
Modulating Neuroinflammation as a Prospective Therapeutic Target in Alzheimer’s Disease

Abstract
:1. Alzheimer’s Disease
2. Neuroinflammation
2.1. Microglia
2.2. Astrocytes
3. Receptors Associated with Microglia in Neuroinflammation and AD
3.1. Toll-like Receptors
3.2. Purinergic Receptors
3.3. TREM2
4. Signaling Associated with Microglia in Neuroinflammation and AD
4.1. JAK/STAT Signaling Pathway
4.2. NOD-like Receptor Signaling
4.3. MAPK Signaling
4.4. PI3K/AKT Signaling
4.5. Cyclic Nucleotide Signaling
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Santos, C.Y.; Snyder, P.J.; Wu, W.-C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 7, 69–87. [Google Scholar] [CrossRef]
- Anjum, I.; Fayyaz, M.; Wajid, A.; Sohail, W.; Ali, A. Does obesity increase the risk of dementia: A literature review. Cureus 2018, 10, e2660. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hyman, B.T.; Serrano-Pozo, A. Multifaceted roles of APOE in Alzheimer disease. Nat. Rev. Neurol. 2024, 20, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Normal and abnormal biology of the! b-amyloid precursor protein. Annu. Rev. Neurosci. 1994, 17, 489–517. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Alonso, A.d.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Ausó, E.; Gómez-Vicente, V.; Esquiva, G. Biomarkers for Alzheimer’s disease early diagnosis. J. Pers. Med. 2020, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2024. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2024, 10, e12465. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Garbuz, D.; Zatsepina, O.; Evgen’ev, M. Beta amyloid, tau protein, and neuroinflammation: An attempt to integrate different hypotheses of Alzheimer’s disease pathogenesis. Mol. Biol. 2021, 55, 670–682. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Hack, C.; Rozemuller, J.; Stam, F. Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch. B 1988, 56, 259–262. [Google Scholar] [CrossRef]
- Batista, A.F.; Khan, K.A.; Papavergi, M.-T.; Lemere, C.A. The importance of complement-mediated immune signaling in Alzheimer’s disease pathogenesis. Int. J. Mol. Sci. 2024, 25, 817. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.; Stanley, L.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.; White, C., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef]
- Hüll, M.; Berger, M.; Volk, B.; Bauer, J. Occurrence of Interleukin-6 in Cortical Plaques of Alzheimer’s Disease Patients May Precede Transformation of Diffuse into Neuritic Plaques a. Ann. N. Y. Acad. Sci. 1996, 777, 205–212. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.S.M.; McGeer, E.G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology 1996, 47, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.M.; Katsouri, L.; Sastre, M. Modulation of inflammation in transgenic models of Alzheimer’s disease. J. Neuroinflammation 2014, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Jaturapatporn, D.; Isaac, M.G.E.K.N.; McCleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012, 2012, CD006378. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Ríos, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Davis, K.L. Inflammatory mechanisms in Alzheimer’s disease: Implications for therapy. Am. J. Psychiatry 1994, 151, 1105–1113. [Google Scholar] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.; Akiyama, H.; Itagaki, S.; McGeer, E. Immune system response in Alzheimer’s disease. Can. J. Neurol. Sci. 1989, 16, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 80, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Ransohoff, R.M. Crosstalk between astrocytes and microglia: An overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, X.; Qian, L.; Wilson, B.; Lee, C.; Flood, P.; Langenbach, R.; Hong, J.-S. Prostaglandin E2 released from activated microglia enhances astrocyte proliferation in vitro. Toxicol. Appl. Pharmacol. 2009, 238, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhou, X.; Zhang, C.; Wang, K.; Liu, X.; Che, Y.; Sun, F.; Li, H.; Wang, Q.; Zhang, D. NADPH oxidase-derived H2O2 mediates the regulatory effects of microglia on astrogliosis in experimental models of Parkinson’s disease. Redox Biol. 2017, 12, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Fenn, A.M.; Dugan, A.; Godbout, J.P. TGFβ produced by IL-10 redirected astrocytes attenuates microglial activation. Glia 2014, 62, 881–895. [Google Scholar] [CrossRef]
- Tanuma, N.; Sakuma, H.; Sasaki, A.; Matsumoto, Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006, 112, 195–204. [Google Scholar] [CrossRef]
- Jo, M.; Kim, J.-H.; Song, G.J.; Seo, M.; Hwang, E.M.; Suk, K. Astrocytic orosomucoid-2 modulates microglial activation and neuroinflammation. J. Neurosci. 2017, 37, 2878–2894. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Kofler, J.; Wiley, C.A. Microglia: Key innate immune cells of the brain. Toxicol. Pathol. 2011, 39, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A. Regulation of neuroimmune processes by damage-and resolution-associated molecular patterns. Neural Regen. Res. 2021, 16, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Wendimu, M.Y.; Hooks, S.B. Microglia phenotypes in aging and neurodegenerative diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Itriago, A.; Radford, R.A.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, W.; Zhang, J. A richer and more diverse future for microglia phenotypes. Heliyon 2023, 9, e14421. [Google Scholar] [CrossRef]
- Rohan Walker, F.; Nilsson, M.; Jones, K. Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr. Drug Targets 2013, 14, 1262–1276. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Jimenez, S.; Baglietto-Vargas, D.; Caballero, C.; Moreno-Gonzalez, I.; Torres, M.; Sanchez-Varo, R.; Ruano, D.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: Age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 2008, 28, 11650–11661. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Gordon, M.N. Diversity of transcriptomic microglial phenotypes in aging and Alzheimer’s disease. Alzheimer’s Dement. 2022, 18, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; Carrier, M.; Tremblay, M.-È. Morphology of microglia across contexts of health and disease. Microglia Methods Protoc. 2019, 2034, 13–26. [Google Scholar]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.Y.; Ma, C.H.E. Bipolar/rod-shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Sci. Rep. 2014, 4, 7279. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.Y.; Au, N.P.B.; Ma, C.H.E. The association between laminin and microglial morphology in vitro. Sci. Rep. 2016, 6, 28580. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia polarization from M1 to M2 in neurodegenerative diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, X. Different phenotypes of microglia in animal models of Alzheimer disease. Immun. Ageing 2022, 19, 44. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Fan, Y.; Zhou, K.; Blomgren, K.; Harris, R.A. Uncovering sex differences of rodent microglia. J. Neuroinflammation 2021, 18, 74. