholar] [CrossRef]
- Reid, S.E.; Kolapalli, S.P.; Nielsen, T.M.; Frankel, L.B. Canonical and Non-Canonical Roles for ATG8 Proteins in Autophagy and beyond. Front. Mol. Biosci. 2022, 9, 1074701. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Huang, S.-K. Metformin Inhibits IgE- and Aryl Hydrocarbon Receptor-Mediated Mast Cell Activation in Vitro and in Vivo. Eur. J. Immunol. 2018, 48, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Jin, Q.; Guo, H.; Han, X.; Xu, L.; Lu, S.; Wu, C. Metformin Ameliorates Inflammation and Airway Remodeling of Experimental Allergic Asthma in Mice by Restoring AMPK Activity. Front. Pharmacol. 2022, 13, 780148. [Google Scholar] [CrossRef]
- Smrž, D.; Kim, M.-S.; Zhang, S.; Mock, B.A.; Smržová, Š.; DuBois, W.; Simakova, O.; Maric, I.; Wilson, T.M.; Metcalfe, D.D.; et al. mTORC1 and mTORC2 Differentially Regulate Homeostasis of Neoplastic and Non-Neoplastic Human Mast Cells. Blood 2011, 118, 6803–6813. [Google Scholar] [CrossRef]
- Noh, S.H.; Kim, Y.J.; Lee, M.G. Autophagy-Related Pathways in Vesicular Unconventional Protein Secretion. Front. Cell Dev. Biol. 2022, 10, 892450. [Google Scholar] [CrossRef]
- Li, J.; Ullah, M.A.; Jin, H.; Liang, Y.; Lin, L.; Wang, J.; Peng, X.; Liao, H.; Li, Y.; Ge, Y.; et al. ORMDL3 Functions as a Negative Regulator of Antigen-Mediated Mast Cell Activation an ATF6-UPR-Autophagy-Dependent Pathway. Front. Immunol. 2021, 12, 604974. [Google Scholar] [CrossRef]
- James, B.; Milstien, S.; Spiegel, S. ORMDL3 and Allergic Asthma: From Physiology to Pathology. J. Allergy Clin. Immunol. 2019, 144, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Su, R.; Gu, Q.; Hu, Y.; Yang, H. PI3K/AKT-Mediated Autophagy Inhibition Facilitates Mast Cell Activation to Enhance Severe Inflammatory Lung Injury in Influenza A Virus- and Secondary Staphylococcus Aureus-Infected Mice. Antivir. Res. 2023, 209, 105502. [Google Scholar] [CrossRef]
- Hirasawa, N.; Sato, Y.; Fujita, Y.; Ohuchi, K. Involvement of a Phosphatidylinositol 3-Kinase-p38 Mitogen Activated Protein Kinase Pathway in Antigen-Induced IL-4 Production in Mast Cells. Biochim. Biophys. Acta 2000, 1456, 45–55. [Google Scholar] [CrossRef]
- Park, J.-W.; Jeon, Y.J.; Lee, J.C.; Ahn, S.R.; Ha, S.W.; Bang, S.Y.; Park, E.K.; Yi, S.A.; Lee, M.G.; Han, J.-W. Destabilization of TNF-α mRNA by Rapamycin. Biomol. Ther. 2012, 20, 43–49. [Google Scholar] [CrossRef][Green Version]
- Chelombitko, M.A.; Chernyak, B.V.; Fedorov, A.V.; Zinovkin, R.A.; Razin, E.; Paruchuru, L.B. The Role Played by Mitochondria in FcεRI-Dependent Mast Cell Activation. Front. Immunol. 2020, 11, 584210. [Google Scholar] [CrossRef] [PubMed]
- Graef, M.; Nunnari, J. Mitochondria Regulate Autophagy by Conserved Signalling Pathways. EMBO J. 2011, 30, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Lippincott-Schwartz, J. Mechanisms of Mitochondria and Autophagy Crosstalk. Cell Cycle 2011, 10, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Mohr, F.C.; Fewtrell, C. The Relative Contributions of Extracellular and Intracellular Calcium to Secretion from Tumor Mast Cells. Multiple Effects of the Proton Ionophore Carbonyl Cyanide M-Chlorophenylhydrazone. J. Biol. Chem. 1987, 262, 10638–10643. [Google Scholar] [CrossRef]
