Highlights
What are the main findings?
- PPARs regulate cancer aggressiveness and stemness.
- PPAR functions in cancer are tissue- and context-dependent.
What is the implication of the main finding?
- PPARs represent potential therapeutic targets for CSC targeting.
- Pleiotropic effects of PPAR modulation may hinder their efficiency.
Abstract
The peroxisome proliferator-activated receptors (PPAR-α, PPAR-δ, and PPAR-γ) are transcription factors that belong to the nuclear hormone receptor superfamily. Upon activation by specific lipids, they regulate gene expression by directly binding to PPAR response elements (PPREs) in the DNA. Although the functions of the different PPARs are specific to the isoform, tissue, and context, all three PPARs are generally involved in energy homeostasis through lipid sensing in physiological conditions. Importantly, there is increasing evidence linking PPARs with malignant behavior in cancer, regulating features frequently attributed to the aggressive subpopulation of cancer stem cells (CSCs): self-renewal, tumor initiation, chemoresistance, metastasis, and immune evasion. However, contradictory effects have been described for each isoform in various cancer types, and their implication in these malignant features may not consistently follow a pro- or anti-tumoral pattern. In this review, we revise the current knowledge on the role of the PPAR family members in cancer, with a special focus on cancer stemness, and discuss the potential for PPARs as therapeutic targets in CSC-driven relapse and resistance.
1. Peroxisome Proliferator-Activated Receptors
The peroxisome proliferator-activated receptors (PPARs) comprise a family of proteins belonging to the nuclear hormone receptor superfamily of transcription factors (TFs) that regulate gene expression by directly binding to PPAR response elements (PPREs) in the DNA, upon activation by specific ligands [1]. Three different PPAR isoforms have been described to date: PPAR-α (NR1C1), PPAR-δ (also known as PPAR-β or NR1C2), and PPAR-γ (NR1C3). The PPAR structure consists of four main regions: (1) A/B or ligand-independent domain, which includes the AF1 region, important for subtype-specific activity; (2) C or DNA-binding domain (DBD); (3) D or docking cofactors domain; and (4) E or ligand-binding domain (LBD). The PPAR LBD domain is larger than in other nuclear receptors, facilitating the interaction with a wider range of compounds, all of them harboring an acidic head group [2]. Moreover, while PPAR-α and PPAR-γ have a similar ligand-binding pocket in terms of size and shape, that of PPAR-δ is significantly smaller, explaining the observed ligand restriction in PPAR-δ versus the other two isoforms. The activity of the different PPARs can be modulated by endogenous ligands such as fatty acids (FAs) and FA-derived molecules or synthetic compounds [1]. Upon ligand-dependent activation, all PPARs heterodimerize with the retinoid X receptor (RXR), which belongs to the same receptor superfamily, and the LBD acquires a conformation that allows the binding of coactivator or corepressor proteins. Then, a complex formed by the PPAR:RXR heterodimer plus the coactivator or corepressor binds to the PPREs present in the promoters of the target genes, activating or repressing their expression [3]. Although the function of the different PPARs is isotype-specific and tissue- and context-dependent, in general all three PPARs are involved in energy homeostasis through lipid sensing, while PPAR-δ additionally regulates cell proliferation, differentiation, survival, and tissue repair (Figure 1). Importantly, mounting evidence associates the PPARs with tumorigenesis, rendering them excellent candidates for therapeutic intervention with the different targeted pharmacological strategies currently available [1,4].
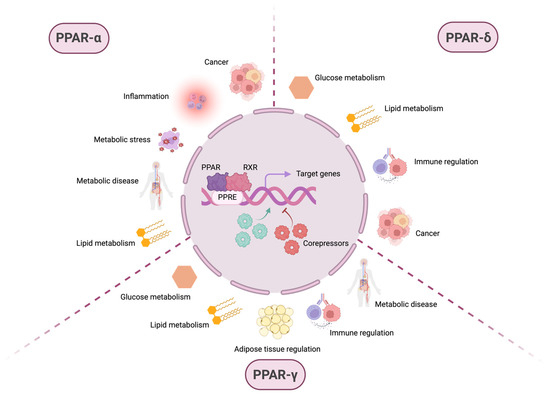
Figure 1.
PPARs are involved in various physiological and pathophysiological processes. After ligand-dependent activation and recruitment of retinoic X receptor (RXR), the PPAR:RXR heterodimer binds to PPAR response elements (PPREs) located in the promoter regions of the DNA of target genes. The transcriptional activation or repression of these genes depends not only on this binding but also on the subsequent recruitment of coactivator or corepressor molecules, which modulate gene expression in response to different cellular contexts. Although PPARs are primarily known for their role in metabolism-related pathways, such as glucose and lipid metabolism, they also regulate inflammation, immune homeostasis, and pathological conditions, including metabolic diseases and cancer. While some of these processes involve overlapping functions among different PPAR isoforms, others are specific to individual PPARs. PPAR-α is mainly associated with lipid metabolism, but also plays a role in metabolic stress, metabolic diseases, inflammation, and cancer. PPAR-δ regulates both glucose and lipid metabolism, contributes to immune system modulation, and exhibits protumorigenic activity. PPAR-γ is involved in glucose and lipid metabolism, adipose tissue regulation, immune homeostasis, and metabolic disease. These observations highlight the broad spectrum of biological processes in which members of the PPAR family—primarily recognized as key regulators of cellular metabolism—are implicated.
1.1. PPAR-Alpha
PPAR-α primarily acts as a nutritional sensor and controls the expression of genes involved in lipid transport and fatty acid oxidation (FAO) to maintain lipid homeostasis, which is essential for overall bodily function [5]. It is predominantly expressed in tissues with high FA catabolism rates, such as the liver, heart, kidney, and muscle. For instance, it plays a crucial role in maintaining lipid homeostasis in the liver, where it regulates apolipoprotein expression, enhances FAO, reduces triglyceride levels, and promotes ketogenesis. In the muscle, PPAR-α facilitates the use of FAs as an energy source.
The expression of PPARA is influenced by lipidic metabolites. Indeed, saturated and monounsaturated FAs, as well as docosahexaenoic acid (DHA), increase PPARA mRNA expression, while linolenic and eicosapentaenoic acids decrease it [6,7]. Knockout studies of liver-specific fatty acid synthase (FAS) have shown that PPAR-α agonists can reverse non-alcoholic steatohepatitis, indicating that FAS products act as PPAR-α activators [8]. Furthermore, it has been shown that disruption of ACOX1 (peroxisomal acyl-coenzyme A oxidase 1), the first enzyme of FAO, led to elevated expression of PPAR-α target genes, suggesting that ACOX1 substrates also function as PPAR-α agonists [9]. Finally, hydrolysis of triglycerides also produces PPAR-α ligands [10].
PPAR-α regulates FA uptake by controlling the expression of FA transporters, such as FATP1, SLC27A1, and FAT/CD36 [11,12,13]. In addition, it controls FAO in mitochondria and peroxisomes by directly regulating the expression of mitochondrial acyl-CoA dehydrogenases (e.g., VLCAD, LCAD, and MCAD) [14] or peroxisomal enzymes (e.g., ACOX1 and EHHADH), respectively [15]. PPARA expression can also be upregulated by the metabolic stress sensor AMPK, which then induces the expression of PPAR-α target genes, such as CPT1 and PGC-1, and increases FAO in skeletal muscle [16,17].
PPAR-α may play a dual role in cancer, demonstrating both protumorigenic and anti-tumorigenic effects depending on the cellular context and tumor type. As mentioned earlier, PPAR-α regulates lipid metabolism, FA homeostasis, and oxidative stress, processes that are crucial for cancer development and progression [6,13,18]. PPAR-α has also been shown to play a critical role in the regulation of genes involved in the initiation and progression of hormone-dependent tumors, including breast cancer, through the biotransformation of endogenous estrogens and environmental carcinogens [19]. Elevated PPARA expression has been demonstrated in several cancers, such as ampullary cancer [20] and stomach adenocarcinomas [21].
On the other hand, PPAR-α exhibits anticancer effects. The repression of PPARA mediated by protein tyrosine phosphatase receptor type O in colon cancer has been associated with a worse prognosis [22]. Moreover, loss of PPAR-α in the intestine promoted colon carcinogenesis by increasing DNA methyltransferase 1-mediated methylation of P21 and protein arginine methyltransferase 6-mediated methylation of P27 in mice [23]. Similar effects have been observed in breast cancer, where PPAR-α agonists enhanced the efficacy of immunotherapy [24] and chemotherapy [25,26], while mitigating side effects.
1.2. PPAR-Delta
PPAR-δ exerts a wide range of physiological functions in a tissue-specific manner, most of which are related to lipid metabolism. It is expressed broadly, especially in organs with high FA metabolism, reproductive organs, and the cardiovascular, endocrine, and immune systems, with increased basal levels in the gastrointestinal tract and skeletal muscle [2].
Unsaturated FAs, including derivatives of linoleic acid, are natural ligands for PPAR-δ. However, the effect of endogenous ligand-dependent activation of PPAR-δ is, in fact, tissue-specific, also depending on the relative presence of coactivators and corepressors. For instance, 13-HODE (13-hydroxyoctadecadienoic acid) has been reported to inhibit PPAR-δ in colorectal cancer cells to induce apoptosis [27], while in pre-adipocytes, it acts as an agonist, enhancing lipid detoxification [28]. Additionally, PPAR-δ may be exogenously activated with synthetic ligands. Interestingly, PPAR-δ activity can be positively regulated via phosphorylation by protein kinase A (PKA) or inhibited by ubiquitin-induced proteolysis [29].
The main function of PPAR-δ involves the regulation of the balance between glucose and lipid metabolism. In the pancreas, PPARD knockout increased insulin secretion, while systemic treatment of obese (ob/ob) [30] and diabetic (db/db) [31] mice with PPAR-δ agonists enhanced glucose-stimulated insulin secretion and normalized pancreatic islet hypertrophy. In the liver, one of the most important organs implicated in energy homeostasis, PPAR-δ promotes lipogenesis and activates the pentose phosphate pathway (PPP) to increase glucose utilization, while Ppard null mice exhibited glucose intolerance [32].
The role of PPAR-δ in cancer is certainly controversial, as both pro- and anti-tumoral functions have been reported in different cancer types. For instance, while ligand-independent activation of PPAR-δ exerted tumor suppressive functions in prostate cancer by repressing trefoil factor 1 (TFF1) [33], PPARD expression has been correlated with cancer progression, angiogenesis, and metastasis in several cancers [34,35]. In liver cancer, Ppard−/− mice were more susceptible to developing induced hepatocellular carcinoma (HCC) and, accordingly, PPARD overexpression in HCC cell lines inhibited proliferation, migration, and invasion while enhancing apoptosis in these cells [36]. A different study correlated increased PPAR-δ activity to treatment resistance in HCC [37]. Importantly, contradictory roles have been described for PPAR-δ even within the same malignancy, probably explained by the different experimental models used [38]. For instance, PPARD knockdown promoted proliferation and resistance to treatment in vitro in the established colon cancer cell line KM12C [39]. Contrastingly, PPAR-δ activation endowed intestinal organoid- and tumor-initiating properties induced by a high-fat diet (HFD) or treatment with synthetic ligands in colon cancer [40]. Contradictory results have also been published for breast cancer cell lines, reporting both PPAR-δ-dependent inhibition of proliferation and tumorigenicity [41], and proliferative, survival, and metastatic advantages in normal and harsh metabolic conditions [42]. In these reports, a similar overexpression system in MCF7 cells and other established cell lines was used, so differences could be attributed to experimental design. Likewise, PPAR-δ was initially found to inhibit invasion in MiaPaca and BXPC3 pancreatic cancer cells [43] in response to TNF-α in vitro, but different studies revealed its implication in epithelial-to-mesenchymal transition (EMT), migration, and invasion, as well as in vivo metastasis, in pancreatic and other cancers [34,44]. Interestingly, recent studies demonstrate a protumoral role for PPAR-δ by creating an immunosuppressive microenvironment via transcriptional regulation of cytokines and chemokines that altered the recruitment of immune cells [45,46].
1.3. PPAR-Gamma
PPAR-γ is recognized as the master regulator of adipose tissue biology. The gene PPARG encodes four isoforms, with PPAR-γ1 and PPAR-γ2 being the most studied. PPARG1 is expressed in a number of tissues, including adipose tissue, liver, colon, heart, skeletal muscle, and various immune cells such as monocytes/macrophages, dendritic cells, and T lymphocytes [47]. In contrast, PPARG2 is almost exclusively expressed in adipose tissue [48]. However, recent studies have identified low levels of PPAR-γ2 expression in T cells, particularly regulatory T cells (Tregs) [49]. PPAR-γΔ5 and PPAR-γ2Δ5 are two additional isoforms with truncated sequences resulting in non-functional LBDs. These variants are hypothesized to negatively regulate PPAR-γ by competing for binding sites in the DNA, without activity per se on gene expression. Indeed, the natural presence of PPAR-γΔ5 isoforms impairs adipocyte precursor cells, contributing to adipose tissue dysfunction [50].