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H. Inflammatory response in Alzheimer’s disease. Tohoku J. Exp. Med. 1994, 174, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C. Early and late CNS inflammation in Alzheimer’s disease: Two extremes of a continuum? Trends Pharmacol. Sci. 2017, 38, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.; Kettenmann, H. Microglia in physiology and disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 2023, 14, 1117172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Z.; Hu, H.; Zhao, M.; Sun, L. Microglia in Alzheimer’s disease: A target for therapeutic intervention. Front. Cell. Neurosci. 2021, 15, 749587. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s disease: Exploring how genetics and phenotype influence risk. J. Mol. Biol. 2019, 431, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The role of astrocytes in CNS inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Franke, H.; Verkhratsky, A.; Burnstock, G.; Illes, P. Pathophysiology of astroglial purinergic signalling. Purinergic Signal. 2012, 8, 629–657. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.-B.; Song, L.-J.; Wang, Q.; Kumar, G.; Yan, Y.-Q.; Ma, C.-G. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1702–1710. [Google Scholar]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef]
- Benedet, A.L.; Milà-Alomà, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; Karikari, T.K.; Hourregue, C.; Cognat, E.; Dumurgier, J. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021, 78, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Oeckl, P.; Halbgebauer, S.; Anderl-Straub, S.; Steinacker, P.; Huss, A.M.; Neugebauer, H.; von Arnim, C.A.; Diehl-Schmid, J.; Grimmer, T.; Kornhuber, J. Glial fibrillary acidic protein in serum is increased in Alzheimer’s disease and correlates with cognitive impairment. J. Alzheimer’s Dis. 2019, 67, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Shin, K.Y.; Chang, K.-A. GFAP as a potential biomarker for Alzheimer’s disease: A systematic review and meta-analysis. Cells 2023, 12, 1309. [Google Scholar] [CrossRef]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013, 27, 187. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Monterey, M.D.; Wei, H.; Wu, X.; Wu, J.Q. The many faces of astrocytes in Alzheimer’s disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef]
- Dallérac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of microglia and astrocytes in Alzheimer’s disease: From neuroinflammation to Ca2+ homeostasis dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.; Lombardero, L.; Rodríguez-Puertas, R.; De Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological ablation of astrocytes reduces Aβ degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J. Neuroinflammation 2021, 18, 73. [Google Scholar] [CrossRef] [PubMed]
- Pihlaja, R.; Koistinaho, J.; Malm, T.; Sikkilä, H.; Vainio, S.; Koistinaho, M. Transplanted astrocytes internalize deposited β-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 2008, 56, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Mallach, A.; Zielonka, M.; van Lieshout, V.; An, Y.; Khoo, J.H.; Vanheusden, M.; Chen, W.-T.; Moechars, D.; Arancibia-Carcamo, I.L.; Fiers, M. Microglia-astrocyte crosstalk in the amyloid plaque niche of an Alzheimer’s disease mouse model, as revealed by spatial transcriptomics. Cell Rep. 2024, 43, 114216. [Google Scholar] [CrossRef] [PubMed]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ryu, S.H.; Hyun, J.; Cho, Y.S.; Jung, Y.K. TLR2 immunotherapy suppresses neuroinflammation, tau spread, and memory loss in rTg4510 mice. Brain Behav. Immun. 2024, 121, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.J.; Jiang, X.Y.; Du, S.Y.; Zhang, K.; Chen, Z. miR-107-5p ameliorates neurological damage, oxidative stress, and immune responses in mice with Alzheimer’s disease by suppressing the Toll-like receptor 4 (TLR4)/nuclear factor-kappaB (NF-κB) pathway. Kaohsiung J. Med. Sci. 2024, 40, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, W.W.; Skalicka-Woźniak, K.; Budzyńska, B.; El Sayed, N.S. NLRP3 inflammasome inhibition and M1-to-M2 microglial polarization shifting via scoparone-inhibited TLR4 axis in ovariectomy/D-galactose Alzheimer’s disease rat model. Int. Immunopharmacol. 2023, 119, 110239. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, T.; Ni, W.; Zhou, C.; Zhou, H.; Lin, L.; Hu, Y.; Sun, X.; Han, J.; Zhou, Y.; et al. Early activation of Toll-like receptor-3 reduces the pathological progression of Alzheimer’s disease in APP/PS1 mouse. Alzheimers Res. Ther. 2023, 15, 33. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, X.; Yang, C.; Jiang, Y.; Zhou, W.; Zheng, W. Artemisinin Attenuates Amyloid-Induced Brain Inflammation and Memory Impairments by Modulating TLR4/NF-κB Signaling. Int. J. Mol. Sci. 2022, 23, 6354. [Google Scholar] [CrossRef]
- Oliveros, G.; Wallace, C.H.; Chaudry, O.; Liu, Q.; Qiu, Y.; Xie, L.; Rockwell, P.; Figueiredo-Pereira, M.E.; Serrano, P.A. Repurposing ibudilast to mitigate Alzheimer’s disease by targeting inflammation. Brain 2023, 146, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Alvarez-Castelao, B.; Sebastián-Serrano, Á.; Di Lauro, C.; Soria-Tobar, L.; Nicke, A.; Engel, T.; Díaz-Hernández, M. P2X7 receptor inhibition ameliorates ubiquitin–proteasome system dysfunction associated with Alzheimer’s disease. Alzheimer’s Res. Ther. 2023, 15, 105. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.C.; Soares, M.S.P.; Blödorn, E.B.; Domingues, W.B.; Reichert, K.P.; Zago, A.M.; Carvalho, F.B.; Gutierres, J.M.; Gonçales, R.A.; da Cruz Fernandes, M.; et al. Investigating the Effect of Inosine on Brain Purinergic Receptors and Neurotrophic and Neuroinflammatory Parameters in an Experimental Model of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 841–855. [Google Scholar] [CrossRef]
- Wang, K.; Zan, S.; Xu, J.; Sun, W.; Li, C.; Zhang, W.; Ni, D.; Cheng, R.; Li, L.; Yu, Z.; et al. Yishen Huazhuo decoction regulates microglial polarization to reduce Alzheimer’s disease-related neuroinflammation through TREM2. Heliyon 2024, 10, e35800. [Google Scholar] [CrossRef]
- Ren, M.; Zhang, M.; Zhang, X.; Wang, C.; Zheng, Y.; Hu, Y. Hydroxysafflor Yellow A Inhibits Aβ(1-42)-Induced Neuroinflammation by Modulating the Phenotypic Transformation of Microglia via TREM2/TLR4/NF-κB Pathway in BV-2 Cells. Neurochem. Res. 2022, 47, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Cheng, X.; Xu, J.; Liu, Y.; Zhou, J.; Jiang, L.; Gu, X.; Xia, T. Activation of TREM2 attenuates neuroinflammation via PI3K/Akt signaling pathway to improve postoperative cognitive dysfunction in mice. Neuropharmacology 2022, 219, 109231. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Yu, W.; Liu, Z.; Zhao, D.; Lin, S.; Szalóki, D.; Kicsák, M.; Kurtán, T.; Zhang, H. JE-133 Suppresses LPS-Induced Neuroinflammation Associated with the Regulation of JAK/STAT and Nrf2 Signaling Pathways. ACS Chem. Neurosci. 2024, 15, 258–267. [Google Scholar] [CrossRef]
