
Single-Cell RNA-Sequencing Reveals Heterogeneity and Transcriptional Dynamics in Porcine Circulating CD8+ T Cells

 ,
,
Abstract
: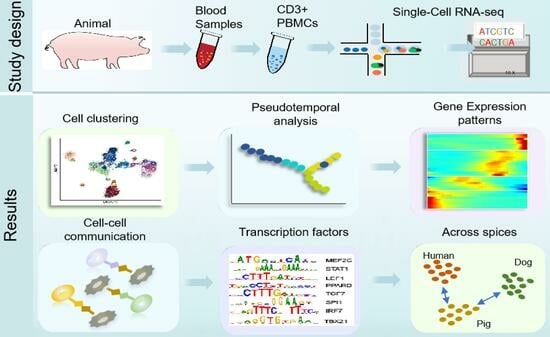
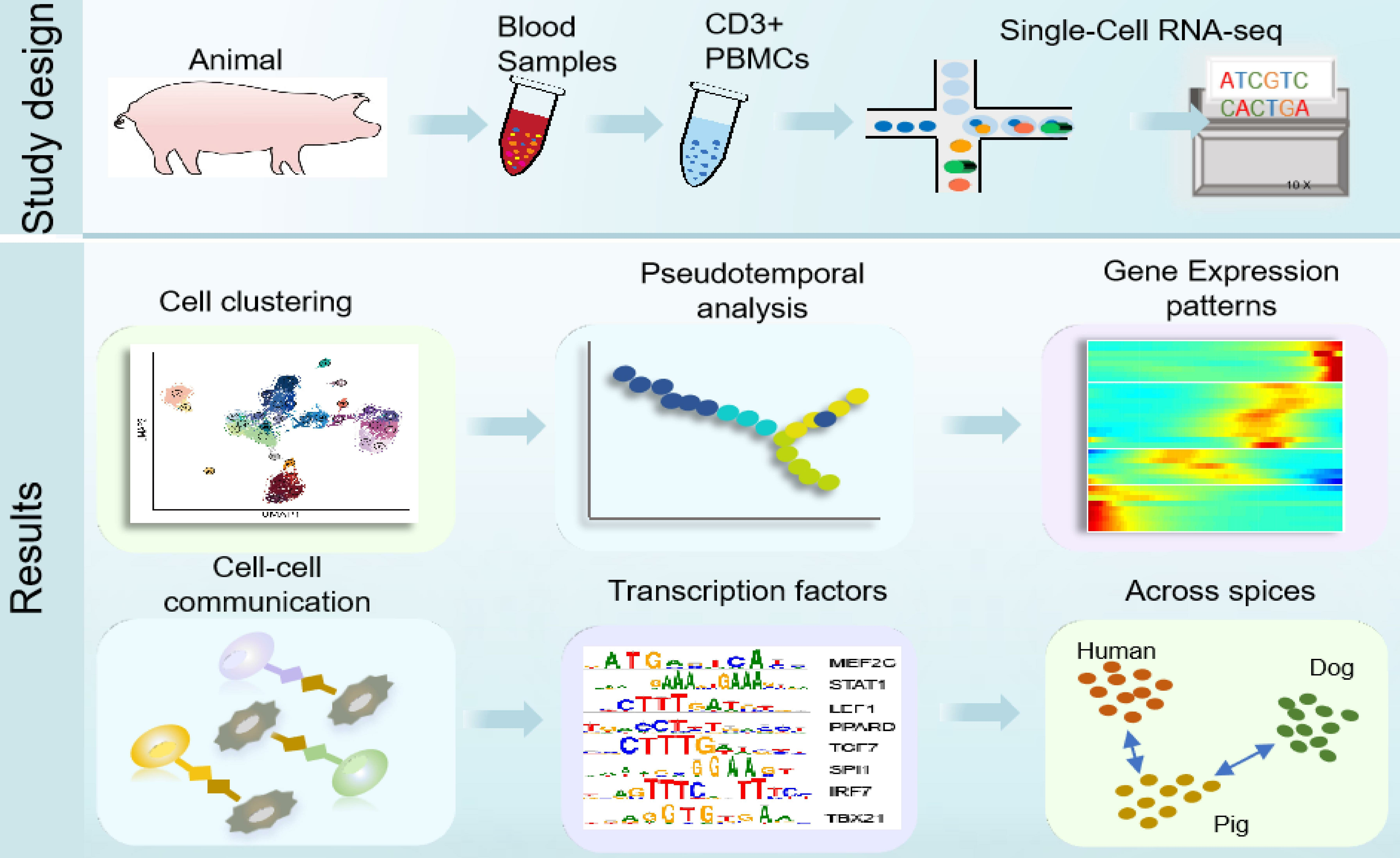{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Processing of scRNA-Seq Data
2.3. Identification of Cell Clusters
2.4. Differential Gene Expression Analysis
2.5. GO Enrichment Analyses
2.6. Gene Set Variation Analysis of CD8 T Cell Subtypes
2.7. Pseudotime Trajectory Analysis
2.8. Cell-Cell Communication Analysis with CellPhoneDB
2.9. Transcription Factor Analysis
2.10. Porcine-Human and Porcine-Canine Homology Analysis
3. Results
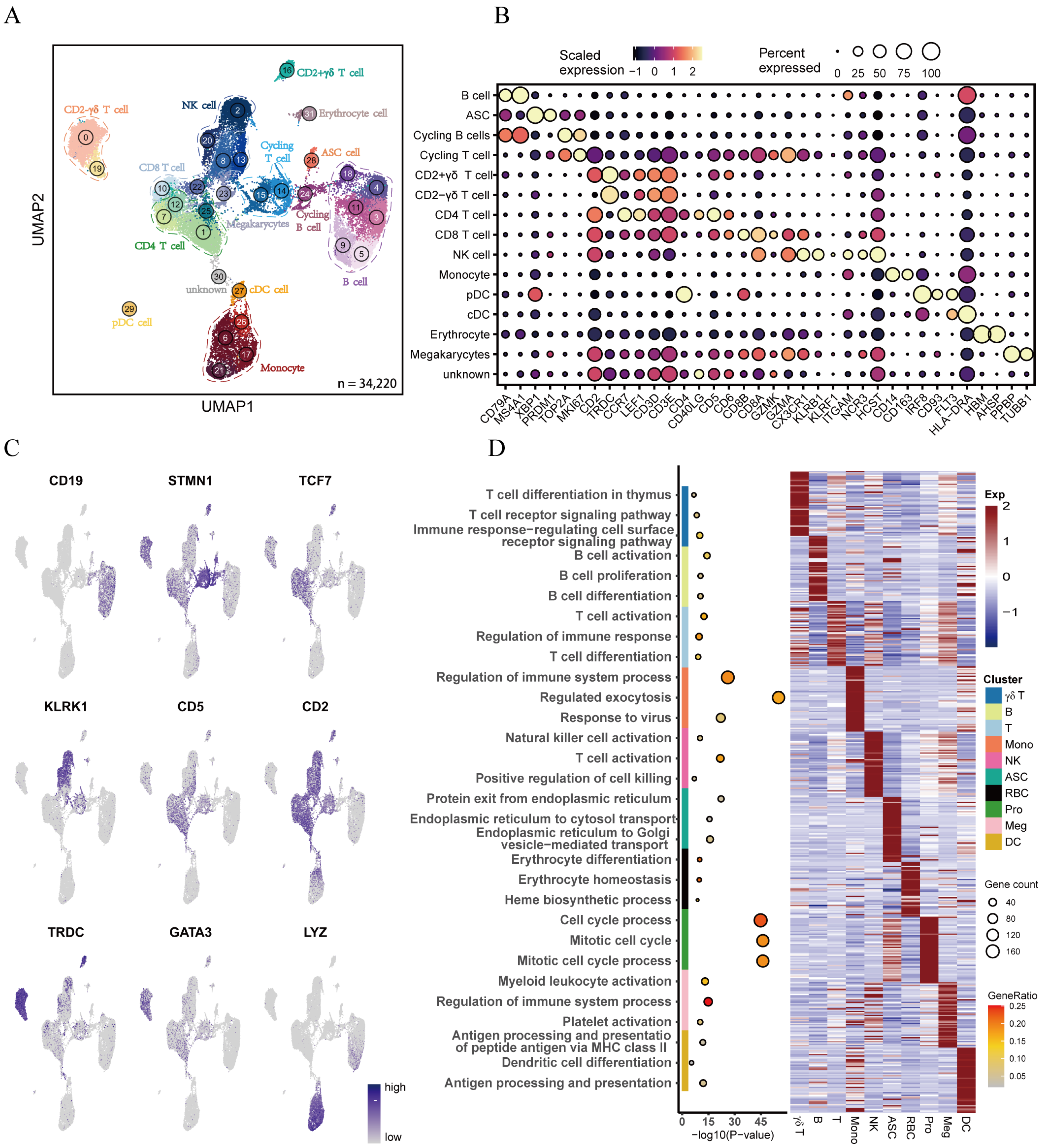
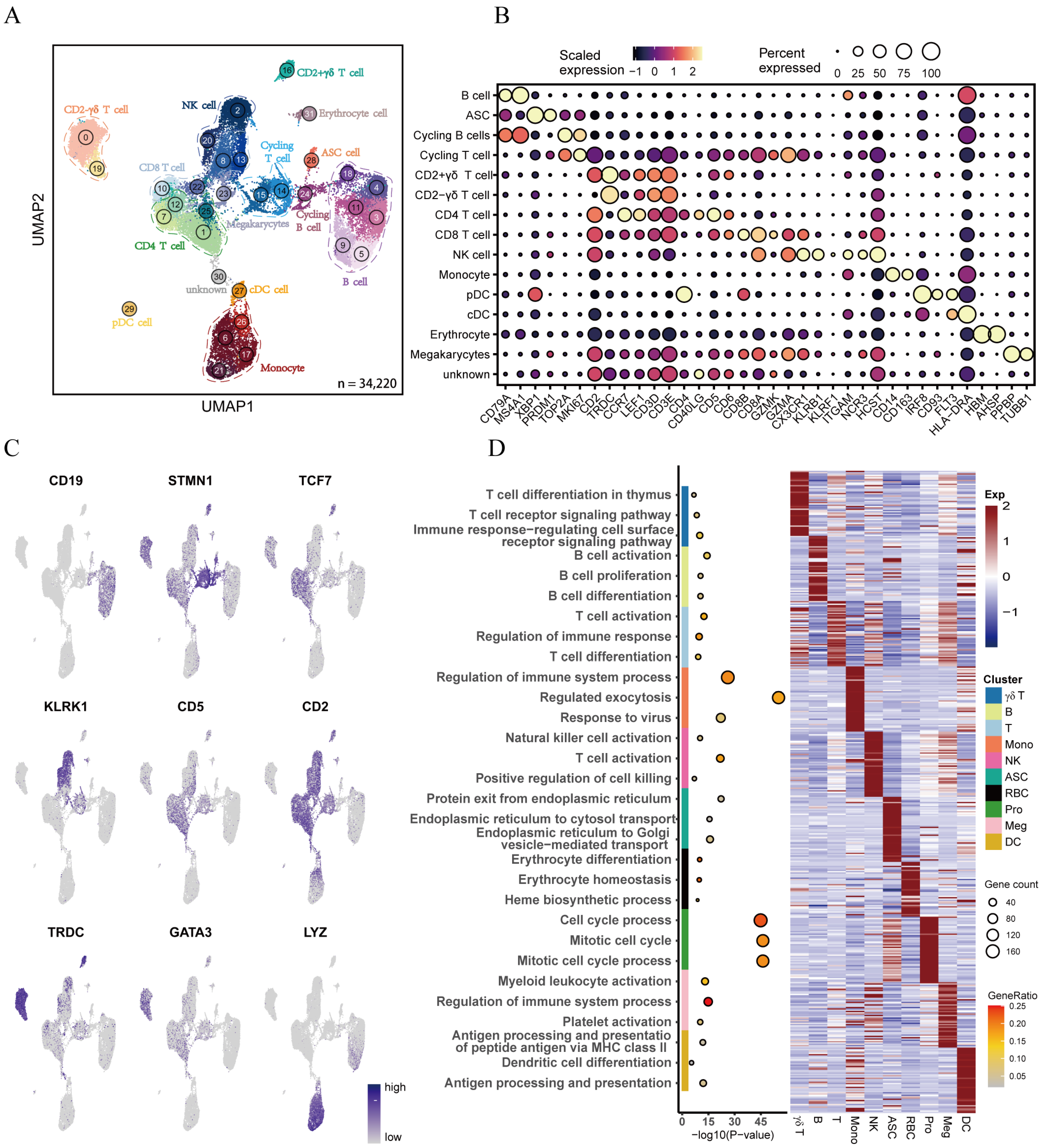
3.1. Transcriptional Landscape Reveals Heterogeneity of Porcine Peripheral Blood Monocytes
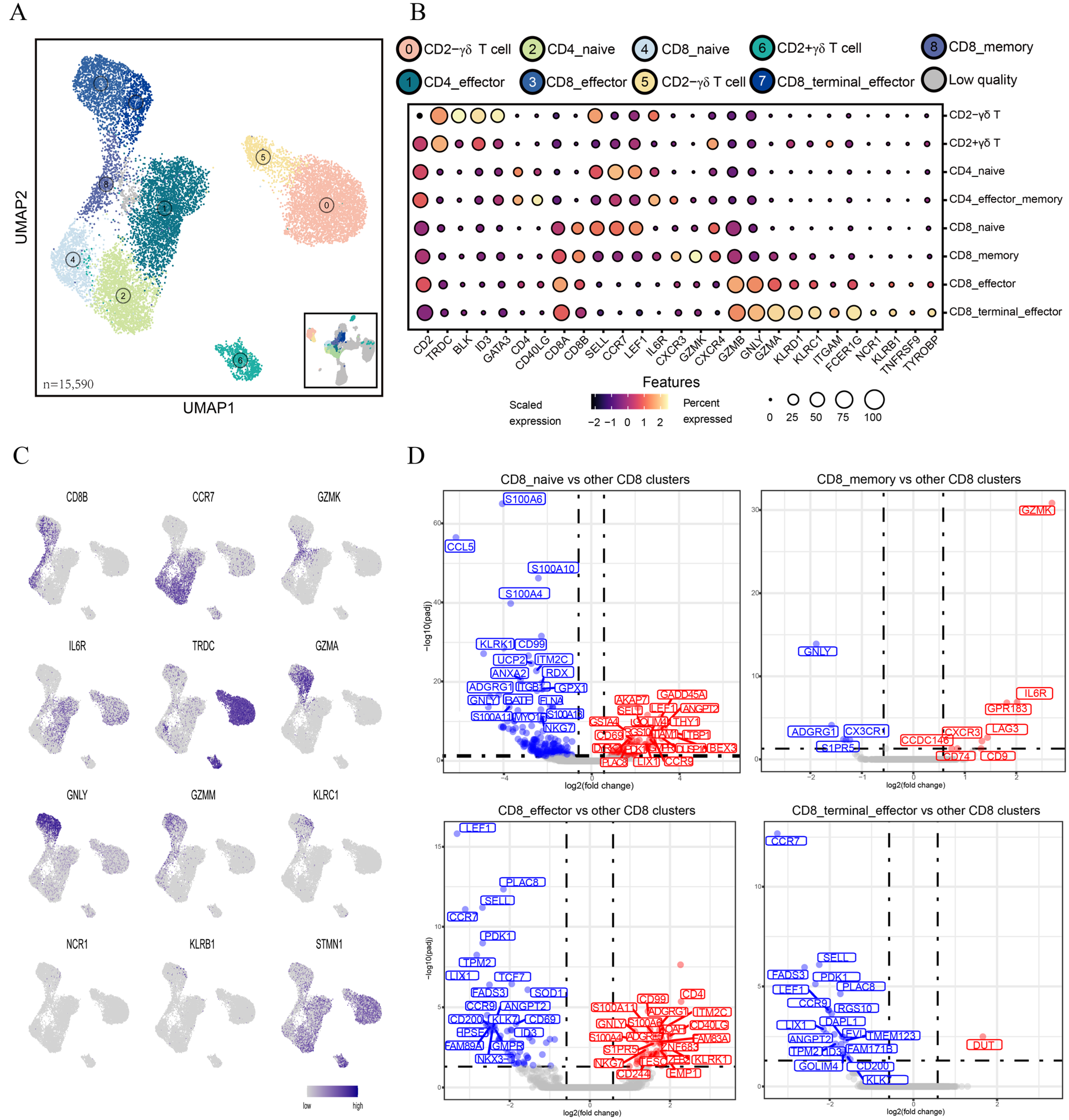
3.2. Clustering Analysis Reveals Heterogeneity of T Lymphocytes in Peripheral Blood

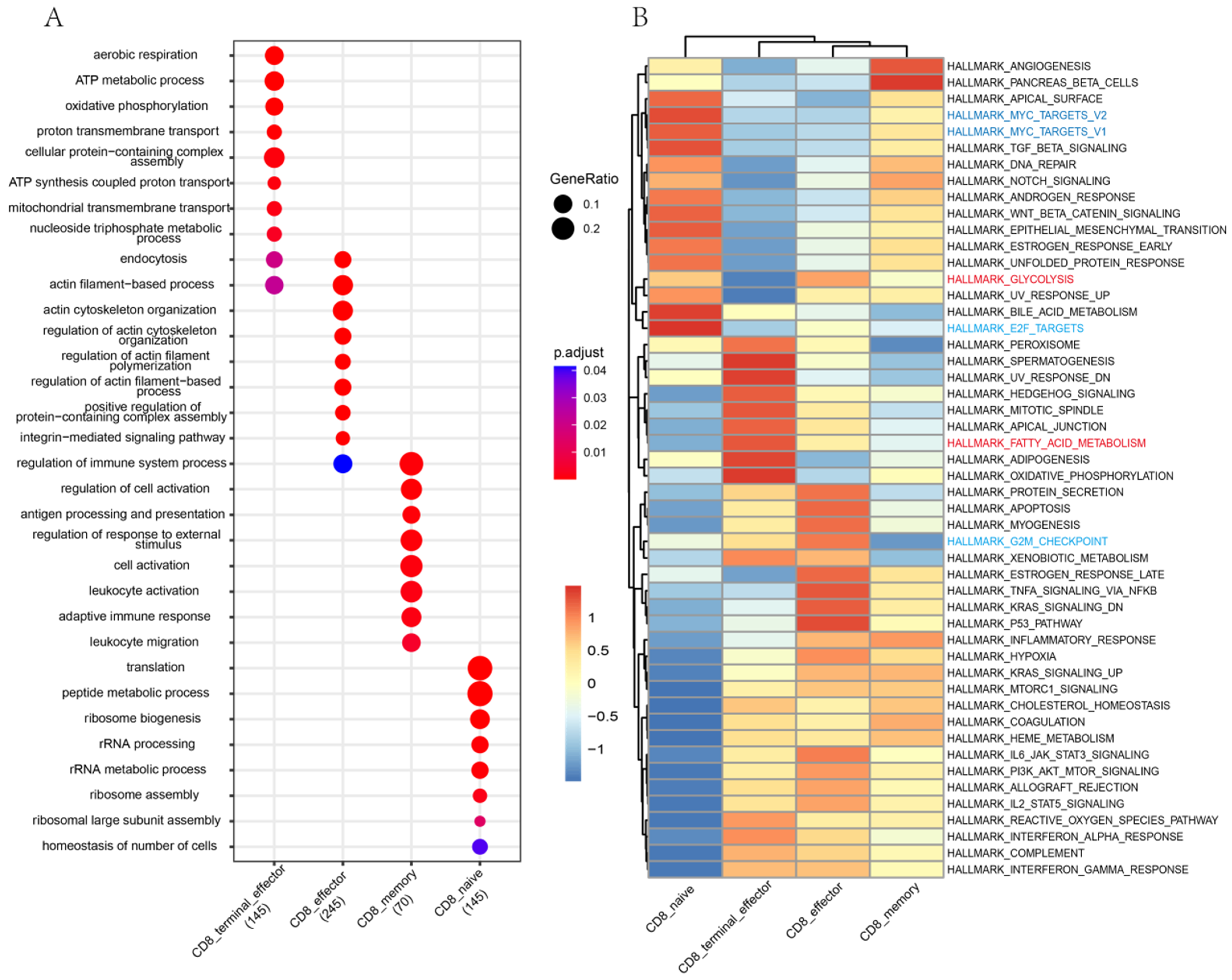
3.3. Differences in Functions and Immunometabolic Patterns among CD8 T Cell Subtypes
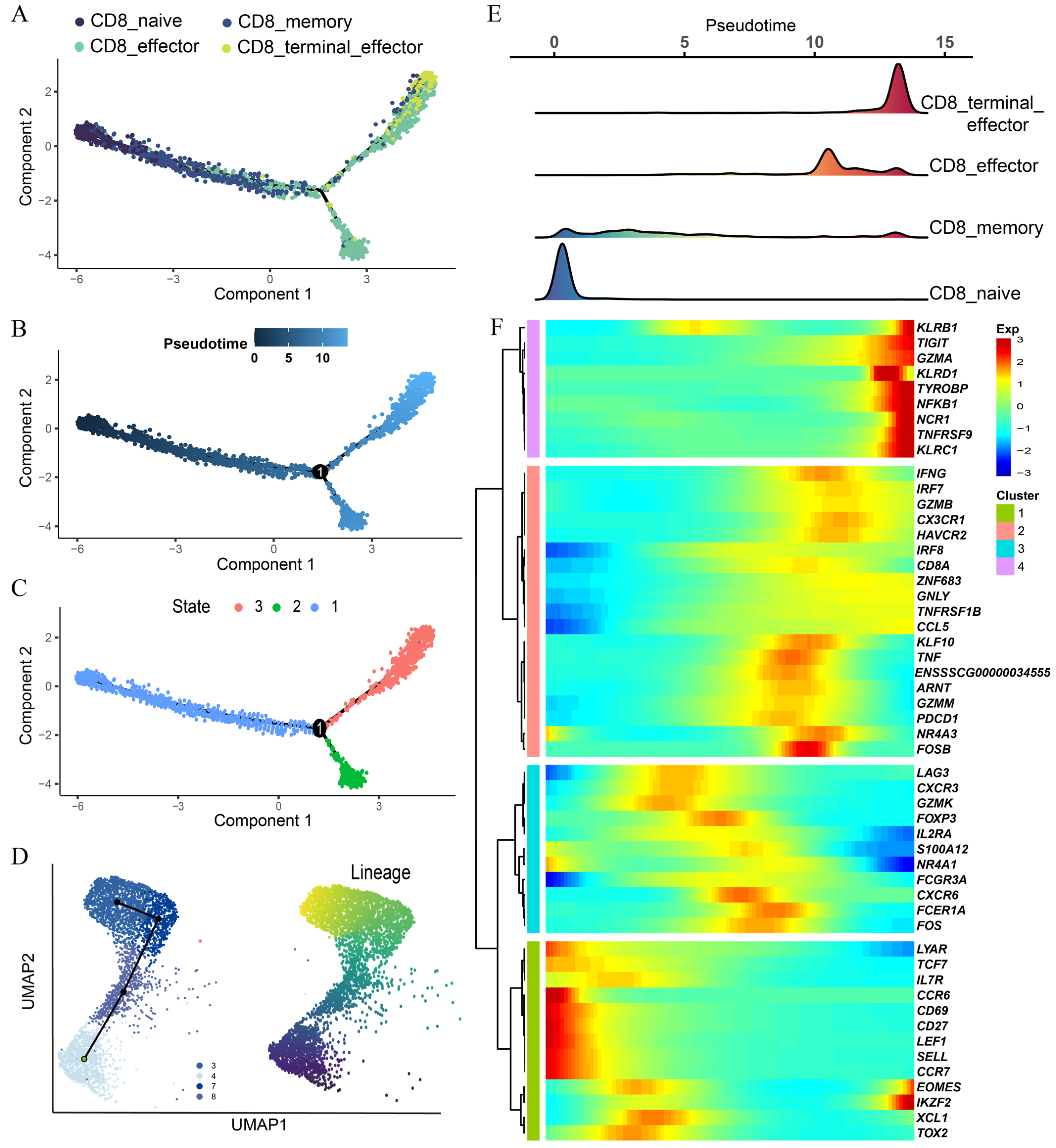
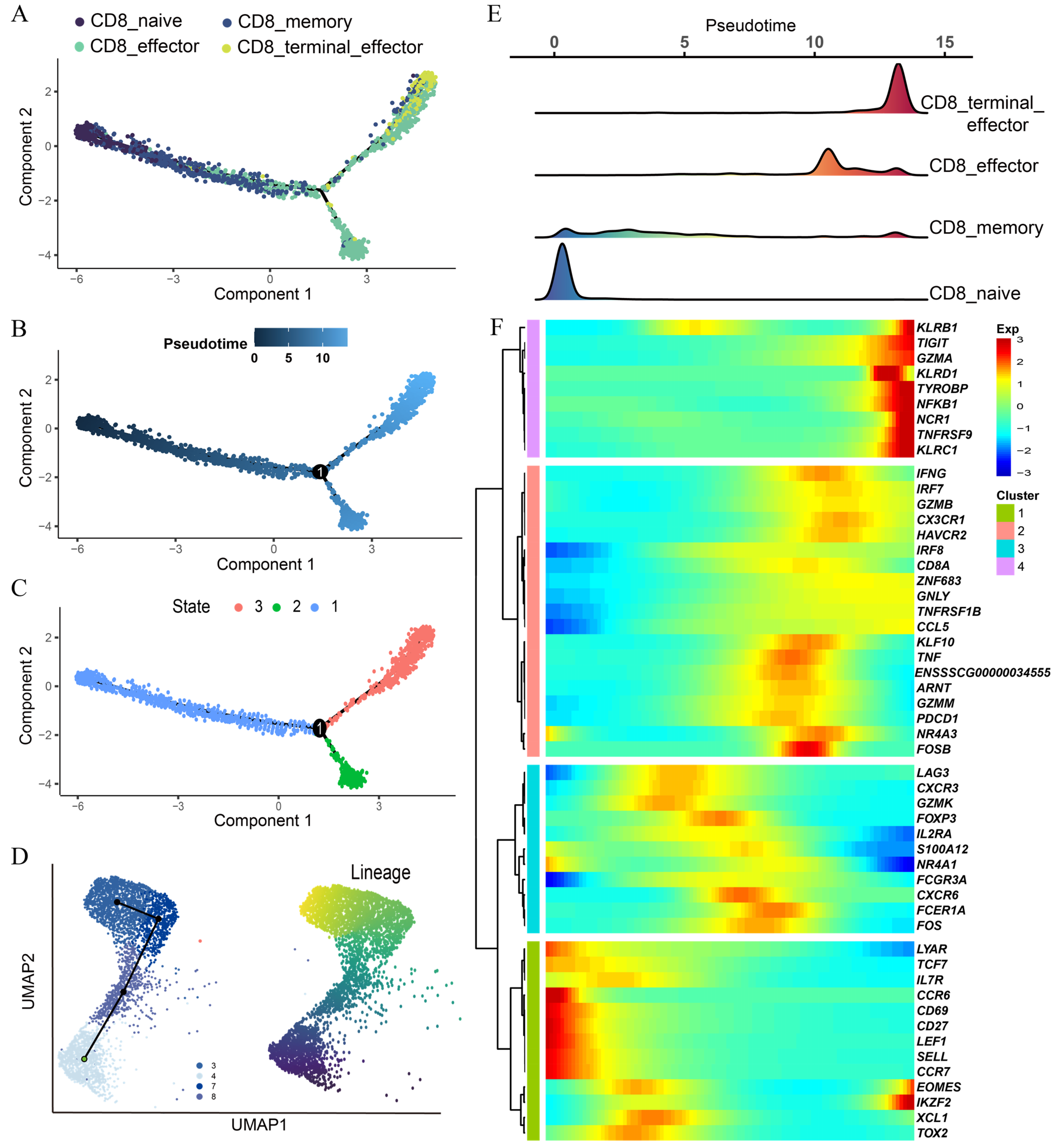
3.4. Pseudotime Trajectory Analysis Reveals Dynamic Heterogeneity among CD8 T Subtypes

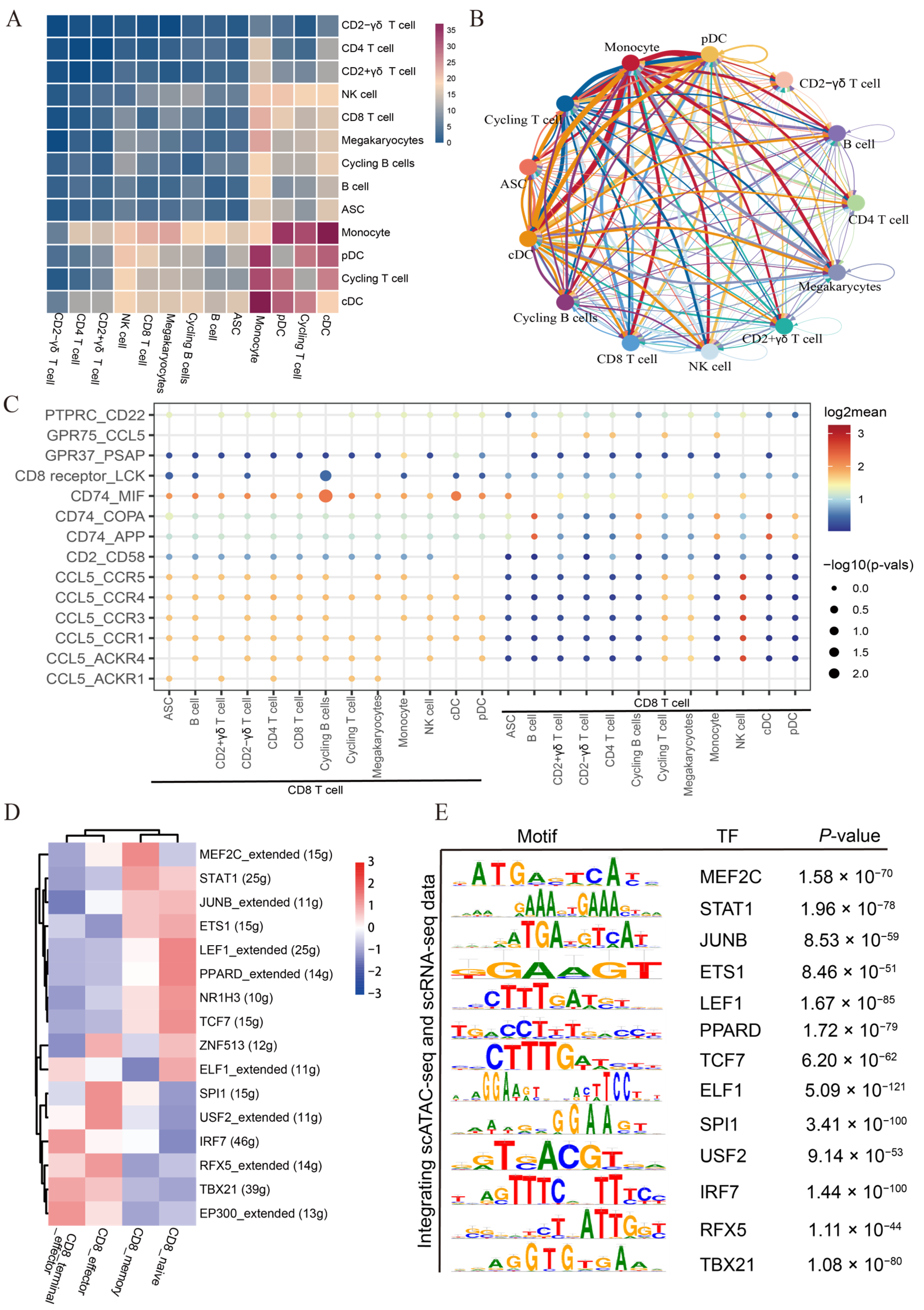