As mentioned earlier, PPAR-γ plays a fundamental role in the adipocyte lifecycle, being crucial for the initiation of the adipogenic differentiation program and maintaining the phenotype, integrity, and function of adipocytes [51,52]. Indeed, Pparg knockout mice showed a reduction in brown adipocytes and diminished lipid droplets compared to wild-type animals [53,54,55]. Furthermore, patients with familial partial lipodystrophy subtype 3 (FPLD3), a rare autosomal dominant condition resulting from loss-of-function mutations in the PPARG gene, display a deficiency of subcutaneous adipose tissue in specific regions [56,57]. Besides this adipogenic activity, PPAR-γ modulates lipid and glucose metabolism, as it controls the expression of genes participating in the release (e.g., lipoprotein lipase, LPL), transport (e.g., CD36), and storage of FAs, as well as gluconeogenesis (e.g., FABP4) [58,59].
PPAR-γ directly regulates the expression of genes involved in lipid transport and metabolism in myeloid cells, with direct implications in their differentiation and polarization. For instance, PPARG expression is highly induced during monocyte differentiation into macrophages [60,61,62] or dendritic cells [63,64,65]. Additionally, PPAR-γ activation drives the acquisition of an alternative M2 macrophage phenotype [66,67,68], which could result in enhanced metastasis in lung [69,70] and prostate [71] cancers. PPAR-γ is also crucial for the differentiation of Treg cells in visceral adipose tissue, driving a unique Treg population that controls inflammation in adipose tissue and influences insulin sensitivity [49,72].
However, except for specific contexts, the role of PPAR-γ when expressed or activated in cancer cells is mainly anti-tumoral, leading to a reduction in cell growth or invasiveness. For example, administration of the synthetic ligand troglitazone inhibited prostate cancer cell proliferation in vitro and in vivo [73]. Similarly, PPAR-γ activation reduced proliferation through estrogen receptor signaling inhibition [74]. In thyroid cancer mouse models, PPAR-γ activation via rosiglitazone decreased cell proliferation and delayed tumor progression by inducing apoptosis [75]. Interestingly, treatment with rosiglitazone repressed tumor metastatic potential in gastric cancer [76], inhibited EMT and metastasis by antagonizing TGF-β signaling in lung cancer [77], and suppressed metastasis in hepatocellular carcinoma by regulating metalloproteinases (MMPs) and E-cadherin [78]. Moreover, treatment with PPAR-γ agonists increases sensitivity to chemotherapy in various types of cancer, including lung [79,80,81], pancreas [5], and breast cancer [82]. Although these anti-tumoral effects highlight PPAR-γ activation as a potential therapeutic option for diverse cancers, a consistent protumorigenic role for PPAR-γ has been suggested in bladder cancer [83,84,85], indicating a tissue-specific role for this transcription factor.
2. Cancer Heterogeneity and Cancer Stem Cells
Next-generation sequencing technologies have allowed us to recognize cancer as independent and unique entities across tumor types (intertumor heterogeneity) and patients (intratumor heterogeneity). Nowadays, it is well acknowledged that intratumor heterogeneity is mainly responsible for therapeutic failure and recurrence [86,87,88,89], thus becoming a major priority in cancer biology research. Indeed, three main causes explain the underlying mechanisms of such heterogeneity: genetic driver mutations, hierarchical organization of tumors, and the influence of the tumor microenvironment (TME). While their nature and mechanisms are divergent, these three factors are not mutually exclusive [90].
First proposed by Peter Nowell in 1976 [91], the “stochastic” clonal evolution model implies that cancer initiation, development, and progression follow Darwinian selection rules. This implies that differentiated cancer cells acquire heritable mutations, called oncogenic drivers, that confer a survival advantage to their progeny. These mutations are cumulative and beneficial for cancer cells, ultimately promoting the expansion of the fittest clones across different tumor regions [92,93,94] and contributing to intratumor heterogeneity [90].
Subsequent studies in leukemia by Tsvee Lapidot [95] demonstrated the existence of non-genetic subclonal heterogeneity in cancer, driven by the hierarchical organization of tumoral cells. The “hierarchical” cancer stem cell (CSC) model implies that intratumoral heterogeneity is sustained by a small subpopulation of cells with tumor-initiating properties undergoing symmetrical (self-renewal) and asymmetrical (differentiation) divisions. As a consequence, the cells within the tumor show differences in morphology, state of differentiation, proliferation, gene expression, metabolism, and invasive, metastatic, and angiogenic potential. CSCs have similar features to their non-tumoral counterparts, as they retain the unlimited ability of self-renewal and dedifferentiation characteristics of stem cells, though in a deregulated manner. Likewise, each subclone is an (epi-) genetically distinct entity that will lead to tumor growth in the context of such heterogeneity [96]. This early study in leukemia served as a precedent to demonstrate the CSC hypothesis in solid malignancies such as breast [97], brain [98], head and neck [99], pancreas [100,101], lung [102], prostate [103,104], colon [105,106], and sarcoma [107] cancers.
The origin of CSCs still remains unclear, and, in fact, it can vary in different cancer types. On the one hand, adult tissue resident stem cells (SCs) may undergo malignant transformation during the physiological regeneration processes that maintain tissue homeostasis [108]. On the other hand, differentiated cancer cells may acquire stemness-related properties, mediated by either EMT [109] or dedifferentiation triggered by microenvironmental signals from stromal cells [110,111]. A dual scenario in which tissue resident SCs and differentiated cancer cells originate new CSCs is present in chemoresistant pancreatic [112] and lung [113] cancer cells.
Considering all these findings, the theory explaining the origin of intratumor heterogeneity has evolved into a plastic CSC model, which implies the existence of hybrid intermediate cellular states [114]. Therefore, CSCs either self-renew or differentiate into plastic hybrid cancer cells that further differentiate or dedifferentiate according to specific signals from their niche. In this scenario, hybrid cancer cells and CSCs are continuously sensing a selective pressure that will ultimately force them to either adapt and progress or extinguish completely, thus promoting the survival of the fittest clones. For example, as malignant cells grow and the TME expands, diverse nutrients such as glucose and oxygen levels diminish in the milieu, the pH becomes acidic, and reactive oxygen species (ROS) and inflammatory mediators accumulate. Considering that differentiated tumor cells are fully glycolytic to cope with their enhanced proliferative demands (i.e., Warburg effect), this scarce scenario forces CSCs to become metabolically and functionally plastic in order to survive and detoxify their microenvironment from ROS. However, CSCs may preferentially use oxidative phosphorylation (OXPHOS) or glycolysis depending on the tumor type and experimental model used [115], and this lack of consensus could be explained, at least in part, by the intrinsic plasticity of CSCs.
Therefore, although the “hierarchical” model was controversial for more than a century [116], mounting evidence over the past decades demonstrates the presence of highly tumorigenic CSCs with self-renewal capacity and functional plasticity. CSCs bear unique features such as increased mitochondrial metabolism (i.e., OXPHOS) linked to metabolic plasticity (i.e., ability to use alternative metabolic substrates for energy production), enhanced mesenchymal-like phenotype primed to invade, increased immunoevasive properties, and an inherent ability to induce chemotherapeutic failure. For these reasons, there is an increasing interest in the study and characterization of CSCs, allowing for the design of therapeutic strategies to target this aggressive subpopulation.
3. PPARs in Cancer Stemness
Considering the implication of PPARs in different cancer types mentioned in the first section, and the crucial role of metabolism for CSC maintenance described above, it is not surprising that an increasing number of reports link this family of transcription factors with cancer stemness. Indeed, several studies have shown that activation of the different PPARs is higher in CSCs compared to non-CSCs, as observed in pancreatic and colorectal cancer models [117,118,119]. Importantly, a complex regulatory network involving PPAR signaling and various canonical CSC pathways modulates stemness and affects tumor progression through both metabolism-dependent and independent mechanisms. For instance, several studies have documented molecular interactions between the Wnt/β-catenin and PPAR-γ signaling pathways, indicating the possibility of cross-regulation occurring at various levels [120]. On the other hand, PPAR-γ expression is directly controlled by Notch or Sonic Hedgehog (Shh) pathways, promoting pancreatic cancer progression in murine models [121] or tumor initiation in medulloblastoma [122], respectively. Finally, direct interaction of PPAR-δ and the Hippo coactivator YAP1 induced SOX9 transcription and gastric cancer progression [123].
In this section, we will review the current knowledge about the role of PPARs in cancer stemness, focusing on specific CSC features.
3.1. Self-Renewal and Tumor Initiation
3.1.1. Introduction
Similar to their non-tumoral counterparts, CSCs express high levels of specific transcription factors, whose activity orchestrates the pluripotent state and self-renewal by regulating the expression of several other crucial genes involved in this specific cell state. For instance, NANOG, OCT3/4, and SOX2 are crucial for embryonic stem cells (ESCs) during the early stage of embryonic development [124]. Indeed, these three pluripotency factors are commonly expressed in all four types of pluripotent SCs: ESCs, adult stem cells (ASCs), induced pluripotent stem cells (iPSCs), and CSCs [124]. Additionally, some CSCs, such as pancreatic CSCs [125,126,127,128], overexpress KLF4, one of the four Yamanaka factors essential for the formation of iPSCs, together with OCT3/4, SOX2, and MYC [129].
Assessment of self-renewal ability is considered the gold standard to evaluate the presence and functionality of CSCs both in vitro and in vivo, as a surrogate marker for their tumor-initiating properties. Indeed, their resistance to anoikis and ability to form spheres when cultured as single-cell suspensions in vitro is one of the most widely used techniques to evaluate this feature. Moreover, the tumor-initiating potential of CSCs is evaluated by studying the tumor engraftment when injected in concentrations as low as a single cell (tumorigenicity assay), after initial studies by Lapidot [95].
3.1.2. PPARs in Self-Renewal and Tumorigenicity
Different studies have reported that the expression and activity of PPAR-α promote the proliferation of liver [130], glioblastoma [131], and glioma [132] CSCs. In fact, knockdown of PPARA in human glioma stem cells not only reduced in vitro proliferation but also inhibited orthotopic xenograft tumor growth, suggesting a role in tumor initiation [132]. Moreover, incubation with specific PPAR-α agonists and knockdown assays have shown that this factor was essential for the expression of pluripotency genes and sphere-forming abilities of pancreatic and colorectal CSCs [117].
So far, the reports implicating PPAR-δ in cancer stemness are mostly restricted to intestinal cancer in animal models of HFD. Indeed, this was first reported by Beyaz et al. [40], who demonstrated that either HFD or in vivo treatment with the PPAR-δ agonist GW501516 enhanced the stemness of intestinal cells and their capacity to initiate tumors [40]. Several studies have later shown that HFD increases the self-renewal and proliferation of intestinal CSCs through PPAR-δ activation [40,133,134]. Additionally, PPAR-δ, activated by HFD, induced the expansion of colonic CSCs and induced liver metastasis by triggering the pluripotency factor NANOG [118].
Interestingly, and in clear contrast with the α and δ isoforms, PPAR-γ activity in cancer cells seems to inhibit self-renewal and pluripotency gene expression in different cancer types. Indeed, a study performed in MCF7 breast cancer cells demonstrated that activation of PPAR-γ or PPAR-α with pharmacological agonists showed opposite effects on the survival of mammospheres by regulating genes implicated in stemness via the NF-κB/IL-6 axis [135]: while treatment with PPAR-γ agonists or siRNA against PPARA reduced MCF7 mammosphere formation, PPAR-α agonists enhanced their number. Similarly, treatment with the PPAR-γ agonist 15d-PGJ2 significantly suppressed spheroid formation and reduced the expression of stemness-related genes in bladder cancer cells [136], as well as in glioma and glioblastoma stem cells [137,138]. Indeed, PPAR-γ agonists also decreased the expression of the stemness surface marker CD133 and induced glial differentiation markers in glioma CSCs. Interestingly, PPAR-γ agonists showed differential effects on the expression of stemness genes, as the authors observed downregulation of SOX2 and upregulation of NANOG [138]. Additionally, molecular iodine reduced sphere formation and the expression of pluripotency genes and the stemness markers CD49f and CK17 in cervical CSCs, through the activation of PPAR-γ [139].