- Cui, X.; Zong, S.; Song, W.; Wang, C.; Liu, Y.; Zhang, L.; Xia, P.; Wang, X.; Zhao, H.; Wang, L.; et al. Omaveloxolone ameliorates cognitive dysfunction in APP/PS1 mice by stabilizing the STAT3 pathway. Life Sci. 2023, 335, 122261. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Liao, Y.; Zhang, Y.; Lu, H.; Huang, Z.; Huang, F.; Zhang, Z.; Dong, Y.; Wang, Z.; Huang, Y. Levistilide A ameliorates neuroinflammation via inhibiting JAK2/STAT3 signaling for neuroprotection and cognitive improvement in scopolamine-induced Alzheimer’s disease mouse model. Int. Immunopharmacol. 2023, 124, 110783. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.H.; Hong, S.M.; Ko, E.J.; Jeon, R.O.; Kim, S.Y. Anti-neuroinflammatory Effects of Active Compound SPA1413 via Suppression of the MAPK and JAK/STAT Signaling Pathways. Biol. Pharm. Bull. 2023, 46, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; He, Z.; Pei, H.; Zhai, L.; Guan, Q.; Wang, G. Aurantiamide promotes M2 polarization of microglial cells to improve the cognitive ability of mice with Alzheimer’s disease. Phytother. Res. 2023, 37, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Tao, W.; Li, J.; Tao, W.; Li, D.; Zhou, L.; Yang, X.; Dong, C.; Huang, S.; Chu, X.; et al. Kai-Xin-San ameliorates Alzheimer’s disease-related neuropathology and cognitive impairment in APP/PS1 mice via the mitochondrial autophagy-NLRP3 inflammasome pathway. J. Ethnopharmacol. 2024, 329, 118145. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yan, Y.; Yi, T.; Wei, Y.; Gao, J.; Gong, Q. Lithocarpus polystachyus Rehd. leaves aqueous extract inhibits learning and memory impairment in Alzheimer’s disease rats: Involvement of the SIRT6/NLRP3 signaling pathway. Ibrain 2024. [Google Scholar] [CrossRef]
- Singh, G.; Kumar, S.; Panda, S.R.; Kumar, P.; Rai, S.; Verma, H.; Singh, Y.P.; Kumar, S.; Srikrishna, S.; Naidu, V.G.M.; et al. Design, Synthesis, and Biological Evaluation of Ferulic Acid-Piperazine Derivatives Targeting Pathological Hallmarks of Alzheimer’s Disease. ACS Chem. Neurosci. 2024, 15, 2756–2778. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Huang, L.; Huang, S.; Chen, W.; Huang, H.; Chi, L.; Su, F.; Liu, X.; Yuan, K.; Jiang, Q.; et al. MSC-Derived Extracellular Vesicles Alleviate NLRP3/GSDMD-Mediated Neuroinflammation in Mouse Model of Sporadic Alzheimer’s Disease. Mol. Neurobiol. 2024, 61, 5494–5509. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Z.; Niu, T.T.; Yu, Q.; Xu, B.L.; Li, X.Q.; Yuan, B.Y.; Yuan, G.B.; Yang, T.T.; Li, H.Q.; Sun, Y. Ginkgolide attenuates memory impairment and neuroinflammation by suppressing the NLRP3/caspase-1 pathway in Alzheimer’s disease. Aging 2023, 15, 10237–10252. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhou, X.; Hong, X.; Wang, S.; Wei, J.; Huang, J.; Ji, L.; Yang, Y.; Efferth, T.; Hong, C.; et al. Essential oil of Acorus tatarinowii Schott inhibits neuroinflammation by suppressing NLRP3 inflammasome activation in 3 × Tg-AD transgenic mice. Phytomedicine 2023, 112, 154695. [Google Scholar] [CrossRef]
- Shen, H.; Pei, H.; Zhai, L.; Guan, Q.; Wang, G. Aurantiamide suppresses the activation of NLRP3 inflammasome to improve the cognitive function and central inflammation in mice with Alzheimer’s disease. CNS Neurosci. Ther. 2023, 29, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Du, L.; Zhang, T.; Chu, Y.; Wang, Y.; Wang, Y.; Ji, X.; Kang, Y.; Cui, R.; Zhang, G.; et al. Edaravone Dexborneol ameliorates the cognitive deficits of APP/PS1 mice by inhibiting TLR4/MAPK signaling pathway via upregulating TREM2. Neuropharmacology 2024, 255, 110006. [Google Scholar] [CrossRef] [PubMed]
- Samsuzzaman, M.; Subedi, L.; Hong, S.M.; Lee, S.; Gaire, B.P.; Ko, E.J.; Choi, J.W.; Seo, S.Y.; Kim, S.Y. A Synthetic Derivative SH 66 of Homoisoflavonoid from Liliaceae Exhibits Anti-Neuroinflammatory Activity against LPS-Induced Microglial Cells. Molecules 2024, 29, 3037. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wen, H.; Liu, L.; Aisa, H.A.; Xin, X. A Pair of Epimers of Lignan Alleviate Neuroinflammatory Effects by Modulating iNOS/COX-2 and MAPK/NF-κB Signaling Pathways. Inflammation 2024, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cui, X.; Dong, Z.; Liu, Y.; Xia, P.; Wang, X.; Zhang, Z.; Yu, S.; Wu, S.; Liu, H. Attenuated memory impairment and neuroinflammation in Alzheimer’s disease by aucubin via the inhibition of ERK-FOS axis. Int. Immunopharmacol. 2024, 126, 111312. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Shao, S.; Wang, Y.; Su, W. Ginsenoside Rg2 alleviates neurovascular damage in 3xTg-AD mice with Alzheimer’s disease through the MAPK-ERK pathway. J. Chem. Neuroanat. 2023, 133, 102346. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhou, Y.; Shi, J.; Wang, F.; Yang, X.; Zheng, X.; Wang, Y.; He, Y.; Xie, X.; Pang, X. Myricetin improves pathological changes in 3×Tg-AD mice by regulating the mitochondria-NLRP3 inflammasome-microglia channel by targeting P38 MAPK signaling pathway. Phytomedicine 2023, 115, 154801. [Google Scholar] [CrossRef] [PubMed]
- Son, S.H.; Lee, N.R.; Gee, M.S.; Song, C.W.; Lee, S.J.; Lee, S.K.; Lee, Y.; Kim, H.J.; Lee, J.K.; Inn, K.S.; et al. Chemical Knockdown of Phosphorylated p38 Mitogen-Activated Protein Kinase (MAPK) as a Novel Approach for the Treatment of Alzheimer’s Disease. ACS Cent. Sci. 2023, 9, 417–426. [Google Scholar] [CrossRef]
- Lv, X.; Zhan, L.; Ye, T.; Xie, H.; Chen, Z.; Lin, Y.; Cai, X.; Yang, W.; Liao, X.; Liu, J.; et al. Gut commensal Agathobacter rectalis alleviates microglia-mediated neuroinflammation against pathogenesis of Alzheimer disease. iScience 2024, 27, 111116. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, N.; Al Saihati, H.A.; Alali, Z.; Aleniz, F.Q.; Mahmoud, S.Y.M.; Badr, O.A.; Dessouky, A.A.; Mostafa, O.; Hussien, N.I.; Farid, A.S.; et al. Exploring the molecular mechanisms of MSC-derived exosomes in Alzheimer’s disease: Autophagy, insulin and the PI3K/Akt/mTOR signaling pathway. Biomed Pharmacother. 2024, 176, 116836. [Google Scholar] [CrossRef]
- Gouveia, F.; Fonseca, C.; Silva, A.; Camins, A.; Teresa Cruz, M.; Ettcheto, M.; Fortuna, A. Intranasal irbesartan reverts cognitive decline and activates the PI3K/AKT pathway in an LPS-induced neuroinflammation mice model. Int. Immunopharmacol. 2024, 128, 111471. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Hu, Y.; Qin, X.; Sun, J.; Miao, Z.; Wang, L.; Ke, Z.; Zheng, Y. Micheliolide attenuates neuroinflammation to improve cognitive impairment of Alzheimer’s disease by inhibiting NF-κB and PI3K/Akt signaling pathways. Heliyon 2023, 9, e17848. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Li, S.; Cheng, H.; Luo, Z.; Qi, X.; Guan, F.; Dong, W.; Gao, S.; Liu, N.; Gao, X.; et al. Discovery of an evodiamine derivative for PI3K/AKT/GSK3β pathway activation and AD pathology improvement in mouse models. Front. Mol. Neurosci. 2022, 15, 1025066. [Google Scholar] [CrossRef]
- Essam, R.M.; Kandil, E.A. p-CREB and p-DARPP-32 orchestrating the modulatory role of cAMP/PKA signaling pathway enhanced by Roflumilast in rotenone-induced Parkinson’s disease in rats. Chem. Biol. Interact. 2023, 372, 110366. [Google Scholar] [CrossRef] [PubMed]