3.5. Single-Cell Network Inference and Cell Communication Analysis Unveil Candidate Regulators and Extensive Intercellular Communication in CD8 T Subtypes
3.6. Cross-Species Comparison Shows Similarities and Differences between Species

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lagumdzic, E.; Pernold, C.; Viano, M.; Olgiati, S.; Schmitt, M.W.; Mair, K.H.; Saalmuller, A. Transcriptome Profiling of Porcine Naive, Intermediate and Terminally Differentiated CD8(+) T Cells. Front. Immunol. 2022, 13, 849922. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Invest. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Halle, S.; Halle, O.; Forster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef]
- Pauly, T.; Elbers, K.; Konig, M.; Lengsfeld, T.; Saalmuller, A.; Thiel, H.J. Classical swine fever virus-specific cytotoxic T lymphocytes and identification of a T cell epitope. J. Gen. Virol. 1995, 76, 3039–3049. [Google Scholar] [CrossRef] [PubMed]
- Patch, J.R.; Kenney, M.; Pacheco, J.M.; Grubman, M.J.; Golde, W.T. Characterization of cytotoxic T lymphocyte function after foot-and-mouth disease virus infection and vaccination. Viral. Immunol. 2013, 26, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.J.; Cha, S.H.; Grimm, A.L.; Ajithdoss, D.; Rzepka, J.; Chung, G.; Yu, J.; Davis, W.C.; Ho, C.S. Pigs that recover from porcine reproduction and respiratory syndrome virus infection develop cytotoxic CD4+CD8+ and CD4+CD8− T-cells that kill virus infected cells. PLoS ONE 2018, 13, e0203482. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Zippelius, A.; Kurth, I.; Pittet, M.J.; Touvrey, C.; Iancu, E.M.; Corthesy, P.; Devevre, E.; Speiser, D.E.; Rufer, N. Four functionally distinct populations of human effector-memory CD8+ T lymphocytes. J. Immunol. 2007, 178, 4112–4119. [Google Scholar] [CrossRef]
- Appay, V.; Rowland-Jones, S.L. Lessons from the study of T-cell differentiation in persistent human virus infection. Semin. Immunol. 2004, 16, 205–212. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Rufer, N.; Zippelius, A.; Batard, P.; Pittet, M.J.; Kurth, I.; Corthesy, P.; Cerottini, J.C.; Leyvraz, S.; Roosnek, E.; Nabholz, M.; et al. Ex vivo characterization of human CD8+ T subsets with distinct replicative history and partial effector functions. Blood 2003, 102, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Book, B.K.; Aasted, B.; Dominguez, J.; Ezquerra, A.; Trebichavsky, I.; Novikov, B.; Valpotic, I.; Sver, L.; Nielsen, J.; et al. Summary of workshop findings for antibodies reacting with porcine T-cells and activation antigens: Results from the Second International Swine CD Workshop. Vet. Immunol. Immunopathol. 1998, 60, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Talker, S.C.; Kaser, T.; Reutner, K.; Sedlak, C.; Mair, K.H.; Koinig, H.; Graage, R.; Viehmann, M.; Klingler, E.; Ladinig, A.; et al. Phenotypic maturation of porcine NK- and T-cell subsets. Dev. Comp. Immunol. 2013, 40, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Uribe, J.; Wiarda, J.E.; Sivasankaran, S.K.; Daharsh, L.; Liu, H.; Byrne, K.A.; Smith, T.P.L.; Lunney, J.K.; Loving, C.L.; Tuggle, C.K. Reference Transcriptomes of Porcine Peripheral Immune Cells Created Through Bulk and Single-Cell RNA Sequencing. Front. Genet. 2021, 12, 689406. [Google Scholar] [CrossRef] [PubMed]
- Ammons, D.T.; Harris, R.A.; Hopkins, L.S.; Kurihara, J.; Weishaar, K.; Dow, S. A single-cell RNA sequencing atlas of circulating leukocytes from healthy and osteosarcoma affected dogs. Front. Immunol. 2023, 14, 1162700. [Google Scholar] [CrossRef] [PubMed]
- Squair, J.W.; Gautier, M.; Kathe, C.; Anderson, M.A.; James, N.D.; Hutson, T.H.; Hudelle, R.; Qaiser, T.; Matson, K.J.E.; Barraud, Q.; et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021, 12, 5692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fang, L.; Baldwin, R.L.; Connor, E.E.; Cole, J.B.; Van Tassell, C.P.; Ma, L.; Li, C.J.; Liu, G.E. Single-cell transcriptomic analyses of dairy cattle ruminal epithelial cells during weaning. Genomics 2021, 113, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Fan, H.; Xu, C.; Du, C.; Wang, H.; Wang, Y.; Zhao, B.; Chen, C.; Shang, S.; Wu, S. Single-Cell Transcriptomic and Chromatin Accessibility Atlas of Peripheral Blood Mononuclear Cells Reveal the Immune Cell Heterogeneity of Pigs. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martinez-Colon, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Sedlak, C.; Patzl, M.; Saalmuller, A.; Gerner, W. CD2 and CD8alpha define porcine gammadelta T cells with distinct cytokine production profiles. Dev. Comp. Immunol. 2014, 45, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, K.; Sinkora, M. The expression of CD25, CD11b, SWC1, SWC7, MHC-II, and family of CD45 molecules can be used to characterize different stages of gammadelta T lymphocytes in pigs. Dev. Comp. Immunol. 2012, 36, 728–740. [Google Scholar] [CrossRef]
- Gu, W.; Madrid, D.M.C.; Joyce, S.; Driver, J.P. A single-cell analysis of thymopoiesis and thymic iNKT cell development in pigs. Cell Rep. 2022, 40, 111050. [Google Scholar] [CrossRef]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Na, N.; Zhang, X.; Zhao, Y. The biological function and significance of CD74 in immune diseases. Inflamm. Res. 2017, 66, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.V. The chromatin landscape and transcription factors in T cell programming. Trends Immunol. 