- Erlich, T.H.; Yagil, Z.; Kay, G.; Peretz, A.; Migalovich-Sheikhet, H.; Tshori, S.; Nechushtan, H.; Levi-Schaffer, F.; Saada, A.; Razin, E. Mitochondrial STAT3 Plays a Major Role in IgE-Antigen-Mediated Mast Cell Exocytosis. J. Allergy Clin. Immunol. 2014, 134, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Sharkia, I.; Hadad Erlich, T.; Landolina, N.; Assayag, M.; Motzik, A.; Rachmin, I.; Kay, G.; Porat, Z.; Tshori, S.; Berkman, N.; et al. Pyruvate Dehydrogenase Has a Major Role in Mast Cell Function, and Its Activity Is Regulated by Mitochondrial Microphthalmia Transcription Factor. J. Allergy Clin. Immunol. 2017, 140, 204–214.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Weng, Z.; Sismanopoulos, N.; Asadi, S.; Therianou, A.; Alysandratos, K.-D.; Angelidou, A.; Shirihai, O.; Theoharides, T.C. Mitochondria Distinguish Granule-Stored from de Novo Synthesized Tumor Necrosis Factor Secretion in Human Mast Cells. Int. Arch. Allergy Immunol. 2012, 159, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Chelombitko, M.A.; Fedorov, A.V.; Ilyinskaya, O.P.; Zinovkin, R.A.; Chernyak, B.V. Role of Reactive Oxygen Species in Mast Cell Degranulation. Biochemistry 2016, 81, 1564–1577. [Google Scholar] [CrossRef]
- Chelombitko, M.A.; Averina, O.A.; Vasilyeva, T.V.; Pletiushkina, O.Y.; Popova, E.N.; Fedorov, A.V.; Chernyak, B.V.; Shishkina, V.S.; Ilinskaya, O.P. Mitochondria-Targeted Antioxidant SkQ1 (10-(6′-Plastoquinonyl)decyltriphenylphosphonium Bromide) Inhibits Mast Cell Degranulation in Vivo and in Vitro. Biochemistry 2017, 82, 1493–1503. [Google Scholar] [CrossRef]
- Pavlyuchenkova, A.N.; Zinovkin, R.A.; Makievskaya, C.I.; Galkin, I.I.; Chelombitko, M.A. Mitochondria-Targeted Triphenylphosphonium-Based Compounds Inhibit FcεRI-Dependent Degranulation of Mast Cells by Preventing Mitochondrial Dysfunction through Erk1/2. Life Sci. 2022, 288, 120174. [Google Scholar] [CrossRef] [PubMed]
- Pletjushkina, O.Y.; Lyamzaev, K.G.; Popova, E.N.; Nepryakhina, O.K.; Ivanova, O.Y.; Domnina, L.V.; Chernyak, B.V.; Skulachev, V.P. Effect of Oxidative Stress on Dynamics of Mitochondrial Reticulum. Biochim. Biophys. Acta 2006, 1757, 518–524. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Antonenko, Y.N.; Cherepanov, D.A.; Chernyak, B.V.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; Korshunova, G.A.; Lyamzaev, K.G.; Pletjushkina, O.Y.; et al. Prevention of Cardiolipin Oxidation and Fatty Acid Cycling as Two Antioxidant Mechanisms of Cationic Derivatives of Plastoquinone (SkQs). Biochim. Biophys. Acta 2010, 1797, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lam, G.Y.; Brumell, J.H. Autophagy Signaling through Reactive Oxygen Species. Antioxid. Redox Signal. 2011, 14, 2215–2231. [Google Scholar] [CrossRef]
- Agostini, F.; Bisaglia, M.; Plotegher, N. Linking ROS Levels to Autophagy: The Key Role of AMPK. Antioxidants 2023, 12, 1406. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-Activated Protein Kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.R.S.; Chandel, N.S. Hypoxic Activation of AMPK Is Dependent on Mitochondrial ROS but Independent of an Increase in AMP/ATP Ratio. Free Radic. Biol. Med. 2009, 46, 1386–1391. [Google Scholar] [CrossRef]