- Yanai, S.; Tago, T.; Toyohara, J.; Arasaki, T.; Endo, S. Reversal of spatial memory impairment by phosphodiesterase 3 inhibitor cilostazol is associated with reduced neuroinflammation and increased cerebral glucose uptake in aged male mice. Front. Pharmacol. 2022, 13, 1031637. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Wang, X.; Feng, J.; Wang, P.; Xu, H. Inhibitory effect of PDE2 on inflammation and apoptosis in cerebral ischemia-reperfusion injury. Int. J. Mol. Med. 2022, 50, 109. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef]
- Wu, L.; Xian, X.; Xu, G.; Tan, Z.; Dong, F.; Zhang, M.; Zhang, F. Toll-Like Receptor 4: A Promising Therapeutic Target for Alzheimer’s Disease. Mediat. Inflamm. 2022, 2022, 7924199. [Google Scholar] [CrossRef] [PubMed]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Xu, C.; Zhang, H.; Lin, C. TLR4 targeting as a promising therapeutic strategy for Alzheimer disease treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.C.; Rasley, A.; Tranguch, S.L.; Marriott, I. Cultured astrocytes express toll-like receptors for bacterial products. Glia 2003, 43, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 2021, 91, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Tahara, K.; Kim, H.-D.; Jin, J.-J.; Maxwell, J.A.; Li, L.; Fukuchi, K.-I. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Jin, J.; Lim, J.-E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.-D.; Tahara, K.; Lalonde, R.; Fukuchi, K.-i. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflammation 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Liu, Y.; Hao, W.; Decker, Y.; Tomic, I.; Menger, M.D.; Liu, C.; Fassbender, K. Stimulation of TLR4 attenuates Alzheimer’s disease–related symptoms and pathology in tau-transgenic mice. J. Immunol. 2016, 197, 3281–3292. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Perry, G.; Rezaei, N. Toll-like receptors in Alzheimer’s disease. J. Neuroimmunol. 2020, 348, 577362. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tian, D.-Y.; Wang, Y.-J. Peripheral clearance of brain-derived Aβ in Alzheimer’s disease: Pathophysiology and therapeutic perspectives. Transl. Neurodegener. 2020, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, R.; Wan Yaacob, W.; Othman, Z.; Long, I.; Ahmad, A.; Al-Rahbi, B. Lipopolysaccharide-induced memory impairment in rats: A model of Alzheimer’s disease. Physiol. Res. 2017, 66, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Schirmbeck, G.H.; Seady, M.; Fróes, F.T.; Taday, J.; Da Ré, C.; Souza, J.M.; Gonçalves, C.A.; Leite, M.C. Long-term LPS systemic administration leads to memory impairment and disturbance in astrocytic homeostasis. Neurotoxicology 2023, 99, 322–331. [Google Scholar] [CrossRef]
- Ali, W.; Ikram, M.; Park, H.Y.; Jo, M.G.; Ullah, R.; Ahmad, S.; Abid, N.B.; Kim, M.O. Oral administration of alpha linoleic acid rescues Aβ-induced glia-mediated neuroinflammation and cognitive dysfunction in C57BL/6N mice. Cells 2020, 9, 667. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 induces M2 microglial polarization and provides neuroprotection via the NLRP3 inflammasome in Alzheimer’s disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef]
- Balu, D.; Valencia-Olvera, A.C.; Nguyen, A.; Patnam, M.; York, J.; Peri, F.; Neumann, F.; LaDu, M.J.; Tai, L.M. A small-molecule TLR4 antagonist reduced neuroinflammation in female E4FAD mice. Alzheimer’s Res. Ther. 2023, 15, 181. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Pei, Z.; Lee, K.-C.; Lopez-Brignoni, E.; Nikolov, B.; Crowley, C.; Marsman, M.; Barbier, R.; Friedmann, N.; Burns, L.H. PTI-125 reduces biomarkers of Alzheimer’s disease in patients. J. Prev. Alzheimer’s Dis. 2020, 7, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-X.; Tseng, J.-C.; Yu, G.-Y.; Luo, Y.; Huang, C.-Y.F.; Hong, Y.-R.; Chuang, T.-H. Recent advances in the development of toll-like receptor agonist-based vaccine adjuvants for infectious diseases. Pharmaceutics 2022, 14, 423. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, P.; Chen, K.; Hu, J.; Gong, W.; Cho, E.H.; Lockert, S.; Uranchimeg, B.; Wang, J.M. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid β 1–42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2. FASEB J. 2005, 19, 2032–2034. [Google Scholar] [CrossRef]
- Scholtzova, H.; Kascsak, R.J.; Bates, K.A.; Boutajangout, A.; Kerr, D.J.; Meeker, H.C.; Mehta, P.D.; Spinner, D.S.; Wisniewski, T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J. Neurosci. 2009, 29, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Scholtzova, H.; Chianchiano, P.; Pan, J.; Sun, Y.; Goñi, F.; Mehta, P.D.; Wisniewski, T. Amyloid β and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol. Commun. 2014, 2, 101. [Google Scholar] [CrossRef]
- Lalo, U.; Verkhratsky, A.; Pankratov, Y. Ionotropic ATP Receptors in Neuronal–Glial Communication. Semin. Cell Dev. Biol. 2011, 22, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M. ATP: A ubiquitous gliotransmitter integrating neuron–glial networks. Semin. Cell Dev. Biol. 2011, 22, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Illes, P. Purinergic modulation of microglial cell activation. Purinergic Signal. 2007, 3, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Calovi, S.; Mut-Arbona, P.; Sperlágh, B. Microglia and the purinergic signaling system. Neuroscience 2019, 405, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of microglial functions by purinergic mechanisms in the healthy and diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef]
- Gómez Morillas, A.; Besson, V.C.; Lerouet, D. Microglia and neuroinflammation: What place for P2RY12? Int. J. Mol. Sci. 2021, 22, 1636. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.-g.; Hu, D.-x.; Zuo, C.; Zhang, W.-j. G protein-coupled P2Y12 receptor is involved in the progression of neuropathic pain. Biomed. Pharmacother. 2023, 162, 114713. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kohyama, K.; Moriyama, K.; Ozaki, M.; Hasegawa, S.; Ueno, T.; Saitoe, M.; Morio, T.; Hayashi, M.; Sakuma, H. Extracellular ADP augments microglial inflammasome and NF-κB activation via the P2Y12 receptor. Eur. J. Immunol. 2020, 50, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Collo, G.; Neidhart, S.; Kawashima, E.; Kosco-Vilbois, M.; North, R.; Buell, G. Tissue distribution of the P2X7 receptor. Neuropharmacology 1997, 36, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.D.; Soltoff, S.P. P2X7 receptors activate protein kinase D and p42/p44 mitogen-activated protein kinase (MAPK) downstream of protein kinase C. Biochem. J. 2002, 366, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Sun, X.; Bai, A.; Zhang, C.; Li, L.; Enjyoji, K.; Junger, W.G.; Robson, S.C.; Wu, Y. P2X7 integrates PI3K/AKT and AMPK-PRAS40-mTOR signaling pathways to mediate tumor cell death. PLoS ONE 2013, 8, e60184. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiao, Y.; Li, Z. P2X7 receptor positively regulates MyD88-dependent NF-κB activation. Cytokine 2011, 55, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.; de Andrade Mello, P.; Da Silva, C.G.; Coutinho-Silva, R. The P2X7 receptor in inflammatory diseases: Angel or demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Dubyak, G.R. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 2004, 286, C1100–C1108. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Pelegrin, P.; Surprenant, A. Facilitation of P2X7 receptor currents and membrane blebbing via constitutive and dynamic calmodulin binding. J. Neurosci. 2008, 28, 6393–6401. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, P.A.; Estacion, M.; Schilling, W.; Dubyak, G.R. P2X7 receptor-dependent blebbing and the activation of Rho-effector kinases, caspases, and IL-1β release. J. Immunol. 2003, 170, 5728–5738. [Google Scholar] [CrossRef] [PubMed]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.J.; Saunders, B.M.; Petrou, S.; Wiley, J.S. P2X7 is a scavenger receptor for apoptotic cells in the absence of its ligand, extracellular ATP. J. Immunol. 2011, 187, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends Pharmacol. Sci. 2014, 35, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ronning, K.E.; Déchelle-Marquet, P.-A.; Che, Y.; Guillonneau, X.; Sennlaub, F.; Delarasse, C. The P2X7 receptor, a multifaceted receptor in Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 11747. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid β requires P2X7 receptor expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G.; Ryu, J.K.; Walker, D.G.; Choi, H.B. Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Han, J.; Resing, D.; Liu, H.; Yue, X.; Miller, R.L.; Schoch, K.M.; Miller, T.M.; Perlmutter, J.S.; Egan, T.M. Synthesis and in vitro characterization of a P2X7 radioligand [123I] TZ6019 and its response to neuroinflammation in a mouse model of Alzheimer disease. Eur. J. Pharmacol. 2018, 820, 8–17. [Google Scholar] [CrossRef]
- Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; León-Otegui, M.; Hontecillas-Prieto, L.; Del Puerto, A.; Trejo, J.L.; Lucas, J.J.; Garrido, J.J.; Gualix, J.; Miras-Portugal, M.T. In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3β and secretases. Neurobiol. Aging 2012, 33, 1816–1828. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Brückner, M.; Prigent, A.; Sazdovitch, V.; Halle, A. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 2019, 24, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Cao, L.; Wang, J.; Zhuang, J.; Liu, Y.; Bi, L.; Qiu, Y.; Hou, Y.; Huang, Q.; Xie, F. Increased Cerebral Level of P2X7R in a Tauopathy Mouse Model by PET Using [18F] GSK1482160. ACS Chem. Neurosci. 2024, 15, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.; Martin, E.; Ces, A.; Sarrazin, N.; Lagouge-Roussey, P.; Nous, C.; Boucherit, L.; Youssef, I.; Prigent, A.; Faivre, E. P2X7-deficiency improves plasticity and cognitive abilities in a mouse model of Tauopathy. Prog. Neurobiol. 2021, 206, 102139. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, H.; Wang, H.; Yang, K.; Luan, J.; Wang, S. TREM2: Potential therapeutic targeting of microglia for Alzheimer’s disease. Biomed. Pharmacother. 2023, 165, 115218. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Koike, M.; Spusta, S.C.; Niemi, E.C.; Yenari, M.; Nakamura, M.C.; Seaman, W.E. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 2009, 109, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 2018, 97, 1023–1031.e1027. [Google Scholar] [CrossRef]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sudan, R.; Peng, V.; Zhou, Y.; Du, S.; Yuede, C.M.; Lei, T.; Hou, J.; Cai, Z.; Cella, M. TREM2 drives microglia response to amyloid-β via SYK-dependent and-independent pathways. Cell 2022, 185, 4153–4169.e4119. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Taso, O.; Wang, R.; Bayram, S.; Graham, A.C.; Garcia-Reitboeck, P.; Mallach, A.; Andrews, W.D.; Piers, T.M.; Botia, J.A. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum. Mol. Genet. 2020, 29, 3224–3248. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, G.; Young-Pearse, T.L. Microglial function, INPP5D/SHIP1 signaling, and NLRP3 inflammasome activation: Implications for Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tan, L.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Tan, M.-S.; Gu, L.-Z.; Wang, H.-F.; Ding, Z.-Z.; Zhang, Y.-D. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Finn, M.B.; Wang, Y.; Shen, A.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Piccio, L.; Colonna, M.; Holtzman, D.M. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 2014, 9, 20. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’loughlin, E.; Xu, Y.; Fanek, Z. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [PubMed]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.; Long, H.; Schwabe, T.; Rhinn, H.; Tassi, I.; Salazar, S.V.; Rychkova, A.; Tu, G.; Barner, C.; Yeh, F.L. A phase 1 study of AL002 in healthy volunteers. Alzheimer’s Dement. 2021, 17, e054669. [Google Scholar] [CrossRef]
- Long, H.; Simmons, A.; Mayorga, A.; Burgess, B.; Nguyen, T.; Budda, B.; Rychkova, A.; Rhinn, H.; Tassi, I.; Ward, M. Preclinical and first-in-human evaluation of AL002, a novel TREM2 agonistic antibody for Alzheimer’s disease. Alzheimer’s Res. Ther. 2024, 16, 235. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT signaling: A double-edged sword of immune regulation and cancer progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Singh, M.K.; Shyam, H.; Mishra, A.; Kumar, S.; Kumar, A.; Kushwaha, J. Role of JAK/STAT in the neuroinflammation and its association with neurological disorders. Ann. Neurosci. 2021, 28, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK–STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H.; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J. Neurosci. 2016, 36, 5144–5159. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Sun, Z.; Zhang, S.; Yang, G.; Jiang, X.; Wang, G.; Li, R.; Wang, Q.; Tian, X. SOCS modulates JAK-STAT pathway as a novel target to mediate the occurrence of neuroinflammation: Molecular details and treatment options. Brain Res. Bull. 2024, 213, 110988. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Zhang, W.; Cao, Q.; Zou, L.; Fan, X.; Qi, C.; Yan, Y.; Song, B.; Wu, B. JAK2/STAT3 pathway regulates microglia polarization involved in hippocampal inflammatory damage due to acute paraquat exposure. Ecotoxicol. Environ. Saf. 2022, 234, 113372. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.V.; Chen, J.; Lyu, H.; Lam, S.Y.E.; Lu, G.; Chan, W.Y.; Wong, G.K. Novel role of STAT3 in microglia-dependent neuroinflammation after experimental subarachnoid haemorrhage. Stroke Vasc. Neurol. 2022, 7, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.; Cianciulli, A.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Curcumin regulates anti-inflammatory responses by JAK/STAT/SOCS signaling pathway in BV-2 microglial cells. Biology 2019, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Gu, L.; Ye, Y.; Zhu, H.; Pu, B.; Wang, J.; Li, Y.; Qiu, S.; Xiong, X.; Jian, Z. JAK2/STAT3 axis intermediates microglia/macrophage polarization during cerebral ischemia/reperfusion injury. Neuroscience 2022, 496, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Nevado-Holgado, A.J.; Ribe, E.; Thei, L.; Furlong, L.; Mayer, M.-A.; Quan, J.; Richardson, J.C.; Cavanagh, J.; Consortium, N.; Lovestone, S. Genetic and real-world clinical data, combined with empirical validation, nominate Jak-Stat signaling as a target for Alzheimer’s disease therapeutic development. Cells 2019, 8, 425. [Google Scholar] [CrossRef] [PubMed]