In liver hepatoblastoma cells, a recent study demonstrated that spheroid formation was rescued by treatment with the PPAR-γ antagonist GW9662 in BEX1 knockout cells, suggesting that BEX1 promoted the maintenance of hepatoblastoma stemness by inhibiting PPAR-γ [140]. The suggested mechanism involves metabolic regulation of cellular functions, since BEX1 promoted glycolysis to sustain self-renewal by downregulating PPARG/PDK1. Interestingly, a negative interplay between BEX1 and PPAR-γ has also been reported during liver regeneration. Gain- and loss-of-function experiments by pharmacological and genetic means demonstrated that PPAR-γ downstream BEX1 negatively regulated the expansion of liver progenitor cells, modulating Myc expression [141].
3.2. Chemoresistance
3.2.1. Introduction
Overcoming resistance to chemotherapy remains a major challenge for cancer management in the pursuit of long-lasting treatment responses. While common chemotherapeutic agents are relatively efficient in eliminating bulk differentiated tumor cells, CSCs are able to escape from the treatment due to their inherent chemoresistance. Different mechanisms define this CSC feature and include, but are not limited to, slow proliferation, overexpression of ATP-binding cassette (ABC) transporters, and hyperactivation of CSC-related signaling pathways [142].
CSCs have been traditionally identified as semi-quiescent slow-proliferating cancer cells, a feature contributing to their resistance to conventional anti-proliferative chemotherapeutic drugs [143,144,145]. For example, Domenichini et al. [146] demonstrated that only slow-cycling pancreatic cancer cells capable of forming tumor spheroids in vitro exhibited resistance to therapies. Interestingly, it has been reported that colon CSCs expressed miR-215 to slow down cell proliferation, thereby evading chemotherapy-induced damage until they receive signals for proliferation and differentiation [147]. Furthermore, conventional chemotherapy is especially ineffective against CSCs when they remain dormant for extended periods [148,149].
ABC transporters are known for their ability to efflux toxic agents such as chemotherapeutic agents out of cancer cells, thereby contributing to cancer chemoresistance. Importantly, ABC transporters expression is enhanced in CSCs [150], thus representing both potential biomarkers for CSC detection and targets for anti-CSC therapy. Nevertheless, ABC transporters are not exclusively located in the cell membrane and can also be found in the membranes of cytoplasmic vesicles. For example, ABCG2 was found to be overexpressed in CSCs from pancreatic cancer and other tumoral malignancies, where it pumps in and accumulates ABCG2-dependent substrates and drugs, such as Mitoxantrone [151].
Activation of several signaling pathways implicated in stemness, such as Hedgehog or the Notch pathways, has also been linked to CSC-intrinsic chemoresistance. For instance, the enzyme TET1, the most important demethylating enzyme implicated in chemoresistance, downregulated the Hedgehog pathway, thus favoring chemosensitivity [152]. Moreover, stromal cells mediated chemotherapy failure in pancreatic cancer by promoting the expression of the stemness regulator transcription factor HES1 via Notch-1 receptor activation [153]. Finally, enhanced WNT/α-catenin signaling promoted increased resistance to doxorubicin in osteosarcoma by inducing the expression of the ABCB1 transporter [154].
3.2.2. PPARs in Chemoresistance
Different studies link the expression and/or activity of both PPAR-δ and PPAR-γ with stemness and chemoresistance, although their functions implicate different mechanisms that can both promote or inhibit tumoral responses to chemotherapy.
The few studies investigating the contribution of PPAR-δ in the acquisition of chemoresistance are concentrated on colon cancer and suggest contradictory roles for this transcription factor. On the one hand, it has been reported that PPAR-δ accelerated cancer cell metabolism in established cell lines via induction of GLUT1 and SLC1A5 expression. The authors demonstrated that this metabolic switch induced tumor progression after subcutaneous injection of colon cancer SW480 cells. Conversely, combined treatment with the PPAR-δ antagonist GSK0660 abrogated the enhanced tumor growth and sensitized cancer cells to cisplatin in vivo [155]. On the other hand, a study performed in primary patient-derived colorectal cancer cells demonstrated that an acidic TME promoted stemness and chemoresistance by downregulating PPARD expression in colorectal CSCs. Indeed, downregulation of PPARD reduced vitamin D receptor (VDR) expression and subsequently mitigated its inhibitory effect on the SOX2 promoter, thus promoting SOX2 expression and resulting in tumor growth and drug resistance of colorectal CSCs [156]. In fact, the authors proved that VDR overexpression significantly inhibited both stemness and resistance to oxaliplatin of colorectal CSCs under acidic conditions [156]. This study also presents confirmatory experiments conducted using the SW480 cell line, which yielded results consistent with those observed in primary patient-derived cells. This suggests that, most likely, environmental signals may influence the contribution of PPAR-δ to stemness and chemoresistance in colon cancer cells.
PPAR-γ can either promote or counteract chemoresistance depending on the cancer type. First, a study suggests a potential pharmacologic benefit from targeting PPAR-γ in chemoresistant ovarian cancer. Gene set enrichment analyses performed in samples from ovarian cancer patients indicated that PPAR signaling was increased after chemotherapy, correlating with chemoresistance. Indeed, in vitro experiments showed protection against cisplatin- and paclitaxel-induced apoptosis in ovarian cancer cell lines cocultured with human adipose tissue extracts (HATEs). Mechanistically, chemoresistance was mediated by the regulation of ABCG2 expression by PPAR-γ: while activation of PPAR-γ with troglitazone induced ABCG2 expression, pharmacologic targeting using GW9662 resulted in decreased ABCG2 promoter activity and protein expression, an outcome enhanced in the presence of HATEs. Further in vivo experiments demonstrated that the combination of cisplatin with GW0662 resulted in decreased metastatic foci measured as intestinal nodules. Importantly, PPARG and ABCG2 expression colocalized in human tissues from chemoresistant ovarian cancer specimens, while the expression of both proteins was reduced in the chemosensitive ones [157].
However, PPAR-γ has also been reported to counteract chemoresistance in lung cancer models. For instance, in non-small cell lung cancer (NSCLC), hypoxia-inducible factor alpha (HIF1α) downregulated PPARG expression, which, subsequently, decreased the expression of uncoupling protein 2 (UCP2). UCP2 disruption resulted in ROS accumulation and NRF2 stabilization, leading to ABCG2 upregulation to enhance drug efflux. Conversely, activation of PPAR-γ with rosiglitazone promoted UCP2 expression and sensitized cells to cisplatin and docetaxel [158]. Another study in lung adenocarcinoma described that PPARG expression was reduced in tumors resistant to targeted therapy with the EGFR inhibitor gefitinib. Indeed, the combination of gefitinib and the PPAR-γ agonist efatutazone resulted in increased caspase activity and decreased expression of the antiapoptotic protein BCL2, causing enhanced cell death. This combinatory approach also resulted in cell cycle arrest, led by PTEN and P21 upregulation [159].
3.3. Epithelial-to-Mesenchymal Transition and Metastasis
3.3.1. Introduction
The metastatic process is intimately linked to cancer stemness in different aspects. Metastasis involves a series of dynamic and reversible steps initiated by the escape of cancer cells from the primary tumor site, followed by intravasation and dissemination through the blood and/or lymphatic system, extravasation at distant organs, metastatic seeding, colonization, and tumor outgrowth in the metastatic niche. This exceptionally complex sequence of events is finely regulated by the initiation of the EMT program, which is essential in physiological conditions for the embryonic developmental process and tissue repair. This program comprises different transitory intermediate states (partial/hybrid EMT) upon activation of certain transcription factors such as Snail, Slug, Twist, and Zeb1 [160]. The cells then undergo morphological changes concomitant to increased motility and resistance to apoptosis, as well as a higher ability to degrade the extracellular matrix [161,162,163]. In this sense, the EMT program is an important contributor to tumor heterogeneity and stemness, as some studies have demonstrated that non-CSCs undergoing EMT acquire CSC-related properties during the process. This associates the EMT program with the de novo CSC emergence mentioned in the previous section [109].
However, while both non-CSCs and CSCs have the ability to undergo EMT and MET (mesenchymal-to-epithelial) processes, only CSCs have the capacity to colonize the secondary site and successfully establish a metastasis due to their inherent self-renewal and tumor-initiating properties [163]. Indeed, a significant percentage of circulating tumor cells (CTCs) express CSC markers on their surface [164,165]. For these reasons, metastasis onset can also be considered a readout of cancer stemness.
3.3.2. PPARs in EMT and Metastasis
Different reports demonstrated that PPAR-α promotes EMT and metastasis onset in different cancer types. Indeed, the ketogenic enzyme HMGCS2 (3-hydroxy-3-methylglutaryl-CoA synthase) facilitates PPAR-α activation to promote SRC expression in colorectal and oral squamous cell carcinoma (CRC and OSCC, respectively) [166], thus inducing invasion and metastasis. Interestingly, this non-canonical role of HMGCS2 was independent of its metabolic activity, since incubation with HMGCS2 metabolites on HMGCS2-deficient cells did not restore migration nor invasion in vitro [166]. On the other hand, PPAR-α was involved in the metastatic spread of hormone-dependent cancers induced by endocrine-disrupting chemicals such as mono-(2-ethylhexyl) phthalate (MEHP). Indeed, low-dose MEHP activated the PI3K/Akt/mTOR pathway to enhance EMT-related gene expression concomitant with increased migration and invasion in vitro and metastasis in vivo in a PPAR-α-dependent manner. Interestingly, in vivo treatment with the PPAR-α antagonist GW6471 diminished the number of mesenteric metastases induced by MEHP [167].
PPAR-δ has been linked to metastatic dissemination in different cancers. Indeed, a comprehensive study interrogated the role of PPAR-δ in the metastatic process in different in vivo models across several cancer types: from tail vein experimental metastasis assays with melanoma, lung carcinoma, and colon cancer cells, to orthotopic spontaneous metastasis assays with pancreatic and breast cancer cells, as well as an intrasplenic experimental metastasis assay with colon cancer cells. In all these experiments, PPARD knockout cells were used to demonstrate a decrease in metastatic dissemination [34]. A very recent study by our group demonstrated that different environmental factors, such as nutrient stress or signals from stromal cells, induced ligand-dependent activation of PPAR-δ in pancreatic cancer, causing a profound metabolic switch [48]. Both genetic and pharmacological approaches targeting PPAR-δ inhibited expression of EMT-related factors, invasion in vitro, and metastasis in vivo, demonstrating its metabolism-related prometastatic activity. Moreover, PPAR-δ has also been shown to mediate EMT induction by stromal-derived factor 1 (SDF-1) in lung adenocarcinoma cells, via activation of CXCR4/β-catenin/PPAR-δ signaling pathway [168]. In clear contrast with the report by Zuo et al. mentioned above [34], inhibition of PPAR-δ with the antagonist 10 h induced the expression of mesenchymal genes (fibronectin and N-cadherin) to increase cell migration in vitro in B16/F10 mouse melanoma cells. These effects were further validated in vivo in a pulmonary metastasis assay by tail vein injection in Ppard knockout mice, where mice treated with 10 h showed enhanced extravasation of melanoma cells, translating into increased lung metastases [169]. Both studies utilized the same cellular model (B16/F10 cells) and in vivo metastasis induction method (tail vein injection). The differing results observed may be attributed to the distinct mechanisms of action between the small molecule drug h10, which combines ligand target and off-target effects, and the genetic approach employed by Zuo et al. [34].
While PPAR-γ may elicit different outcomes in terms of chemoresistance depending on the tumor type, the majority of studies on the topic indicate that PPAR-γ counteracts EMT, invasion, and metastasis. For instance, low PPARG expression correlated with clinicopathological parameters such as macroscopic vascular invasion and TNM (tumor, node, metastasis) stage in HCC patients [170]. Moreover, PPAR-γ activation with either rosiglitazone or troglitazone rescued the effects of TGF-β in lung cancer cells, preventing E-cadherin repression and mesenchymal markers upregulation, and thus leading to a reduced mesenchymal-like morphology. These molecular and phenotypic changes translated into decreased cell invasion in vitro and metastases in vivo, induced by TFG-β-primed cells [77]. In gastric cancer, the inhibition of PPAR-γ nuclear translocation and function by FABP4 has been described as an important mechanism regulating migration and metastasis. Indeed, while FABP4 deficiency resulted in increased invading cells in vitro, PPAR-γ activation with rosiglitazone blocked invasion and lung metastases, increasing the survival of mice injected intrasplenically with gastric cancer cells [86]. Finally, PPAR-γ activation with rosiglitazone downregulated the expression of metalloproteinase-2 (MMP-2) via PTEN upregulation, blocking invasion in vitro [171].