2014, 35, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, X.; Chen, S.; Liu, H.; Hong, N.; Zhong, H.; Chen, X.; Jin, W. Reinvestigation of Classic T Cell Subsets and Identification of Novel Cell Subpopulations by Single-Cell RNA Sequencing. J. Immunol. 2022, 208, 396–406. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.H.; Liu, Y.; Li, Y.Q.; Chen, H.T.; Xu, J.H.; Peng, W.; Lin, G.W.; Wei, P.P.; Li, B.; et al. Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol. 2020, 21, 294. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Fitch, M.; Edupuganti, S.; Yang, S.; Kissick, H.T.; Li, K.W.; Youngblood, B.A.; Abdelsamed, H.A.; McGuire, D.J.; Cohen, K.W.; et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017, 552, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Hale, J.S.; Kissick, H.T.; Ahn, E.; Xu, X.; Wieland, A.; Araki, K.; West, E.E.; Ghoneim, H.E.; Fan, Y.; et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017, 552, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Jin, W.; Zoppi, G.; Vogel, I.A.; Akhmedov, M.; Bleck, C.K.E.; Beltraminelli, T.; Rieckmann, J.C.; Ramirez, N.J.; Benevento, M.; et al. Dynamics in protein translation sustaining T cell preparedness. Nat. Immunol. 2020, 21, 927–937. [Google Scholar] [CrossRef]