- Haim, L.B.; Ceyzériat, K.; Carrillo-de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef]
- Wen, X.; Hu, J. Targeting STAT3 signaling pathway in the treatment of Alzheimer’s disease with compounds from natural products. Int. Immunopharmacol. 2024, 141, 112936. [Google Scholar] [CrossRef] [PubMed]
- Millot, P.; San, C.; Bennana, E.; Porte, B.; Vignal, N.; Hugon, J.; Paquet, C.; Hosten, B.; Mouton-Liger, F. STAT3 inhibition protects against neuroinflammation and BACE1 upregulation induced by systemic inflammation. Immunol. Lett. 2020, 228, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Kim, H.; Yang, E.-J.; Kim, H.-S. Inhibition of STAT3 phosphorylation attenuates impairments in learning and memory in 5XFAD mice, an animal model of Alzheimer’s disease. J. Pharmacol. Sci. 2020, 143, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Ruganzu, J.B.; Zheng, Q.; Wu, X.; He, Y.; Peng, X.; Jin, H.; Zhou, J.; Ma, R.; Ji, S.; Ma, Y. TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp. Neurol. 2021, 336, 113506. [Google Scholar] [CrossRef]
- Iwahara, N.; Hisahara, S.; Kawamata, J.; Matsumura, A.; Yokokawa, K.; Saito, T.; Fujikura, M.; Manabe, T.; Suzuki, H.; Matsushita, T. Role of suppressor of cytokine signaling 3 (SOCS3) in altering activated microglia phenotype in APPswe/PS1dE9 mice. J. Alzheimer’s Dis. 2017, 55, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Sasabe, J.; Terashita, K.; Shimoda, M.; Matsuoka, M.; Aiso, S. Amyloid-β causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatry 2009, 14, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Aiso, S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin. Ther. Targets 2009, 13, 1155–1167. [Google Scholar] [CrossRef]
- Hindam, M.O.; Ahmed, L.A.; Sayed, N.S.E.; Khattab, M.; Sallam, N.A. Repositioning of baricitinib for management of memory impairment in ovariectomized/D-galactose treated rats: A potential role of JAK2/STAT3-PI3K/AKT/mTOR signaling pathway. Life Sci. 2024, 351, 122838. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yuan, Z.; Cheng, J. The function of NOD-like receptors in central nervous system diseases. J. Neurosci. Res. 2017, 95, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Hanslik, K.L.; Ulland, T.K. The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef] [PubMed]
- Kodi, T.; Sankhe, R.; Gopinathan, A.; Nandakumar, K.; Kishore, A. New insights on NLRP3 inflammasome: Mechanisms of activation, inhibition, and epigenetic regulation. J. Neuroimmune Pharmacol. 2024, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, C.; Araiz, A.R.; Bryson, K.; Finucane, O.; Larkin, C.; Mills, E.; Robertson, A.; Cooper, M.; O’Neill, L.; Lynch, M. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Lodder, C.; Botella Lucena, P.; Vanherle, S.; Gutiérrez de Ravé, M.; Terwel, D.; Bottelbergs, A.; Dewachter, I. The NLRP3 inflammasome modulates tau pathology and neurodegeneration in a tauopathy model. Glia 2022, 70, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Zuppe, H.; Reed, E. Common cytokine receptor gamma chain family cytokines activate MAPK, PI3K, and JAK/STAT pathways in microglia to influence Alzheimer’s Disease. Front. Mol. Neurosci. 2024, 17, 1441691. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wei, Y.-Z.; Wang, G.-Q.; Li, D.-D.; Shi, J.-S.; Zhang, F. Targeting MAPK pathways by naringenin modulates microglia M1/M2 polarization in lipopolysaccharide-stimulated cultures. Front. Cell. Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef]
- Qiu, Z.; Lu, P.; Wang, K.; Zhao, X.; Li, Q.; Wen, J.; Zhang, H.; Li, R.; Wei, H.; Lv, Y. Dexmedetomidine inhibits neuroinflammation by altering microglial M1/M2 polarization through MAPK/ERK pathway. Neurochem. Res. 2020, 45, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Plastira, I.; Bernhart, E.; Joshi, L.; Koyani, C.N.; Strohmaier, H.; Reicher, H.; Malle, E.; Sattler, W. MAPK signaling determines lysophosphatidic acid (LPA)-induced inflammation in microglia. J. Neuroinflammation 2020, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O. A comprehensive review on MAPK: A promising therapeutic target in cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, L.; Zeng, Q.; He, J.; Tang, Q.; Wang, K.; He, G. MKP-1 regulates the inflammatory activation of microglia against Alzheimer’s disease. CNS Neurosci. Ther. 2024, 30, e14409. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J. Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1704–1721. [Google Scholar] [CrossRef] [PubMed]
- Bodles, A.M.; Barger, S.W. Secreted β-amyloid precursor protein activates microglia via JNK and p38-MAPK. Neurobiol. Aging 2005, 26, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Monteleone, D.; Giliberto, L.; Fornaro, M.; Borghi, R.; Tamagno, E.; Tabaton, M. Amyloid-β 42 activates the expression of BACE1 through the JNK pathway. J. Alzheimer’s Dis. 2011, 27, 871–883. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. [Google Scholar] [CrossRef]
- Savage, M.J.; Lin, Y.G.; Ciallella, J.R.; Flood, D.G.; Scott, R.W. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J. Neurosci. 2002, 22, 3376–3385. [Google Scholar] [CrossRef]
- Solas, M.; Vela, S.; Smerdou, C.; Martisova, E.; Martínez-Valbuena, I.; Luquin, M.R.; Ramírez, M.J. JNK Activation in Alzheimer’s Disease Is Driven by Amyloid β and Is Associated with Tau Pathology. ACS Chem. Neurosci. 2023, 14, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Qin, Y.; Liu, M.; Sun, J.; Tang, H.; Zeng, Y.; Zhang, J.; Wang, W.; Liang, G.; Zhao, X. Magnoflorine improves cognitive deficits and pathology of Alzheimer’s disease via inhibiting of JNK signaling pathway. Phytomedicine 2023, 112, 154714. [Google Scholar] [CrossRef] [PubMed]
- Melchiorri, D.; Merlo, S.; Micallef, B.; Borg, J.J.; Dráfi, F. Alzheimer’s disease and neuroinflammation: Will new drugs in clinical trials pave the way to a multi-target therapy? Front. Pharmacol. 2023, 14, 1196413. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Dixon, S.G.; Hu, W.; Hamlett, E.D.; Jin, J.; Ergul, A.; Wang, G.Y. p38 MAPK Is a Major Regulator of Amyloid Beta-Induced IL-6 Expression in Human Microglia. Mol. Neurobiol. 2022, 59, 5284–5298. [Google Scholar] [CrossRef] [PubMed]