3.4. Immune Evasion
3.4.1. Introduction
The immune system plays a dual role in tumorigenesis. On the one hand, natural immunity provides host-protecting mechanisms. On the other hand, the immune system has tumor-shaping effects, defined as the ability to promote tumor progression by escaping from immunosurveillance mechanisms [172]. Interestingly, the strong correlation found between CSC marker expression and the infiltration of immunosuppressive cells such as M2-polarized protumoral macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs) has provided clear insights into the importance of CSCs in the regulation of the tumoral immune landscape [173,174].
For example, pancreatic CSCs exhibit upregulation of specific surface markers linked to immune evasion, such as the effector T cell inhibitor programmed cell death ligand-1 (PD-L1), the antiphagocytic signal CD47, or the more recently described Peptidoglycan Recognition Protein 1, all of which were correlated with poor prognosis [127,175,176]. Additionally, secreted factors and cytokines found in the immunotolerant microenvironment, such as interferons, are also involved in the maintenance of CSC properties. On the one hand, an early study demonstrated that IFN-γ enhanced the expression of CSC markers both in vitro and in vivo while promoting invasiveness and metastasis [177]. On the other hand, a more recent study further reinforced the notion that IFNs modulate pancreatic CSC functionality. In this study, pancreatic CSC-secreted IFN-γ promoted IFN-stimulated gene 15 (ISG15) secretion by TAMs, which in turn fostered CSC maintenance and activity [178].
3.4.2. PPARs and Immune Evasion
A link between PPAR-δ and immunoevasion has been recently reported in pancreatic cancer. On the one hand, PPAR-δ activation with a chemical agonist or with HFD significantly accelerated PDAC progression from PanINs via CCL2-mediated immunosuppression. PPAR-δ mediated the secretion of the chemokine CCL2 in the early stages of preneoplastic lesions in the pancreas, which induced the recruitment of protumoral macrophages and MDSCs, and exclusion of CD8+ T cells [45]. In addition to the above, an elegant study demonstrated a non-canonical function of the mitochondrial glutamic-oxaloacetic transaminase 2 (GOT2) in pancreatic cancer progression. Mechanistically, activation of PPAR-δ via GOT2 inhibited T cell effector recruitment in the TME. This promoted an immunoevasive scenario in vivo [46].
Although immunotherapy is currently approved for the treatment of muscle-invasive bladder cancer (MIBC), specific immunoevasive mechanisms driven by PPAR-γ can impair the outcome. Indeed, the expression of PPARGhigh/RXRAS427Y, a mutation that leads to PPAR-γ ligand-independent activation, correlates inversely with cytokine and IFN signaling, antigen processing and presentation, T cell markers, and immune checkpoints expression in MIBC patients. Moreover, PPARG expression in human MIBC tissues inversely correlated with CD8 infiltration, further confirming an association of PPAR-γ with immune evasion in bladder cancer [179].
Although not directly related to the main scope of this review, we would like to highlight that PPARs play an important role in mediating immune suppression in the TME when expressed directly in immune cells. For example, an elegant study on breast cancer demonstrated that PPARA expression in B cells promoted their transformation into tBregs, a unique type of regulatory B cells able to convert CD4 T cells into Tregs with prometastatic capability [180]. Another study on cervical cancer described that the accumulation of immunoevasive lipid-laden tumor-infiltrating dendritic cells (TIDCs) was mediated by PPAR-α, and a combination of the PPAR-α antagonist GW6471 with a PD-L1 blocking antibody led to decreased tumor burden and increased survival in mice [181]. Moreover, PPAR-δ was responsible for hepatic resident macrophage polarization towards an M2 protumoral phenotype [182]. Finally, a recent study described that PPAR-γ inhibition by GW9662 in MDSCs resulted in increased CD8 T cells infiltration due to reduced free fatty acid receptor 2 (FFAR2)-induced immunosuppressive activity [183].
4. PPARs Targeting in the Clinical Context
As described throughout this review, PPAR agonists and antagonists have been widely used in preclinical studies in vitro and in vivo in the context of cancer. The application of these agents has contributed both to elucidating the mechanisms by which PPARs function in various pathophysiological settings and to providing proof of concept for their potential therapeutic utility as repurposed drugs. Some of these compounds with agonistic activity are used in the management of conditions including type 2 diabetes, dyslipidemia, and metabolic syndrome. However, their clinical application has been limited by toxic or systemic side effects in several cases, and, consequently, safety considerations have moderated the enthusiasm for adapting these drugs for oncology applications.
One of the most well-known classes of PPAR agonists is the TZDs, i.e., pioglitazone or rosiglitazone, initially used for the treatment of several metabolic diseases. Supported by early preclinical data, several clinical trials have explored the role of these compounds for cancer treatment (Table 1). For instance, efatutazone has been tested as a single treatment or in combination with other chemotherapeutic agents for the treatment of metastatic and advanced solid tumors, with promising results for thyroid cancer [184]. On the other hand, pioglitazone has been tested in small, single-arm, or exploratory phase II trials in oral premalignant lesions, thyroid carcinoma, non-small cell lung cancer, and pancreatic cancer. The most consistent clinical activity was reported in chronic myeloid leukemia (CML), where the addition of pioglitazone to imatinib enhanced the depth of molecular responses in residual disease, as shown in the NCT02730195 trial. However, the U.S. Food and Drug Administration raised a warning due to an increased risk of developing bladder cancer in patients treated with pioglitazone, and it was withdrawn from the French market [185].

Table 1.
Overview of clinical studies involving drugs that target PPARs.
Indeed, certain TZDs have shown significant systemic toxicity, and some of their adverse effects were substantial enough to limit or even lead to the withdrawal of certain agents from the market. Among their most important side effects is fluid retention, which is strongly associated with an increased risk of new or worsening heart failure [186,187,188]. Early clinical trials with rosiglitazone reported a potential increase in ischemic cardiovascular events, such as myocardial infarction and heart failure [189]. Additional safety concerns included weight gain and bone fractures [190]. Similarly, another TZD, troglitazone, was withdrawn from the market after a growing number of case reports and epidemiological studies linked it to severe liver toxicity and acute liver failure [191,192].
Considering the toxic side effects associated with TZDs, research is underway to develop new PPAR-targeting compounds with different mechanisms of action and improved safety. However, concerns remain; indeed, a Phase II trial of the dual PPAR-α/γ agonist saroglitazar (NCT02609048) ended early because of increased serum transaminase levels [193], and bezafibrate, a PPAR-α/δ/γ agonist, has been linked to higher serum creatinine, indicating possible renal risks [194].
On the other hand, there is an increasing interest in the development and evaluation of molecules with inhibitory effects on the PPAR signaling pathway. For instance, the compound FX-909 is a covalent inverse PPAR-γ agonist [195] currently tested in a first-in-human, multicenter, open-label Phase 1 study (NCT05929235) designed to assess its safety and tolerability, and preliminary clinical activity of FX-909 given orally to patients with advanced solid malignancies, including urothelial carcinoma [196]. Moreover, the PPAR-α antagonist TPST-1120 has entered first-in-human testing. Phase I data in advanced solid tumors demonstrated acceptable safety and preliminary anti-tumor activity, with signs of synergy in combination with PD-1 blockade (NCT03829436 [197]).
Importantly, despite preclinical evidence linking PPAR-δ signaling to tumor biology, no clinical trials have yet been reported that specifically evaluate PPAR-δ agonists or antagonists in oncology. Collectively, while experiences remain fragmented and heterogeneous, emerging results indicate that PPAR modulation may hold value both as a direct anticancer strategy and as a means of enhancing established therapeutic regimens.
5. Conclusions
Increasing evidence suggests a prominent role for the PPAR family of receptors in features and functions tightly linked to cancer stemness, such as self-renewal, tumor initiation, chemoresistance, and immune evasion, through the transcriptional regulation of specific target genes (Figure 2). Despite their predominant involvement in energy homeostasis in physiological conditions, only part of the effects reported in the context of cancer have been directly linked to changes in tumoral metabolism. This fact suggests the crucial (and generally disregarded) implication of factors such as promoter accessibility, availability of activating ligands and the expression and activity of transcriptional co-activators and co-repressors during oncogenic transformation, tumor progression, and differentiation state; factors that will directly influence the transcriptional cascade initiated by the PPARs and that can also explain the often contradictory roles described for PPARs in different cancer types or even within the same tumor entity. On the other hand, we cannot forget that in vivo treatment with synthetic agonists/antagonists will inevitably induce systemic consequences with the potential to influence cancer progression and/or metastatic spread. In this sense, effects previously frequently attributed to PPARs modulation in tumor cells may have been overestimated. Systemic side effects of PPAR-targeting molecules also represent a great challenge for repurposing drugs for cancer treatment.
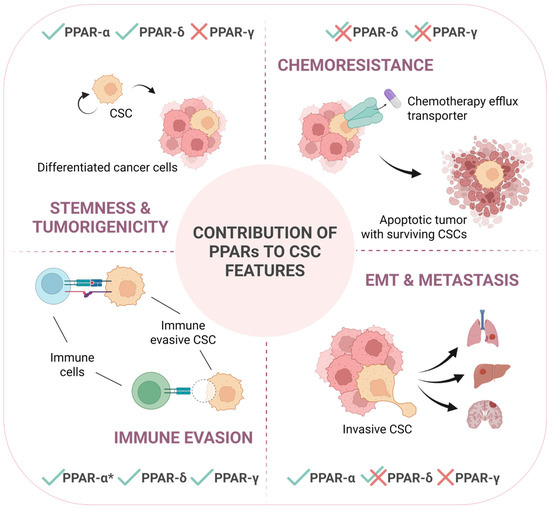
Figure 2.
PPARs contribute to different CSC-intrinsic features in a context-dependent manner. This schematic illustrates the differential involvement of PPARs in regulating key characteristics of cancer stem cells: stemness and tumorigenicity, chemoresistance, immune evasion and epithelial-to-mesenchymal transition (EMT), and metastasis. Checkmarks (✓) and crosses (✗) indicate the positive or negative, respectively, functional contribution by each PPAR isoform to the respective process, while overlapping checkmarks with crosses (✓✗) indicate that the specific isoform is reported to impact in both manners depending on the tumor type or specific circumstances. PPAR-α*: contribution to immune evasion of this isoform is related to its direct expression in immune cells, not in cancer cells. This underscores the context-dependent roles of PPARs in modulating CSC behavior and their potential impact on cancer progression and therapeutic failure.
In conclusion, PPARs influence different aspects of cancer progression, including cancer stemness, through context-dependent transcriptional regulation. However, we are still very far from understanding how different cellular factors and states regulate their activity and thus affect their downstream transcriptional program. Nevertheless, targeting PPARs presents significant potential in oncology, particularly through the development of novel drugs with enhanced selectivity and safety profiles, as well as combination therapies that maximize therapeutic benefits.
Author Contributions
Conceptualization, B.P.-A. and P.S.; writing—original draft preparation, B.P.-A., M.M. and A.R.-G.; writing—review and editing, P.S.; supervision, P.S.; funding acquisition, P.S. All authors have read and agreed to the published version of the manuscript.