- Kumari, S.; Curado, S.; Mayya, V.; Dustin, M.L. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim. Biophys. Acta. 2014, 1838, 546–556. [Google Scholar] [CrossRef]
- Daneshpour, H.; Youk, H. Modeling cell-cell communication for immune systems across space and time. Curr. Opin. Syst. Biol. 2019, 18, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Zhang, M.; Zou, Z.; Shi, W.; Tan, S.; Wang, C.; Xu, W.; Jin, J.; Milton, S.; Chen, Y.; et al. Single-cell transcriptomics reveals zinc and copper ions homeostasis in epicardial adipose tissue of heart failure. Int. J. Biol. Sci. 2023, 19, 4036–4051. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Rowland-Jones, S.L. RANTES: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Freeman, T.; Herbert, M.; Hasegawa, H.; McMichael, A.J.; Callan, M.F. Human naive CD8 T cells down-regulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. J. Immunol. 2006, 176, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shen, J.; Kasmani, M.Y.; Topchyan, P.; Cui, W. Single-Cell Transcriptomics Reveals Core Regulatory Programs That Determine the Heterogeneity of Circulating and Tissue-Resident Memory CD8(+) T Cells. Cells 2021, 10, 2143. [Google Scholar] [CrossRef] [PubMed]
- Harly, C.; Kenney, D.; Wang, Y.; Ding, Y.; Zhao, Y.; Awasthi, P.; Bhandoola, A. A Shared Regulatory Element Controls the Initiation of Tcf7 Expression During Early T Cell and Innate Lymphoid Cell Developments. Front. Immunol. 2020, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, H.; Rothenberg, E.V. How transcription factors drive choice of the T cell fate. Nat. Rev. Immunol. 2021, 21, 162–176. [Google Scholar] [CrossRef]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, P.; Guo, Y.; Zhang, W.; Wang, D.; Wu, Y.; Li, X.; Zhu, M. Single-Cell RNA-Sequencing Reveals Heterogeneity and Transcriptional Dynamics in Porcine Circulating CD8+ T Cells. Cells 2024, 13, 692. https://doi.org/10.3390/cells13080692
Han P, Guo Y, Zhang W, Wang D, Wu Y, Li X, Zhu M. Single-Cell RNA-Sequencing Reveals Heterogeneity and Transcriptional Dynamics in Porcine Circulating CD8+ T Cells. Cells. 2024; 13(8):692. https://doi.org/10.3390/cells13080692
Chicago/Turabian StyleHan, Pingping, Yaping Guo, Wei Zhang, Daoyuan Wang, Yalan Wu, Xinyun Li, and Mengjin Zhu. 2024. "Single-Cell RNA-Sequencing Reveals Heterogeneity and Transcriptional Dynamics in Porcine Circulating CD8+ T Cells" Cells 13, no. 8: 692. https://doi.org/10.3390/cells13080692
APA StyleHan, P., Guo, Y., Zhang, W., Wang, D., Wu, Y., Li, X., & Zhu, M. (2024). Single-Cell RNA-Sequencing Reveals Heterogeneity and Transcriptional Dynamics in Porcine Circulating CD8+ T Cells. Cells, 13(8), 692. https://doi.org/10.3390/cells13080692