- Kim, J.-H.; Choi, T.G.; Park, S.; Yun, H.R.; Nguyen, N.N.Y.; Jo, Y.H.; Jang, M.; Kim, J.; Kim, J.; Kang, I.; et al. Mitochondrial ROS-Derived PTEN Oxidation Activates PI3K Pathway for mTOR-Induced Myogenic Autophagy. Cell Death Differ. 2018, 25, 1921–1937. [Google Scholar] [CrossRef]
- Hwang, S.-L.; Lu, Y.; Li, X.; Kim, Y.D.; Cho, Y.S.; Jahng, Y.; Son, J.-K.; Lee, Y.J.; Kang, W.; Taketomi, Y.; et al. ERK1/2 Antagonize AMPK-Dependent Regulation of FcεRI-Mediated Mast Cell Activation and Anaphylaxis. J. Allergy Clin. Immunol. 2014, 134, 714–721.e7. [Google Scholar] [CrossRef]
- Carriere, A.; Romeo, Y.; Acosta-Jaquez, H.A.; Moreau, J.; Bonneil, E.; Thibault, P.; Fingar, D.C.; Roux, P.P. ERK1/2 Phosphorylate Raptor to Promote Ras-Dependent Activation of mTOR Complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 567–577. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Substance | Target | Cell Line | Effect on the Degranulation | Effect on the Cytokines |
|---|---|---|---|---|
| Autophagy inhibitors | ||||
| Wortmannin (30–100 nM) | PI3K (inhibition) | RBL-2H3 | Suppression of IL-4 transcription and secretion [61] | |
| Wortmannin (100 nM) | BMMCs | Suppression [9,45] | Suppression of IL-6 secretion [9,45], IL-13, and TNF secretion [45] | |
| Wortmannin (500 nM) | RBL-2H3 | No effect on the TNF-α transcription level [62] | ||
| LY294002 (25 nM) | ||||
| LY294002 (5 μM) | BMMCs | Suppression [45] | Suppression of IL-6, IL-13, and TNF secretion [45] | |
| LY294002 (3–30 μM) | RBL-2H3 | Suppression of IL-4 transcription and secretion [61] | ||
| 3-methyladenine (5 mM) | LAD2 | Stimulation [46] | Stimulation of IL-4 and IL-6 cytokine production [46] | |
| MHY148 (2 μM) | mTORC1 (activation); mTORC2 (inhibition) | BMMCs | Suppression [39] | Suppression of TNF and IL-6 transcription and expression [39] |
| 3BDO (50 μM) | BMMCs | Suppression [40] | Suppression of TNF and IL-6 transcription and expression [40] | |
| MRT68921 (1 μM) | ULK1/2 (inhibition) | RBL-2H3 and LAD2 | Suppression [47] | |
| Autophagy activators | ||||
| Rapamycin (10 nM) | mTORC1 (inhibition) | RBL-2H3 | Destabilization of TNF-α mRNA [62] | |
| Rapamycin (100 nM) | BMMCs (IL-6) and MCs from peripheral blood (IL-8) | No effect [9] | Suppression of IL-6 and IL-8 secretion [9] | |
| Metformin (1–10 μM) | AMPK (activation) | BMMCs | Suppression [29] | Suppression of IL-13 and TNF-α secretion [29] |
| AICAR (1 mM) | BMMCs | Suppression [27] | Suppression of LTC4, PGD2, TNF-a, and IL-6 secretion [27] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlyuchenkova, A.N.; Smirnov, M.S.; Chernyak, B.V.; Chelombitko, M.A. The Role Played by Autophagy in FcεRI-Dependent Activation of Mast Cells. Cells 2024, 13, 690. https://doi.org/10.3390/cells13080690
Pavlyuchenkova AN, Smirnov MS, Chernyak BV, Chelombitko MA. The Role Played by Autophagy in FcεRI-Dependent Activation of Mast Cells. Cells. 2024; 13(8):690. https://doi.org/10.3390/cells13080690
Chicago/Turabian StylePavlyuchenkova, Anastasia N., Maxim S. Smirnov, Boris V. Chernyak, and Maria A. Chelombitko. 2024. "The Role Played by Autophagy in FcεRI-Dependent Activation of Mast Cells" Cells 13, no. 8: 690. https://doi.org/10.3390/cells13080690
APA StylePavlyuchenkova, A. N., Smirnov, M. S., Chernyak, B. V., & Chelombitko, M. A. (2024). The Role Played by Autophagy in FcεRI-Dependent Activation of Mast Cells. Cells, 13(8), 690. https://doi.org/10.3390/cells13080690