Funding
The research was supported by the Instituto de Salud Carlos III through the Miguel Servet Program (CPII21/00005 to P.S.; co-financed by European funds (FSE: “El FSE invierte en tu futuro”) and a LAB AECC grant (LABAE223389SANC, to P.S.). M.M. was a recipient of a Margarita Salas fellowship from the Universidad Autónoma de Madrid (CA1/RSUE/202100646). A.R-G. was a recipient of a predoctoral contract from the Spanish AECC (PRDAR222458ROYO).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
All figures were created in BioRender. Parejo, B. (2025) https://BioRender.com/pdkt2n4.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| CSCs | cancer stem cells |
| EMT | epithelial-to-mesenchymal transition |
| FAs | fatty acids |
| FAO | fatty acid oxidation |
| HCC | hepatocellular carcinoma |
| HFD | high-fat diet |
| LBD | ligand-binding domain |
| OXPHOS | oxidative phosphorylation |
| PPARs | peroxisome proliferator-activated receptors |
| TFs | transcription factors |
| SCs | stem cells |
| TME | tumor microenvironment |
References
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef]
- Attianese, G.M.G.; Desvergne, B. Integrative and Systemic Approaches for Evaluating PPARβ/δ (PPARD) Function. Nucl. Recept. Signal. 2015, 13, nrs.13001. [Google Scholar] [CrossRef] [PubMed]
- Gearing, K.L.; Göttlicher, M.; Teboul, M.; Widmark, E.; Gustafsson, J.A. Interaction of the Peroxisome-Proliferator-Activated Receptor and Retinoid X Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 1440–1444. [Google Scholar] [CrossRef]
- Kota, B.; Huang, T.; Roufogalis, B. An Overview on Biological Mechanisms of PPARs. Pharmacol. Res. 2005, 51, 85–94. [Google Scholar] [CrossRef]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in Peroxisome Proliferator-Activated Receptor Alpha-Inducible Fatty Acid Oxidation Determines the Severity of Hepatic Steatosis in Response to Fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty Acids and Eicosanoids Regulate Gene Expression through Direct Interactions with Peroxisome Proliferator-Activated Receptors α and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.; Varricchio, E.; Vito, P.; Turchini, G.M.; Francis, D.S.; Paolucci, M. Fatty Acid-Specific Alterations in Leptin, PPARα, and CPT-1 Gene Expression in the Rainbow Trout. Lipids 2014, 49, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. “New” Hepatic Fat Activates PPARα to Maintain Glucose, Lipid, and Cholesterol Homeostasis. Cell Metab. 2005, 1, 309–322. [Google Scholar] [CrossRef]
- Fan, C.-Y.; Pan, J.; Usuda, N.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Steatohepatitis, Spontaneous Peroxisome Proliferation and Liver Tumors in Mice Lacking Peroxisomal Fatty Acyl-CoA Oxidase. J. Biol. Chem. 1998, 273, 15639–15645. [Google Scholar] [CrossRef]
- Sapiro, J.M.; Mashek, M.T.; Greenberg, A.S.; Mashek, D.G. Hepatic Triacylglycerol Hydrolysis Regulates Peroxisome Proliferator-Activated Receptor α Activity. J. Lipid Res. 2009, 50, 1621–1629. [Google Scholar] [CrossRef]
- Motojima, K.; Passilly, P.; Peters, J.M.; Gonzalez, F.J.; Latruffe, N. Expression of Putative Fatty Acid Transporter Genes Are Regulated by Peroxisome Proliferator-Activated Receptor α and γ Activators in a Tissue- and Inducer-Specific Manner. J. Biol. Chem. 1998, 273, 16710–16714. [Google Scholar] [CrossRef]
- Frohnert, B.I.; Hui, T.Y.; Bernlohr, D.A. Identification of a Functional Peroxisome Proliferator-Responsive Element in the Murine Fatty Acid Transport Protein Gene. J. Biol. Chem. 1999, 274, 3970–3977. [Google Scholar] [CrossRef]
- Martin, G.; Schoonjans, K.; Lefebvre, A.-M.; Staels, B.; Auwerx, J. Coordinate Regulation of the Expression of the Fatty Acid Transport Protein and Acyl-CoA Synthetase Genes by PPARα and PPARγ Activators. J. Biol. Chem. 1997, 272, 28210–28217. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered Constitutive Expression of Fatty Acid-Metabolizing Enzymes in Mice Lacking the Peroxisome Proliferator-Activated Receptor α (PPARα). J. Biol. Chem. 1998, 273, 5678–5684. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Hooiveld, G.; Müller, M.; Kersten, S. Comparative Analysis of Gene Regulation by the Transcription Factor PPARα between Mouse and Human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef]
- Joly, E.; Roduit, R.; Peyot, M.; Habinowski, S.A.; Ruderman, N.B.; Witters, L.A.; Prentki, M. Glucose Represses PPARαgene Expression via AMP-activated Protein Kinase but Not via P38 Mitogen-activated Protein Kinase in the Pancreatic Β-cell. J. Diabetes 2009, 1, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Kim, M.; Park, H.-S.; Kim, H.S.; Jeon, M.J.; Oh, K.S.; Koh, E.H.; Won, J.C.; Kim, M.-S.; Oh, G.T.; et al. AMPK Activation Increases Fatty Acid Oxidation in Skeletal Muscle by Activating PPARα and PGC-1. Biochem. Biophys. Res. Commun. 2006, 340, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, J.; Dong, L.; He, X.; Cheng, R.; Zhou, K.; Liu, J.; Qiu, F.; Li, X.; Ma, J. A Protective Effect of PPARα in Endothelial Progenitor Cells Through Regulating Metabolism. Diabetes 2019, 68, 2131–2142. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Won, S.S.; Jin, S.W.; Lee, G.H.; Pham, T.H.; Choi, J.H.; Kang, K.W.; Jeong, H.G. WY-14643 Regulates CYP1B1 Expression through Peroxisome Proliferator-Activated Receptor α-Mediated Signaling in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 5928. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Chao, Y.-J.; Chen, Y.-L.; Wang, T.-W.; Phan, N.N.; Hsu, H.-P.; Shan, Y.-S.; Lai, M.-D. Upregulation of Peroxisome Proliferator-Activated Receptor-α and the Lipid Metabolism Pathway Promotes Carcinogenesis of Ampullary Cancer. Int. J. Med. Sci. 2021, 18, 256–269. [Google Scholar] [CrossRef]
- Jia, Q.; Li, B.; Wang, X.; Ma, Y.; Li, G. Comprehensive Analysis of Peroxisome Proliferator-Activated Receptors to Predict the Drug Resistance, Immune Microenvironment, and Prognosis in Stomach Adenocarcinomas. PeerJ 2024, 12, e17082. [Google Scholar] [CrossRef]
- Dai, W.; Xiang, W.; Han, L.; Yuan, Z.; Wang, R.; Ma, Y.; Yang, Y.; Cai, S.; Xu, Y.; Mo, S.; et al. PTPRO Represses Colorectal Cancer Tumorigenesis and Progression by Reprogramming Fatty Acid Metabolism. Cancer Commun. 2022, 42, 848–867. [Google Scholar] [CrossRef]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8+ T Cells and Facilitates Anti–PD-1 Therapy. Cancer Immunol. Res. 2018, 6, 1375–1387. [Google Scholar] [CrossRef]
- Chandran, K.; Goswami, S.; Sharma-Walia, N. Implications of a Peroxisome Proliferator-Activated Receptor Alpha (PPARα) Ligand Clofibrate in Breast Cancer. Oncotarget 2016, 7, 15577–15599. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zheng, Z.; Chen, Q.; Pan, Y.; Quan, M.; Dai, Y. Fenofibrate Potentiates Chemosensitivity to Human Breast Cancer Cells by Modulating Apoptosis via AKT/NF-κB Pathway. OncoTargets Ther. 2019, 12, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Shureiqi, I.; Jiang, W.; Zuo, X.; Wu, Y.; Stimmel, J.B.; Leesnitzer, L.M.; Morris, J.S.; Fan, H.-Z.; Fischer, S.M.; Lippman, S.M. The 15-Lipoxygenase-1 Product 13-S-Hydroxyocta- Decadienoic Acid down-Regulates PPAR-δ to Induce Apoptosis in Colorectal Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9968–9973. [Google Scholar] [CrossRef]
- Coleman, J.D.; Prabhu, K.S.; Thompson, J.T.; Reddy, P.S.; Peters, J.M.; Peterson, B.R.; Reddy, C.C.; Vanden Heuvel, J.P. The Oxidative Stress Mediator 4-Hydroxynonenal Is an Intracellular Agonist of the Nuclear Receptor Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ). Free Radic. Biol. Med. 2007, 42, 1155–1164. [Google Scholar] [CrossRef]
- Burns, K.; Vandenheuvel, J. Modulation of PPAR Activity via Phosphorylation. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2007, 1771, 952–960. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of Peroxisome Proliferator-Activated Receptor δ Induces Fatty Acid β-Oxidation in Skeletal Muscle and Attenuates Metabolic Syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef]
- Winzell, M.S.; Wulff, E.M.; Olsen, G.S.; Sauerberg, P.; Gotfredsen, C.F.; Ahrén, B. Improved Insulin Sensitivity and Islet Function after PPARδ Activation in Diabetic Db/Db Mice. Eur. J. Pharmacol. 2010, 626, 297–305. [Google Scholar] [CrossRef]
- Lee, C.-H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.-W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARδ Regulates Glucose Metabolism and Insulin Sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Martín-Martín, N.; Zabala-Letona, A.; Fernández-Ruiz, S.; Arreal, L.; Camacho, L.; Castillo-Martin, M.; Cortazar, A.R.; Torrano, V.; Astobiza, I.; Zúñiga-García, P.; et al. PPARδ Elicits Ligand-Independent Repression of Trefoil Factor Family to Limit Prostate Cancer Growth. Cancer Res. 2018, 78, 399–409. [Google Scholar] [CrossRef]
- Zuo, X.; Xu, W.; Xu, M.; Tian, R.; Moussalli, M.J.; Mao, F.; Zheng, X.; Wang, J.; Morris, J.S.; Gagea, M.; et al. Metastasis Regulation by PPARD Expression in Cancer Cells. JCI Insight 2017, 2, e91419. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, A.; Schwager, C.; Kleeff, J.; Esposito, I.; Domhan, S.; Peschke, P.; Hauser, K.; Hahnfeldt, P.; Hlatky, L.; Debus, J.; et al. Transcriptional Network Governing the Angiogenic Switch in Human Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12890–12895. [Google Scholar] [CrossRef]
- Shen, B.; Li, A.; Wan, Y.-J.Y.; Shen, G.; Zhu, J.; Nie, Y. Lack of PPAR β/δ-Inactivated SGK-1 Is Implicated in Liver Carcinogenesis. BioMed Res. Int. 2020, 2020, 9563851. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Choi, Y.-K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.-G.; Jeon, H.-J.; Kim, J.-H.; Kim, J.-G.; et al. PPARδ Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol. Cancer Res. 2017, 15, 1230–1242. [Google Scholar] [CrossRef]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The Role of Peroxisome Proliferator-Activated Receptors in Carcinogenesis and Chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, J.; Ma, Q.; Wang, C.; Chen, K.; Meng, W.; Yu, Y.; Zhou, Z.; Sun, X. Knockdown of PPAR δ Gene Promotes the Growth of Colon Cancer and Reduces the Sensitivity to Bevacizumab in Nude Mice Model. PLoS ONE 2013, 8, e60715. [Google Scholar] [CrossRef]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.-J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-Fat Diet Enhances Stemness and Tumorigenicity of Intestinal Progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef]
- Yao, P.-L.; Morales, J.L.; Zhu, B.; Kang, B.-H.; Gonzalez, F.J.; Peters, J.M. Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPAR-β/δ) Inhibits Human Breast Cancer Cell Line Tumorigenicity. Mol. Cancer Ther. 2014, 13, 1008–1017. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.-J.; Xu, Z.; Spaner, D.E. PPAR-Delta Promotes Survival of Breast Cancer Cells in Harsh Metabolic Conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef]
- Coleman, J.D.; Thompson, J.T.; Smith, R.W.; Prokopczyk, B.; Vanden Heuvel, J.P. Role of Peroxisome Proliferator-Activated Receptor β/δ and B-Cell Lymphoma-6 in Regulation of Genes Involved in Metastasis and Migration in Pancreatic Cancer Cells. PPAR Res. 2013, 2013, 121956. [Google Scholar] [CrossRef]
- Parejo-Alonso, B.; Barneda, D.; Trabulo, S.M.D.; Courtois, S.; Compte-Sancerni, S.; Zurkovic, J.; Ruiz-Cañas, L.; Zheng, Q.; Tang, J.; Gaida, M.M.; et al. PPARδ Orchestrates a Prometastatic Metabolic Response to Microenvironmental Cues in Pancreatic Cancer. Cancer Res. 2025, 85, 3275–3291. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deguchi, Y.; Wei, D.; Liu, F.; Moussalli, M.J.; Deguchi, E.; Li, D.; Wang, H.; Valentin, L.A.; Colby, J.K.; et al. Rapid Acceleration of KRAS-Mutant Pancreatic Carcinogenesis via Remodeling of Tumor Immune Microenvironment by PPARδ. Nat. Commun. 2022, 13, 2665. [Google Scholar] [CrossRef] [PubMed]
- Abrego, J.; Sanford-Crane, H.; Oon, C.; Xiao, X.; Betts, C.B.; Sun, D.; Nagarajan, S.; Diaz, L.; Sandborg, H.; Bhattacharyya, S.; et al. A Cancer Cell-Intrinsic GOT2-PPARd Axis Suppresses Antitumor Immunity. Cancer Discov. 2022, 12, 2414–2433. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jiang, C.; Kim, M.; Xiao, Y.; Richter, H.J.; Guan, D.; Zhu, K.; Krusen, B.M.; Roberts, A.N.; Miller, J.; et al. Isoform-Specific Functions of PPARγ in Gene Regulation and Metabolism. Genes Dev. 2022, 36, 300–312. [Google Scholar] [CrossRef]
- Li, D.; Zhang, F.; Zhang, X.; Xue, C.; Namwanje, M.; Fan, L.; Reilly, M.P.; Hu, F.; Qiang, L. Distinct Functions of PPARγ Isoforms in Regulating Adipocyte Plasticity. Biochem. Biophys. Res. Commun. 2016, 481, 132–138. [Google Scholar] [CrossRef]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ Is a Major Driver of the Accumulation and Phenotype of Adipose Tissue Treg Cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef]
- Aprile, M.; Cataldi, S.; Ambrosio, M.R.; D’Esposito, V.; Lim, K.; Dietrich, A.; Blüher, M.; Savage, D.B.; Formisano, P.; Ciccodicola, A.; et al. PPARγΔ5, a Naturally Occurring Dominant-Negative Splice Isoform, Impairs PPARγ Function and Adipocyte Differentiation. Cell Rep. 2018, 25, 1577–1592.e6. [Google Scholar] [CrossRef]
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocytes Arise from Multiple Lineages That Are Heterogeneously and Dynamically Distributed. Nat. Commun. 2014, 5, 4099. [Google Scholar] [CrossRef]
- Rosen, E.D.; Hsu, C.-H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPα Induces Adipogenesis through PPARγ: A Unified Pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R.; Barrick, C.; Kim, K.-A.; Lindner, J.; Blondeau, B.; Fujimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of PPARγ in Adipose Tissues of Mice Protects against High Fat Diet-Induced Obesity and Insulin Resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Petrovic, N.; Lindgren, E.M.; Jacobsson, A.; Cannon, B. PPARγ in the Control of Brown Adipocyte Differentiation. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2005, 1740, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Lasar, D.; Rosenwald, M.; Kiehlmann, E.; Balaz, M.; Tall, B.; Opitz, L.; Lidell, M.E.; Zamboni, N.; Krznar, P.; Sun, W.; et al. Peroxisome Proliferator Activated Receptor Gamma Controls Mature Brown Adipocyte Inducibility through Glycerol Kinase. Cell Rep. 2018, 22, 760–773. [Google Scholar] [CrossRef]
- Broekema, M.F.; Savage, D.B.; Monajemi, H.; Kalkhoven, E. Gene-Gene and Gene-Environment Interactions in Lipodystrophy: Lessons Learned from Natural PPARγ Mutants. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2019, 1864, 715–732. [Google Scholar] [CrossRef]
- Chen, X.; Ma, Z.; Chen, P.; Song, X.; Li, W.; Yu, X.; Xie, J. Case Report: A New Peroxisome Proliferator-Activated Receptor Gamma Mutation Causes Familial Partial Lipodystrophy Type 3 in a Chinese Patient. Front. Endocrinol. 2022, 13, 830708. [Google Scholar] [CrossRef]
- Strand, D.W.; Jiang, M.; Murphy, T.A.; Yi, Y.; Konvinse, K.C.; Franco, O.E.; Wang, Y.; Young, J.D.; Hayward, S.W. PPARγ Isoforms Differentially Regulate Metabolic Networks to Mediate Mouse Prostatic Epithelial Differentiation. Cell Death Dis. 2012, 3, e361. [Google Scholar] [CrossRef]
- Kim, S.-H.; Hong, S.H.; Park, Y.-J.; Sung, J.-H.; Suh, W.; Lee, K.W.; Jung, K.; Lim, C.; Kim, J.-H.; Kim, H.; et al. MD001, a Novel Peroxisome Proliferator-Activated Receptor α/γ Agonist, Improves Glucose and Lipid Metabolism. Sci. Rep. 2019, 9, 1656. [Google Scholar] [CrossRef]
- Schneider, C.; Nobs, S.P.; Kurrer, M.; Rehrauer, H.; Thiele, C.; Kopf, M. Induction of the Nuclear Receptor PPAR-γ by the Cytokine GM-CSF Is Critical for the Differentiation of Fetal Monocytes into Alveolar Macrophages. Nat. Immunol. 2014, 15, 1026–1037. [Google Scholar] [CrossRef]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.A.; Thomazy, V.A.; Evans, R.M. PPARγ Promotes Monocyte/Macrophage Differentiation and Uptake of Oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-Inflammatory Properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef]
- Szatmari, I.; Töröcsik, D.; Agostini, M.; Nagy, T.; Gurnell, M.; Barta, E.; Chatterjee, K.; Nagy, L. PPARγ Regulates the Function of Human Dendritic Cells Primarily by Altering Lipid Metabolism. Blood 2007, 110, 3271–3280. [Google Scholar] [CrossRef]
- Erra Diaz, F.; Mazzitelli, I.; Bleichmar, L.; Melucci, C.; Thibodeau, A.; Dalotto Moreno, T.; Marches, R.; Rabinovich, G.A.; Ucar, D.; Geffner, J. Concomitant Inhibition of PPARγ and mTORC1 Induces the Differentiation of Human Monocytes into Highly Immunogenic Dendritic Cells. Cell Rep. 2023, 42, 112156. [Google Scholar] [CrossRef]
- Klotz, L.; Dani, I.; Edenhofer, F.; Nolden, L.; Evert, B.; Paul, B.; Kolanus, W.; Klockgether, T.; Knolle, P.; Diehl, L. Peroxisome Proliferator-Activated Receptor γ Control of Dendritic Cell Function Contributes to Development of CD4+ T Cell Anergy. J. Immunol. 2007, 178, 2122–2131. [Google Scholar] [CrossRef]
- Abdalla, H.B.; Napimoga, M.H.; Lopes, A.H.; De Macedo Maganin, A.G.; Cunha, T.M.; Van Dyke, T.E.; Clemente Napimoga, J.T. Activation of PPAR-γ Induces Macrophage Polarization and Reduces Neutrophil Migration Mediated by Heme Oxygenase 1. Int. Immunopharmacol. 2020, 84, 106565. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-Specific PPARγ Controls Alternative Activation and Improves Insulin Resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Isali, I.; McClellan, P.; Shankar, E.; Gupta, S.; Jain, M.; Anderson, J.M.; Hijaz, A.; Akkus, O. Genipin Guides and Sustains the Polarization of Macrophages to the Pro-Regenerative M2 Subtype via Activation of the pSTAT6-PPAR-Gamma Pathway. Acta Biomater. 2021, 131, 198–210. [Google Scholar] [CrossRef]
- Li, H.; Sorenson, A.L.; Poczobutt, J.; Amin, J.; Joyal, T.; Sullivan, T.; Crossno, J.T.; Weiser-Evans, M.C.M.; Nemenoff, R.A. Activation of PPARγ in Myeloid Cells Promotes Lung Cancer Progression and Metastasis. PLoS ONE 2011, 6, e28133. [Google Scholar] [CrossRef]
- Sippel, T.R.; Johnson, A.M.; Li, H.Y.; Hanson, D.; Nguyen, T.T.; Bullock, B.L.; Poczobutt, J.M.; Kwak, J.W.; Kleczko, E.K.; Weiser-Evans, M.C.; et al. Activation of PPARγ in Myeloid Cells Promotes Progression of Epithelial Lung Tumors through TGFβ1. Mol. Cancer Res. 2019, 17, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone Marrow Adipocytes Promote Tumor Growth in Bone via FABP4-Dependent Mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but Not Obese, Fat Is Enriched for a Unique Population of Regulatory T Cells That Affect Metabolic Parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Koshizuka, K.; Williamson, E.A.; Asou, H.; Said, J.W.; Holden, S.; Miyoshi, I.; Koeffler, H.P. Ligand for Peroxisome Proliferator-Activated Receptor Gamma (Troglitazone) Has Potent Antitumor Effect against Human Prostate Cancer Both in Vitro and in Vivo. Cancer Res. 1998, 58, 3344–3352. [Google Scholar]
- Qin, C.; Burghardt, R.; Smith, R.; Wormke, M.; Stewart, J.; Safe, S. Peroxisome Proliferator-Activated Receptor Gamma Agonists Induce Proteasome-Dependent Degradation of Cyclin D1 and Estrogen Receptor Alpha in MCF-7 Breast Cancer Cells. Cancer Res. 2003, 63, 958–964. [Google Scholar]
- Kato, Y.; Ying, H.; Zhao, L.; Furuya, F.; Araki, O.; Willingham, M.C.; Cheng, S. PPARγ Insufficiency Promotes Follicular Thyroid Carcinogenesis via Activation of the Nuclear Factor-κB Signaling Pathway. Oncogene 2006, 25, 2736–2747. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Y.; Huang, X.-B.; Zhao, Y.-J.; Wang, H.-G.; Wang, J.-B.; Liu, L.-C.; Wang, L.-Q.; Zhong, Q.; Xie, J.-W.; Lin, J.-X.; et al. The Peroxisome Proliferator-Activated Receptor Agonist Rosiglitazone Specifically Represses Tumour Metastatic Potential in Chromatin Inaccessibility-Mediated FABP4-Deficient Gastric Cancer. Theranostics 2022, 12, 1904–1920. [Google Scholar] [CrossRef] [PubMed]
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome Proliferator-Activated Receptor-γ Activation Inhibits Tumor Metastasis by Antagonizing Smad3-Mediated Epithelial-Mesenchymal Transition. Mol. Cancer Ther. 2010, 9, 3221–3232. [Google Scholar] [CrossRef]
- Shen, B.; Chu, E.S.H.; Zhao, G.; Man, K.; Wu, C.-W.; Cheng, J.T.Y.; Li, G.; Nie, Y.; Lo, C.M.; Teoh, N.; et al. PPARgamma Inhibits Hepatocellular Carcinoma Metastases in Vitro and in Mice. Br. J. Cancer 2012, 106, 1486–1494. [Google Scholar] [CrossRef]
- Tsubouchi, Y.; Sano, H.; Kawahito, Y.; Mukai, S.; Yamada, R.; Kohno, M.; Inoue, K.; Hla, T.; Kondo, M. Inhibition of Human Lung Cancer Cell Growth by the Peroxisome Proliferator-Activated Receptor-γ Agonists through Induction of Apoptosis. Biochem. Biophys. Res. Commun. 2000, 270, 400–405. [Google Scholar] [CrossRef]
- Bren-Mattison, Y.; Van Putten, V.; Chan, D.; Winn, R.; Geraci, M.W.; Nemenoff, R.A. Peroxisome Proliferator-Activated Receptor-γ (PPARγ) Inhibits Tumorigenesis by Reversing the Undifferentiated Phenotype of Metastatic Non-Small-Cell Lung Cancer Cells (NSCLC). Oncogene 2005, 24, 1412–1422. [Google Scholar] [CrossRef]
- To, K.K.W.; Wu, W.K.K.; Loong, H.H.F. PPARgamma Agonists Sensitize PTEN-Deficient Resistant Lung Cancer Cells to EGFR Tyrosine Kinase Inhibitors by Inducing Autophagy. Eur. J. Pharmacol. 2018, 823, 19–26. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Cione, E.; Qi, H.; Pingitore, A.; Perri, M.; Catalano, S.; Vizza, D.; Panno, M.L.; Genchi, G.; Fuqua, S.A.W.; et al. Combined Low Doses of PPARγ and RXR Ligands Trigger an Intrinsic Apoptotic Pathway in Human Breast Cancer Cells. Am. J. Pathol. 2009, 175, 1270–1280. [Google Scholar] [CrossRef]
- Rochel, N.; Krucker, C.; Coutos-Thévenot, L.; Osz, J.; Zhang, R.; Guyon, E.; Zita, W.; Vanthong, S.; Hernandez, O.A.; Bourguet, M.; et al. Recurrent Activating Mutations of PPARγ Associated with Luminal Bladder Tumors. Nat. Commun. 2019, 10, 253. [Google Scholar] [CrossRef]
- Sanchez, D.J.; Missiaen, R.; Skuli, N.; Steger, D.J.; Simon, M.C. Cell-Intrinsic Tumorigenic Functions of PPARγ in Bladder Urothelial Carcinoma. Mol. Cancer Res. 2021, 19, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.T.; Berger, A.C.; Shih, J.; Duke, F.F.; Furst, L.; Kwiatkowski, D.J.; Cherniack, A.D.; Meyerson, M.; Strathdee, C.A. Genomic Activation of PPARG Reveals a Candidate Therapeutic Axis in Bladder Cancer. Cancer Res. 2017, 77, 6987–6998. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed Neuroblastomas Show Frequent RAS-MAPK Pathway Mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.-Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and Temporal Homogeneity of Driver Mutations in Diffuse Intrinsic Pontine Glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef]
- Eckert, M.A.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations: Acquired Genetic Lability Permits Stepwise Selection of Variant Sublines and Underlies Tumor Progression. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A Cell Initiating Human Acute Myeloid Leukaemia after Transplantation into SCID Mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a Subpopulation of Cells with Cancer Stem Cell Properties in Head and Neck Squamous Cell Carcinoma. Proc. Natl. Acad. Sci. USA. 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and Expansion of the Tumorigenic Lung Cancer Stem Cell Population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly Purified CD44+ Prostate Cancer Cells from Xenograft Human Tumors Are Enriched in Tumorigenic and Metastatic Progenitor Cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A Human Colon Cancer Cell Capable of Initiating Tumour Growth in Immunodeficient Mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wei, Q.; Utomo, V.; Nadesan, P.; Whetstone, H.; Kandel, R.; Wunder, J.S.; Alman, B.A. Side Population Cells Isolated from Mesenchymal Neoplasms Have Tumor Initiating Potential. Cancer Res. 2007, 67, 8216–8222. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.-L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting Stromal Remodeling and Cancer Stem Cell Plasticity Overcomes Chemoresistance in Triple Negative Breast Cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt Activity Defines Colon Cancer Stem Cells and Is Regulated by the Microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of Epithelial-Mesenchymal Transition Phenotype of Gemcitabine-Resistant Pancreatic Cancer Cells Is Linked with Activation of the Notch Signaling Pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, L.; He, C.; Xu, F.; Liu, J.; Hu, Z.; Zhao, L.; Tian, Y. Activation of Notch-1 Enhances Epithelial-Mesenchymal Transition in Gefitinib-Acquired Resistant Lung Cancer Cells. J. Cell Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef]
- O’Brien-Ball, C.; Biddle, A. Reprogramming to Developmental Plasticity in Cancer Stem Cells. Dev. Biol. 2017, 430, 266–274. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of Cancer Stem Cell Metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Virchow, R. An Address on the Value of Pathological Experiments. Br. Med. J. 1881, 2, 198–203. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Togashi, K.; Sanomachi, T.; Kitanaka, C.; Okada, M. Inhibition of the Lipid Droplet-Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties. Genes 2021, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, L.; Wei, J.; Xiong, Y.; DuBois, R.N. PPARδ Mediates the Effect of Dietary Fat in Promoting Colorectal Cancer Metastasis. Cancer Res. 2019, 79, 4480–4490. [Google Scholar] [CrossRef]
- Mascaraque, M.; Courtois, S.; Royo-García, A.; Barneda, D.; Stoian, A.M.; Villaoslada, I.; Espiau-Romera, P.; Bokil, A.; Cano-Galiano, A.; Jagust, P.; et al. Fatty Acid Oxidation Is Critical for the Tumorigenic Potential and Chemoresistance of Pancreatic Cancer Stem Cells. J. Transl. Med. 2024, 22, 797. [Google Scholar] [CrossRef]
- Sabatino, L.; Pancione, M.; Votino, C.; Colangelo, T.; Lupo, A.; Novellino, E.; Lavecchia, A.; Colantuoni, V. Emerging Role of the β-Catenin-PPARγ Axis in the Pathogenesis of Colorectal Cancer. World J. Gastroenterol. 2014, 20, 7137–7151. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the Canonical NF-κB and Notch Signaling Pathways Inhibits Pparγ Expression and Promotes Pancreatic Cancer Progression in Mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef]
- Bhatia, B.; Potts, C.R.; Guldal, C.; Choi, S.; Korshunov, A.; Pfister, S.; Kenney, A.M.; Nahlé, Z.A. Hedgehog-Mediated Regulation of PPARγ Controls Metabolic Patterns in Neural Precursors and Shh-Driven Medulloblastoma. Acta Neuropathol. 2012, 123, 587–600. [Google Scholar] [CrossRef]
- Song, S.; Wang, Z.; Li, Y.; Ma, L.; Jin, J.; Scott, A.W.; Xu, Y.; Estrella, J.S.; Song, Y.; Liu, B.; et al. PPARδ Interacts with the Hippo Coactivator YAP1 to Promote SOX9 Expression and Gastric Cancer Progression. Mol. Cancer Res. 2020, 18, 390–402. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Bao, S.; Lee, C.; Nordman, E.; Wang, X.; Lao, K.; Surani, M.A. Tracing the Derivation of Embryonic Stem Cells from the Inner Cell Mass by Single-Cell RNA-Seq Analysis. Cell Stem Cell 2010, 6, 468–478. [Google Scholar] [CrossRef]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Valle, S.; Alcalá, S.; Martin-Hijano, L.; Cabezas-Sáinz, P.; Navarro, D.; Muñoz, E.R.; Yuste, L.; Tiwary, K.; Walter, K.; Ruiz-Cañas, L.; et al. Exploiting Oxidative Phosphorylation to Promote the Stem and Immunoevasive Properties of Pancreatic Cancer Stem Cells. Nat. Commun. 2020, 11, 5265. [Google Scholar] [CrossRef]
- Courtois, S.; de Luxán-Delgado, B.; Penin-Peyta, L.; Royo-García, A.; Parejo-Alonso, B.; Jagust, P.; Alcalá, S.; Rubiolo, J.A.; Sánchez, L.; Sainz, B.; et al. Inhibition of Mitochondrial Dynamics Preferentially Targets Pancreatic Cancer Cells with Enhanced Tumorigenic and Invasive Potential. Cancers 2021, 13, 698. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-L.; Sun, Y.-F.; Wang, B.-L.; Shen, M.-N.; Zhou, Y.; Chen, J.-W.; Hu, B.; Gong, Z.-J.; Zhang, X.; Cao, Y.; et al. Sphere-Forming Culture Enriches Liver Cancer Stem Cells and Reveals Stearoyl-CoA Desaturase 1 as a Potential Therapeutic Target. BMC Cancer 2019, 19, 760. [Google Scholar] [CrossRef]
- Haynes, H.R.; White, P.; Hares, K.M.; Redondo, J.; Kemp, K.C.; Singleton, W.G.B.; Killick-Cole, C.L.; Stevens, J.R.; Garadi, K.; Guglani, S.; et al. The Transcription Factor PPARα Is Overexpressed and Is Associated with a Favourable Prognosis in IDH-Wildtype Primary Glioblastoma. Histopathology 2017, 70, 1030–1043. [Google Scholar] [CrossRef]
- Haynes, H.R.; Scott, H.L.; Killick-Cole, C.L.; Shaw, G.; Brend, T.; Hares, K.M.; Redondo, J.; Kemp, K.C.; Ballesteros, L.S.; Herman, A.; et al. shRNA-Mediated PPARα Knockdown in Human Glioma Stem Cells Reduces in Vitro Proliferation and Inhibits Orthotopic Xenograft Tumour Growth. J. Pathol. 2019, 247, 422–434. [Google Scholar] [CrossRef]
- Tabrizian, T.; Wang, D.; Guan, F.; Hu, Z.; Beck, A.P.; Delahaye, F.; Huffman, D.M. Apc Inactivation, but Not Obesity, Synergizes with Pten Deficiency to Drive Intestinal Stem Cell-Derived Tumorigenesis. Endocr. Relat. Cancer 2017, 24, 253–265. [Google Scholar] [CrossRef]
- Alonso, S.; Yilmaz, Ö.H. Nutritional Regulation of Intestinal Stem Cells. Annu. Rev. Nutr. 2018, 38, 273–301. [Google Scholar] [CrossRef]
- Papi, A.; Guarnieri, T.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; De Carolis, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; et al. Nuclear Receptors Agonists Exert Opposing Effects on the Inflammation Dependent Survival of Breast Cancer Stem Cells. Cell Death Differ. 2012, 19, 1208–1219. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, H.; Xu, D.; Ma, A.; Zhang, L.; Sun, J.; Yang, Z.; Liu, Y.; Shi, G. The Combinatory Effects of PPAR-γ Agonist and Survivin Inhibition on the Cancer Stem-like Phenotype and Cell Proliferation in Bladder Cancer Cells. Int. J. Mol. Med. 2014, 34, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Chearwae, W.; Bright, J.J. PPARgamma Agonists Inhibit Growth and Expansion of CD133+ Brain Tumour Stem Cells. Br. J. Cancer 2008, 99, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Pestereva, E.; Kanakasabai, S.; Bright, J.J. PPARγ Agonists Regulate the Expression of Stemness and Differentiation Genes in Brain Tumour Stem Cells. Br. J. Cancer 2012, 106, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Bigoni-Ordóñez, G.D.; Ortiz-Sánchez, E.; Rosendo-Chalma, P.; Valencia-González, H.A.; Aceves, C.; García-Carrancá, A. Molecular Iodine Inhibits the Expression of Stemness Markers on Cancer Stem-like Cells of Established Cell Lines Derived from Cervical Cancer. BMC Cancer 2018, 18, 928. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, N.; Liu, C.; Li, J.; Bai, Y.; Lei, S.; Huang, Q.; Sun, L.; Tang, L.; Zeng, C.; et al. BEX1 Supports the Stemness of Hepatoblastoma by Facilitating Warburg Effect in a PPARγ/PDK1 Dependent Manner. Br. J. Cancer 2023, 129, 1477–1489. [Google Scholar] [CrossRef]
- Gu, Y.; Wei, W.; Cheng, Y.; Wan, B.; Ding, X.; Wang, H.; Zhang, Y.; Jin, M. A Pivotal Role of BEX1 in Liver Progenitor Cell Expansion in Mice. Stem Cell Res. Ther. 2018, 9, 164. [Google Scholar] [CrossRef]
- Abdullah, L.N.; Chow, E.K. Mechanisms of Chemoresistance in Cancer Stem Cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer Stem Cells (CSCs): Metabolic Strategies for Their Identification and Eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- Takeishi, S.; Nakayama, K.I. To Wake up Cancer Stem Cells, or to Let Them Sleep, That Is the Question. Cancer Sci. 2016, 107, 875–881. [Google Scholar] [CrossRef]
- Domenichini, A.; Edmands, J.S.; Adamska, A.; Begicevic, R.-R.; Paternoster, S.; Falasca, M. Pancreatic Cancer Tumorspheres Are Cancer Stem-like Cells with Increased Chemoresistance and Reduced Metabolic Potential. Adv. Biol. Regul. 2019, 72, 63–77. [Google Scholar] [CrossRef]
- Song, B.; Wang, Y.; Titmus, M.A.; Botchkina, G.; Formentini, A.; Kornmann, M.; Ju, J. Molecular Mechanism of Chemoresistance by miR-215 in Osteosarcoma and Colon Cancer Cells. Mol. Cancer 2010, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Bizzoca, M.E.; Caponio, V.C.A.; Lo Muzio, L.; Claudio, P.P.; Cortese, A. Methods for Overcoming Chemoresistance in Head and Neck Squamous Cell Carcinoma: Keeping the Focus on Cancer Stem Cells, a Systematic Review. Cancers 2024, 16, 3004. [Google Scholar] [CrossRef] [PubMed]
- Loaiza, B.; Rojas, E.; Valverde, M. The New Model of Carcinogenesis: The Cancer Stem Cell Hypothesis. In Carcinogen; IntechOpen: London, UK, 2012; ISBN 978-953-51-0658-6. [Google Scholar][Green Version]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Miranda-Lorenzo, I.; Dorado, J.; Lonardo, E.; Alcala, S.; Serrano, A.G.; Clausell-Tormos, J.; Cioffi, M.; Megias, D.; Zagorac, S.; Balic, A.; et al. Intracellular Autofluorescence: A Biomarker for Epithelial Cancer Stem Cells. Nat. Methods 2014, 11, 1161–1169. [Google Scholar] [CrossRef]
- Li, H.; Jiang, W.; Liu, X.-N.; Yuan, L.-Y.; Li, T.-J.; Li, S.; Xu, S.-S.; Zhang, W.-H.; Gao, H.-L.; Han, X.; et al. TET1 Downregulates Epithelial-Mesenchymal Transition and Chemoresistance in PDAC by Demethylating CHL1 to Inhibit the Hedgehog Signaling Pathway. Oncogene 2020, 39, 5825–5838. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Li, J.; Sun, H.; Liu, S.; Cui, Y.; Li, F. HES 1 Is Essential for Chemoresistance Induced by Stellate Cells and Is Associated with Poor Prognosis in Pancreatic Cancer. Oncol. Rep. 2015, 33, 1883–1889. [Google Scholar] [CrossRef]
- Wu, D.; Liu, L.; Yan, X.; Wang, C.; Wang, Y.; Han, K.; Lin, S.; Gan, Z.; Min, D. Pleiotrophin Promotes Chemoresistance to Doxorubicin in Osteosarcoma by Upregulating P-Glycoprotein. Oncotarget 2017, 8, 63857–63870. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Y.; Xu, Q.; Shi, H.; Shi, J.; Hou, Y. PPARδ Promotes Tumor Progression via Activation of Glut1 and SLC1-A5 Transcription. Carcinogenesis 2017, 38, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.-S.; Li, T.; Lin, J.-F.; Qiu, M.-Z.; Wang, D.-S.; Liu, Z.-X.; Chen, Z.-H.; Yang, L.-P.; Zhang, X.-L.; Zhao, Q.; et al. VDR-SOX2 Signaling Promotes Colorectal Cancer Stemness and Malignancy in an Acidic Microenvironment. Signal Transduct. Target. Ther. 2020, 5, 183. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Z.; Wu, H.; Wang, B.; Ouyang, Y.; Liu, J.; Zheng, X.; Zhang, H.; Li, X.; Feng, X.; et al. Adipocyte-Rich Microenvironment Promotes Chemoresistance via Upregulation of Peroxisome Proliferator-Activated Receptor Gamma/ABCG2 in Epithelial Ovarian Cancer. Int. J. Mol. Med. 2024, 53, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, G.; Yang, Z.; Wang, L.; Zhang, L.; Wang, T.; Zhang, Y.; Zhang, S.; Han, Y.; Jia, L. Uncoupling Protein 2 Downregulation by Hypoxia through Repression of Peroxisome Proliferator-Activated Receptor g Promotes Chemoresistance of Non-Small Cell Lung Cancer. Oncotarget 2016, 8, 8083. [Google Scholar] [CrossRef]
- Ni, J.; Zhou, L.L.; Ding, L.; Zhao, X.; Cao, H.; Fan, F.; Li, H.; Lou, R.; Du, Y.; Dong, S.; et al. PPARγ Agonist Efatutazone and Gefitinib Synergistically Inhibit the Proliferation of EGFR-TKI-Resistant Lung Adenocarcinoma Cells via the PPARγ/PTEN/Akt Pathway. Exp. Cell Res. 2017, 361, 246–256. [Google Scholar] [CrossRef]
- Jolly, M.K. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Dimitrov-Markov, S.; Perales-Patón, J.; Bockorny, B.; Dopazo, A.; Muñoz, M.; Baños, N.; Bonilla, V.; Menendez, C.; Duran, Y.; Huang, L.; et al. Discovery of New Targets to Control Metastasis in Pancreatic Cancer by Single-Cell Transcriptomics Analysis of Circulating Tumor Cells. Mol. Cancer Ther. 2020, 19, 1751–1760. [Google Scholar] [CrossRef]
- Palamaris, K.; Felekouras, E.; Sakellariou, S. Epithelial to Mesenchymal Transition: Key Regulator of Pancreatic Ductal Adenocarcinoma Progression and Chemoresistance. Cancers 2021, 13, 5532. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McCallister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Poruk, K.E.; Blackford, A.L.; Weiss, M.J.; Cameron, J.L.; He, J.; Goggins, M.; Rasheed, Z.A.; Wolfgang, C.L.; Wood, L.D. Circulating Tumor Cells Expressing Markers of Tumor-Initiating Cells Predict Poor Survival and Cancer Recurrence in Patients with Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-W.; Chou, C.-T.; Chang, C.-C.; Li, Y.-J.; Chen, S.-T.; Lin, I.-C.; Kok, S.-H.; Cheng, S.-J.; Lee, J.-J.; Wu, T.-S.; et al. HMGCS2 Enhances Invasion and Metastasis via Direct Interaction with PPARα to Activate Src Signaling in Colorectal Cancer and Oral Cancer. Oncotarget 2017, 8, 22460–22476. [Google Scholar] [CrossRef]
- Leng, J.; Li, H.; Niu, Y.; Chen, K.; Yuan, X.; Chen, H.; Fu, Z.; Zhang, L.; Wang, F.; Chen, C.; et al. Low-Dose Mono(2-Ethylhexyl) Phthalate Promotes Ovarian Cancer Development through PPARα-Dependent PI3K/Akt/NF-κB Pathway. Sci. Total Environ. 2021, 790, 147990. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lan, W.; Xu, M.; Song, J.; Mao, J.; Li, C.; Du, X.; Jiang, Y.; Li, E.; Zhang, R.; et al. Cancer-Associated Fibroblast-Derived SDF-1 Induces Epithelial-Mesenchymal Transition of Lung Adenocarcinoma via CXCR4/β-Catenin/PPARδ Signalling. Cell Death Dis. 2021, 12, 214. [Google Scholar] [CrossRef]
- Lim, J.; Kwan, Y.; Tan, M.; Teo, M.; Chiba, S.; Wahli, W.; Wang, X. The Role of PPARβ/δ in Melanoma Metastasis. Int. J. Mol. Sci. 2018, 19, 2860. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.T.; Sung, M.T.; Lee, C.C.; Kuo, Y.J.; Chi, C.W.; Lee, H.C.; Hsia, C.Y. Peroxisome Proliferator-Activated Receptor γ Expression Is Inversely Associated with Macroscopic Vascular Invasion in Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2016, 17, 1226. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.W.; Lin, D.Q.; Cao, L.Q. Peroxisome Proliferator-activated Receptor-γ Inhibits Pancreatic Cancer Cell Invasion and Metastasis via Regulating MMP-2 Expression through PTEN. Mol. Med. Rep. 2015, 12, 6255–6260. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Romano, S.; Tufano, M.; D’Arrigo, P.; Vigorito, V.; Russo, S.; Romano, M.F. Cell Stemness, Epithelial-to-Mesenchymal Transition, and Immunoevasion: Intertwined Aspects in Cancer Metastasis. Semin. Cancer Biol. 2020, 60, 181–190. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Shiota, G. Immune Evasion by Cancer Stem Cells. Regen. Ther. 2021, 17, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-C.; Chao, Y.-J.; Hsieh, M.-H.; Tung, H.-L.; Wang, H.-C.; Shan, Y.-S. Low CD8+ T Cell Infiltration and High PD-L1 Expression Are Associated with Level of CD44+/CD133+ Cancer Stem Cells and Predict an Unfavorable Prognosis in Pancreatic Cancer. Cancers 2019, 11, 541. [Google Scholar] [CrossRef] [PubMed]
- López-Gil, J.C.; García-Silva, S.; Ruiz-Cañas, L.; Navarro, D.; Palencia-Campos, A.; Giráldez-Trujillo, A.; Earl, J.; Dorado, J.; Gómez-López, G.; Monfort-Vengut, A.; et al. The Peptidoglycan Recognition Protein 1 Confers Immune Evasive Properties on Pancreatic Cancer Stem Cells. Gut 2024, 73, 1489. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Karakhanova, S.; Huang, X.; Deng, S.P.; Werner, J.; Bazhin, A.V. Influence of Interferon-α on the Expression of the Cancer Stem Cell Markers in Pancreatic Carcinoma Cells. Exp. Cell Res. 2014, 324, 146–156. [Google Scholar] [CrossRef]
- Sainz, B.; Martín, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 Is a Critical Microenvironmental Factor for Pancreatic Cancer Stem Cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef]
- Korpal, M.; Puyang, X.; Jeremy Wu, Z.; Seiler, R.; Furman, C.; Oo, H.Z.; Seiler, M.; Irwin, S.; Subramanian, V.; Julie Joshi, J.; et al. Evasion of Immunosurveillance by Genomic Alterations of PPARγ/RXRα in Bladder Cancer. Nat. Commun. 2017, 8, 103. [Google Scholar] [CrossRef]
- Wejksza, K.; Lee-Chang, C.; Bodogai, M.; Bonzo, J.; Gonzalez, F.J.; Lehrmann, E.; Becker, K.; Biragyn, A. Cancer-Produced Metabolites of 5-Lipoxygenase Induce Tumor-Evoked Regulatory B Cells via Peroxisome Proliferator–Activated Receptor α. J. Immunol. 2013, 190, 2575–2584. [Google Scholar] [CrossRef]
- Yin, X.; Zeng, W.; Wu, B.; Wang, L.; Wang, Z.; Tian, H.; Wang, L.; Jiang, Y.; Clay, R.; Wei, X.; et al. PPARα Inhibition Overcomes Tumor-Derived Exosomal Lipid-Induced Dendritic Cell Dysfunction. Cell Rep. 2020, 33, 108278. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 Activation of Kupffer Cells by PPARδ Ameliorates Obesity-Induced Insulin Resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Qin, J.; Qian, Y.; Huang, C.; Liu, X.; Wang, N.; Li, L.; Chao, Y.; Tan, B.; Zhang, N.; et al. FFAR2 Expressing Myeloid-Derived Suppressor Cells Drive Cancer Immunoevasion. J. Hematol. Oncol. 2024, 17, 9. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P.; et al. Efatutazone, an Oral PPAR-γ Agonist, in Combination With Paclitaxel in Anaplastic Thyroid Cancer: Results of a Multicenter Phase 1 Trial. J. Clin. Endocrinol. Metab. 2013, 98, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Shi, W.; Fu, S.; Wang, T.; Zhai, S.; Song, Y.; Han, J. Pioglitazone and Bladder Cancer Risk: A Systematic Review and Meta-analysis. Cancer Med. 2018, 7, 1070–1080. [Google Scholar] [CrossRef]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.A.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefèbvre, P.J.; Murray, G.D.; et al. Secondary Prevention of Macrovascular Events in Patients with Type 2 Diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): A Randomised Controlled Trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Home, P.D.; Pocock, S.J.; Beck-Nielsen, H.; Gomis, R.; Hanefeld, M.; Jones, N.P.; Komajda, M.; McMurray, J.J.V. RECORD Study Group Rosiglitazone Evaluated for Cardiovascular Outcomes—An Interim Analysis. N. Engl. J. Med. 2007, 357, 28–38. [Google Scholar] [CrossRef]
- Lago, R.M.; Singh, P.P.; Nesto, R.W. Congestive Heart Failure and Cardiovascular Death in Patients with Prediabetes and Type 2 Diabetes given Thiazolidinediones: A Meta-Analysis of Randomised Clinical Trials. Lancet 2007, 370, 1129–1136. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of Rosiglitazone on the Risk of Myocardial Infarction and Death from Cardiovascular Causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- Home, P.D.; Pocock, S.J.; Beck-Nielsen, H.; Curtis, P.S.; Gomis, R.; Hanefeld, M.; Jones, N.P.; Komajda, M.; McMurray, J.J.V. RECORD Study Team Rosiglitazone Evaluated for Cardiovascular Outcomes in Oral Agent Combination Therapy for Type 2 Diabetes (RECORD): A Multicentre, Randomised, Open-Label Trial. Lancet 2009, 373, 2125–2135. [Google Scholar] [CrossRef]
- Kohlroser, J.; Mathai, J.; Reichheld, J.; Banner, B.F.; Bonkovsky, H.L. Hepatotoxicity Due to Troglitazone: Report of Two Cases and Review of Adverse Events Reported to the United States Food and Drug Administration. Am. J. Gastroenterol. 2000, 95, 272–276. [Google Scholar] [CrossRef]
- Graham, D.J.; Drinkard, C.R.; Shatin, D. Incidence of Idiopathic Acute Liver Failure and Hospitalized Liver Injury in Patients Treated with Troglitazone. Am. J. Gastroenterol. 2003, 98, 175–179. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Caldwell, S.H.; Pyrsopoulos, N.; deLemos, A.S.; Rossi, S.; Levy, C.; Goldberg, D.S.; Mena, E.A.; Sheikh, A.; Ravinuthala, R.; et al. Proof-of-Concept Study to Evaluate the Safety and Efficacy of Saroglitazar in Patients with Primary Biliary Cholangitis. J. Hepatol. 2022, 76, 75–85. [Google Scholar] [CrossRef]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Sims, R.; Mertz, J.A.; Wilson, J.E.; Audia, J.E.; Williamson, K.E.; Li, Y.; Kuljanin, M.; Motely, W.W.; DeLaBarre, B.; Bowden, M.; et al. Abstract ND08: Discovery of FX-909, a First-in-Class Inverse Agonist of the Peroxisome Proliferator-Activated Receptor Gamma (PPARG) Lineage Transcription Factor, to Potentially Treat Patients with the Luminal Subtype of Advanced Urothelial Cancer (UC). Cancer Res. 2023, 83, ND08. [Google Scholar] [CrossRef]
- Iyer, G.; Gao, X.; Rasco, D.W.; Milowsky, M.I.; Garmezy, B.; Rodriguez Rivera, I.I.; Tepper, J.; Moles, M.A.; Gjini, E.; Bowden, M.; et al. A Phase 1, First-in-Human, Dose-Escalation and Expansion Study of FX-909 in Patients with Advanced Solid Malignancies, Including Advanced Urothelial Carcinoma. J. Clin. Oncol. 2024, 42, TPS709. [Google Scholar] [CrossRef]
- Yarchoan, M.; Powderly, J.D.; Bastos, B.R.; Karasic, T.B.; Crysler, O.V.; Munster, P.N.; McKean, M.A.; Emens, L.A.; Saenger, Y.M.; Ged, Y.; et al. First-in-Human Phase I Trial of TPST-1120, an Inhibitor of PPARα, as Monotherapy or in Combination with Nivolumab, in Patients with Advanced Solid Tumors. Cancer Res. Commun. 2024, 4, 1100–1110. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).