- Kheiri, G.; Dolatshahi, M.; Rahmani, F.; Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: Implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 2018, 30, 9–30. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Bolós, M.; Cuadros, R.; García, E.; García-Escudero, V.; Hernández, F.; McManus, R.M.; Heneka, M.T.; Avila, J. p38 Inhibition Decreases Tau Toxicity in Microglia and Improves Their Phagocytic Function. Mol. Neurobiol. 2022, 59, 1632–1648. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Bachstetter, A.D.; Späni, C.B.; Roy, S.M.; Watterson, D.M.; Van Eldik, L.J. Retention of normal glia function by an isoform-selective protein kinase inhibitor drug candidate that modulates cytokine production and cognitive outcomes. J. Neuroinflammation 2017, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.S.; Son, S.H.; Jeon, S.H.; Do, J.; Kim, N.; Ju, Y.J.; Lee, S.J.; Chung, E.K.; Inn, K.S.; Kim, N.J.; et al. A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimers Res. Ther. 2020, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.M.; Grum-Tokars, V.L.; Schavocky, J.P.; Saeed, F.; Staniszewski, A.; Teich, A.F.; Arancio, O.; Bachstetter, A.D.; Webster, S.J.; Van Eldik, L.J.; et al. Targeting human central nervous system protein kinases: An isoform selective p38αMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem. Neurosci. 2015, 6, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Cianciulli, A.; Calvello, R.; Dragone, T.; Iacobazzi, F.; Panaro, M.A. The PI3K/Akt pathway is required for LPS activation of microglial cells. Immunopharmacol. Immunotoxicol. 2012, 34, 858–865. [Google Scholar] [CrossRef]
- Chu, E.; Mychasiuk, R.; Hibbs, M.L.; Semple, B.D. Dysregulated phosphoinositide 3-kinase signaling in microglia: Shaping chronic neuroinflammation. J. Neuroinflammation 2021, 18, 276. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Porro, C.; Calvello, R.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Microglia Mediated Neuroinflammation: Focus on PI3K Modulation. Biomolecules 2020, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Tarassishin, L.; Suh, H.S.; Lee, S.C. Interferon regulatory factor 3 plays an anti-inflammatory role in microglia by activating the PI3K/Akt pathway. J. Neuroinflammation 2011, 8, 187. [Google Scholar] [CrossRef]
- He, G.L.; Luo, Z.; Shen, T.T.; Wang, Z.Z.; Li, P.; Luo, X.; Yang, J.; Tan, Y.L.; Wang, Y.; Gao, P.; et al. TREM2 Regulates Heat Acclimation-Induced Microglial M2 Polarization Involving the PI3K-Akt Pathway Following EMF Exposure. Front. Cell. Neurosci. 2019, 13, 591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, Y.; Wang, L.; Zhan, H.; Luo, X.; Zeng, Y.; Wu, W.; Zhang, X.; Wang, F. TREM2 ameliorates neuroinflammatory response and cognitive impairment via PI3K/AKT/FoxO3a signaling pathway in Alzheimer’s disease mice. Aging 2020, 12, 20862–20879. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Henry, R.J.; Blanchard, A.C.; Stoica, B.A.; Loane, D.J.; Faden, A.I. Enhanced Akt/GSK-3β/CREB signaling mediates the anti-inflammatory actions of mGluR5 positive allosteric modulators in microglia and following traumatic brain injury in male mice. J. Neurochem. 2021, 156, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Bansal, N. Implications of Phosphoinositide 3-Kinase-Akt (PI3K-Akt) Pathway in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 354–385. [Google Scholar] [CrossRef]
- Pan, J.; Yao, Q.; Wang, Y.; Chang, S.; Li, C.; Wu, Y.; Shen, J.; Yang, R. The role of PI3K signaling pathway in Alzheimer’s disease. Front. Aging Neurosci. 2024, 16, 1459025. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary regulation of PI3K/AKT/GSK-3β pathway in Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 35. [Google Scholar] [CrossRef]
- Yin, G.; Li, L.Y.; Qu, M.; Luo, H.B.; Wang, J.Z.; Zhou, X.W. Upregulation of AKT attenuates amyloid-β-induced cell apoptosis. J. Alzheimers Dis. 2011, 25, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, J.; Malm, T.; Goldsteins, G. Glycogen synthase kinase-3β: A mediator of inflammation in Alzheimer’s disease? Int. J. Alzheimers Dis. 2011, 2011, 129753. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Mármol, R.; Mohannak, N.; Qian, L.; Wang, T.; Gormal, R.S.; Ruitenberg, M.J.; Vanhaesebroeck, B.; Coulson, E.J.; Meunier, F.A. p110δ PI3-Kinase Inhibition Perturbs APP and TNFα Trafficking, Reduces Plaque Burden, Dampens Neuroinflammation, and Prevents Cognitive Decline in an Alzheimer’s Disease Mouse Model. J. Neurosci. 2019, 39, 7976–7991. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Yang, C.Z.; Wang, S.H.; Zhang, R.H.; Lin, J.H.; Tian, Y.H.; Yang, Y.Q.; Liu, J.; Ma, Y.X. Neuroprotective effect of astragalin via activating PI3K/Akt-mTOR-mediated autophagy on APP/PS1 mice. Cell Death Discov. 2023, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yan, J.; Jiang, W.; Yao, X.G.; Chen, J.; Chen, L.; Li, C.; Hu, L.; Jiang, H.; Shen, X. Arctigenin effectively ameliorates memory impairment in Alzheimer’s disease model mice targeting both β-amyloid production and clearance. J. Neurosci. 2013, 33, 13138–13149. [Google Scholar] [CrossRef] [PubMed]
- Bellozi, P.M.Q.; Gomes, G.F.; de Oliveira, L.R.; Olmo, I.G.; Vieira, É.L.M.; Ribeiro, F.M.; Fiebich, B.L.; de Oliveira, A.C.P. NVP-BEZ235 (Dactolisib) Has Protective Effects in a Transgenic Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 1345. [Google Scholar] [CrossRef]
- Sheng, J.; Zhang, S.; Wu, L.; Kumar, G.; Liao, Y.; Gk, P.; Fan, H. Inhibition of phosphodiesterase: A novel therapeutic target for the treatment of mild cognitive impairment and Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1019187. [Google Scholar] [CrossRef] [PubMed]
- Argyrousi, E.K.; Heckman, P.R.A.; Prickaerts, J. Role of cyclic nucleotides and their downstream signaling cascades in memory function: Being at the right time at the right spot. Neurosci. Biobehav. Rev. 2020, 113, 12–38. [Google Scholar] [CrossRef] [PubMed]
- Gerlo, S.; Kooijman, R.; Beck, I.M.; Kolmus, K.; Spooren, A.; Haegeman, G. Cyclic AMP: A selective modulator of NF-κB action. Cell. Mol. Life Sci. 2011, 68, 3823–3841. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Aguirre, V.; Wai, K.; Felfly, H.; Dietrich, W.D.; Pearse, D.D. The interplay between cyclic AMP, MAPK, and NF-κB pathways in response to proinflammatory signals in microglia. Biomed Res. Int. 2015, 2015, 308461. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Garcia-Castillo, D.; Aguirre, V.; Golshani, R.; Atkins, C.M.; Bramlett, H.M.; Dietrich, W.D.; Pearse, D.D. Proinflammatory cytokine regulation of cyclic AMP-phosphodiesterase 4 signaling in microglia in vitro and following CNS injury. Glia 2012, 60, 1839–1859. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, H.; Iwashita, S.; Kuraishi, T.; Goto, A.; Fuse, N.; Ueno, H.; Nimura, M.; Oyama, T.; Tang, C.; Watanabe, R.; et al. cGMP signaling pathway that modulates NF-κB activation in innate immune responses. iScience 2021, 24, 103473. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflammation 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.P.; Bohlen, C.J.; York, E.M.; Choi, H.B.; Kamyabi, A.; Dissing-Olesen, L.; Hefendehl, J.K.; Collins, H.Y.; Stevens, B.; Barres, B.A.; et al. Nanoscale Surveillance of the Brain by Microglia via cAMP-Regulated Filopodia. Cell Rep. 2019, 27, 2895–2908.e2894. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Roloff, F.; Singh, V.; Stangel, M.; Stern, M.; Bicker, G. Nitric oxide/cyclic GMP signaling regulates motility of a microglial cell line and primary microglia in vitro. Brain Res. 2014, 1564, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Cattani-Cavalieri, I.; Musheshe, N.; Nikolaev, V.O.; Schmidt, M. Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol. Ther. 2019, 197, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, N. Inhibitor of Phosphodiestearse-4 improves memory deficits, oxidative stress, neuroinflammation and neuropathological alterations in mouse models of dementia of Alzheimer’s Type. Biomed Pharmacother. 2017, 88, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, J.; Zhao, X.; Chen, Z.; Wang, G.; Liu, A.; Wang, Q.; Zhou, W.; Xu, Y.; Wang, C. Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav. Brain Res. 2013, 250, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Du, K.; Tao, M.; Shan, C.; Ye, R.; Tang, Y.; Pan, H.; Lv, J.; Zhang, M.; Pan, J. Phosphodiesterase-2 Inhibitor Bay 60-7550 Ameliorates Aβ-Induced Cognitive and Memory Impairment via Regulation of the HPA Axis. Front. Cell. Neurosci. 2019, 13, 432. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Vitolo, O.V.; Trinchese, F.; Liu, S.; Shelanski, M.; Arancio, O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Investig. 2004, 114, 1624–1634. [Google Scholar] [CrossRef]
- Kang, B.W.; Kim, F.; Cho, J.Y.; Kim, S.; Rhee, J.; Choung, J.J. Phosphodiesterase 5 inhibitor mirodenafil ameliorates Alzheimer-like pathology and symptoms by multimodal actions. Alzheimers Res. Ther. 2022, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Donders, Z.; Skorupska, I.J.; Willems, E.; Mussen, F.; Van Broeckhoven, J.; Carlier, A.; Schepers, M.; Vanmierlo, T. Beyond PDE4 inhibition: A comprehensive review on downstream cAMP signaling in the central nervous system. Biomed. Pharmacother. 2024, 177, 117009. [Google Scholar] [CrossRef] [PubMed]
- Wiley, N.J.; Madhankumar, A.; Mitchell, R.M.; Neely, E.B.; Rizk, E.; Douds, G.L.; Simmons, Z.; Connor, J.R. Lipopolysaccharide modified liposomes for amyotropic lateral sclerosis therapy: Efficacy in SOD1 mouse model. Adv. Nanoparticles 2012, 1, 44–53. [Google Scholar] [CrossRef]
- Choi, B.; Soh, M.; Manandhar, Y.; Kim, D.; Han, S.I.; Baik, S.; Shin, K.; Koo, S.; Kwon, H.J.; Ko, G. Highly selective microglial uptake of ceria–zirconia nanoparticles for enhanced analgesic treatment of neuropathic pain. Nanoscale 2019, 11, 19437–19447. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Francis, N.L.; Calvelli, H.R.; Moghe, P.V. Microglia-targeting nanotherapeutics for neurodegenerative diseases. APL Bioeng. 2020, 4, 030902. [Google Scholar] [CrossRef] [PubMed]

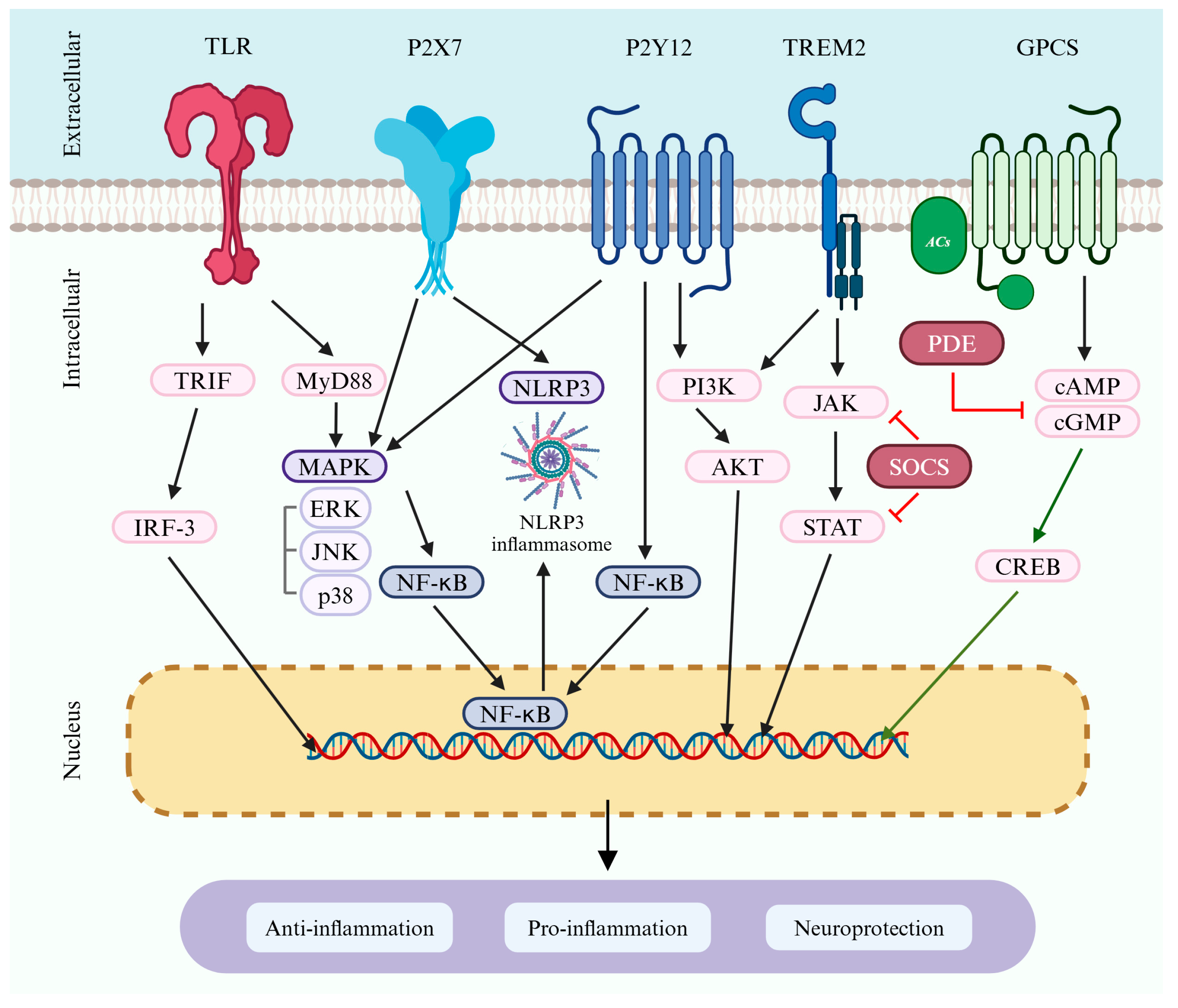

{kind=link}
{kind=link}
| Pathway | Model | Target | Refs. |
|---|---|---|---|
| TLR2/MyD88 | BV-2 cell line | TLR2↓, MyD88↓, NLRP3↓, IL-1β↓, TNF-α↓, IL-6↓, IL-8↓, Iba-1↓ | [96] |
| TLR4/NF-κB | APP/PS1 AD mouse model | ROS↓, IL-1β↓, TNF-α↓, IL-6↓, TUNEL-positive cells↓, APOE↑, TREM2↑, APOE↑, TLR4↓, nuclear NF-κB↓, cytoplasmic NF-κB↓, Iba-1↓ | [97] |
| TLR4/MyD88 | OVX/D-Gal + scopolamine-induced rat model | Histopathological score↓, intact neuron count↓, Aβ42↓, p-tau↓, NF-κB↓, NLRP3↓, caspase-1↓, IL-1β↓, IL-18↓, TLR4↓, MyD88↓, TRAF-6↓, TAK-1↓, p-JNK↓, CD86-positive microglia↓, CD163-positive microglia↑ | [98] |
| TLR3 | APP/PS1 AD mouse model | Aβ↓, Iba-1↓, GFAP↓, CD68↓, NeuN+ cell↑, CD206↓, CD68↓, CD16/32↓, NF-κB↓, IL-1β↓, TNF-α↓, IL-4↑ | [99] |
| TLR4/NF-κB | Aβ–ICV-induced mouse AD model, BV-2 cell line | Survival neurons↑, apoptosis index↓, GFAP↓, Iba-1↓, NF-κB↓, TLR4↓, IκBα↓, NeuN+ cell↑, ROS↓, iNOS↓, COX-2↓, TNF-α↓, IL-1β↓, IL-6↓, apoptotic cells↓ | [100] |
| TLR and ubiquitin–proteasome | TgF344 AD rat model | Aβ↓, tau PHF↓, amoeboid/ramified microglia↓ | [101] |
| P2X7R and PI3K/Akt/GSK3/Nrf2 | Human AD patients, P301S mice, N2a cell line | p-GSK3↑, Nrf2- and FK2-positive intracellular aggregates↓ | [102] |
| A1R/A2AR and BDNF | STZ-ICV-induced AD rat model | A1R↑, A2AR↓, TrkB↑, BDNF↑, BUchE↓, cholesterol↓, IL-4↑, IL-10↑ | [103] |
| TREM2/NF-κB | SAMP8 mice, BV-2 cell line | iNOS↓, Arg-1↑, IL-10↑, PU.1↑, TREM2↑, p-NF-κB↓, TNF-α↓, IL-6↓, IL-1β↓, p-IKKβ↓ | [104] |
| TREM2/TLR4/NF-κB | BV-2 cell line | TREM2↑, TNF-α↓, IL-1β↓, IL-6↓, IL-10↑, CD11b↓, iNOS↓, Arg-1↑ | [105] |
| TREM2/PI3K/AKT | Dysfunction mouse model, BV-2 cell line | TREM2↑, p-NR2B↑, BDNF↑, IL-1β↓, IL-6↓, p-PI3K↑, p-AKT↑, Iba-1↓, iNOS↓ | [106] |
| JAK/STAT | LPS-induced mouse, BV-2 cell line | IL-1β↓, IL-6↓, TNF-α↓, iNOS↓, JAK↓, STAT↓, Nrf2↑, HO-1↑ | [107] |
| NFκB/IL6/STAT3 | APP/PS1 AD mouse model, HT-22, BV2 cell line | NeuN↑, Aβ↓, p-STAT3↑↓, iNOS↓, p65↓, IL-6↓, IL-1β↓, IL-6↓, TNF-α↓ | [108] |
| JAK2/STAT3 | Scopolamine-induced BALB/c mouse, LPS- and Aβ-treated BV-2 cell line | AchE↓, ChAT↓, p-JAK2↓, p-STAT3, iNOS↓, Arg-1↑, IL-1β↓, IL-6↓, TNF-α↓, IL-12↓, IL-4↑, IL-10↑ | [109] |
| JAK/STAT | LPS-treated BV-2 cell line, primary microglial | NO↓, IL-6↓, TNF-α↓, COX-2↓, iNOS↓, p-ERK↑, p-JNK↓, p-p38↓, IL-11↓, JAK2↓, Tyk2↓, STAT1↓, STAT3↓, neurite length↑ | [110] |
| JAK1–STAT6 | APP/PS1 AD mouse model, BV-2 cell line | p-JAK1↑, p-STAT6↑, SOCS3↑, CD206↑, Iba-1↑ | [111] |
| Autophagy–NLRP3 inflammasome, PINK1/Parkin | APP/PS1 AD mouse model | AchE↓, MDA↓, IL-1β↓, MCP-1↓, IL-6↓, IL-18↓, TNF-α↓, SOD↑, Aβ plaques↓, tau↓, LC3B↑, p62↓, PINK1↑, parkin↑, beclin1↑, LC3II/LC3I↑, NLRP3↓, ASC↓, cleaved caspase-1↓ | [112] |
| SIRT6/NLRP3 | Aβ–ICV-induced AD rat model | SIRT6↑, H3K9Ac↓, p-NF-κB↓, IκBα↓, NLRP3↓, ASC↓, caspase-1↓, cleaved caspase-1↓, GSDMD↓, IL-18↓, Iba-1↓, GFAP↓, COX-2↓, TNF-α↓, ROS↓, NO↓, IFN-γ↓, IL-1β↓, IL-6↓, ROS↓, MDA↓, IL-4↑, IL-10↑, TGF-β↑, SOD↑, GSH↑ | [113] |
| NLRP3 | HMC-3 human microglia cell line, scopolamine-induced mouse model | JC-1↓, NLRP3↓, NF-κB↓, vimentin↓, ROS↓, AchE↓, BChE↓, MDA↓, SOD↑, catalase↑ | [114] |
| NLRP3/GSDMD | STZ-ICV-induced mouse model (C57BL/6J) | NLRP3↓, caspase-1↓, IL-1β↓, GSDMD↓, caspase-3↓ | [115] |
| NLRP3/caspase-1 | Aβ-treated BV-2 cell line, APP/PS1 AD mouse model | NLRP3↓, ASC↓, caspase-1↓, IL-1β↓, IL-18↓, ROS↓ | [116] |
| NLRP3 | 3xTg AD mouse model | p-tau↓, Aβ↓, p-GSK3β↑, NeuN↑, Bcl-2↑, Bax↓, Iba-1↓, GFAP↓, IL-1β↓, TNF-α↓, IL-6↓, IL-18↓, p-NF-κB, IKKβ↓, NLRP3↓, ASC↓, caspase-1↓, GSDMD-N↓ | [117] |
| NLRP3 | LPS- + IFN-γ-treated BV-2 cell line | TNF-α↓, IL-6↓, IL-1β↓, NLRP3↓, ASC↓, caspase-1↓, CD11b↓, ROS↓ | [118] |
| TLR4/MAPK | APP/PS1 AD mouse model | Aβ↓, MDA↓, SYP↑, PSD95↑, IL-1β↓, IL-6↓, TNF-α↓, COX-2↓, CD14↓, Fos↓, Igf2↓, MAPK13↓, CCL3↓, CCL4↓, TREM2↑, TLR4↓, MyD88↓, JNK↓, p38↓, Iba-1↓, CD86↓, iNOS↓, ARG1↑, CD206↑, IL-1β, IL-6, TNF-α↓CD86, iNOS↓, ARG1, TREM2↑, TLR4, MyD88, JNK, p38↓ | [119] |
| MAPK | LPS-treated BV-2 cell line | NO↓, iNOS↓, COX-2↓, p-p38↓, p-JNK↓, p-ERK↓, NLRP3↓, ASC↓, cleaved-caspase-1↓, IL-1β↓, pro-IL-1β↑, PGE2↓, TNF-α↓, IL-6↓ | [120] |
| MAPK/NF-κB | LPS-treated BV-2 cell line | NO↓, PGE2↓, IL-6↓, TNF-α↓, MCP-1↓, COX-2↓, p-ERK↓, p-JNK↓, p-p38↓, p-IκB↓, p-p65↓ | [121] |
| MAPK/ERK | APP/PS1 AD mouse model, LPS-treated BV-2 cell line | Aβ plaques↓, Iba-1↓, GFAP↓, p-ERK1/2↓, c-FOS↓, FOSB↓, FOSL1↓, ROS↓, iNOS↓, NO↓, IL-1β↓, IL-6↓, TNF-α↓, COX-2↓ | [122] |
| MAPK/ERK | 3xTg AD mouse model | Aβ25-35↓, TNF-α, ↓ IL-1β↓, IL-6↓NeuN↑, CD31↑, GFAP↓, VCAM-1↓, ICAM-1↓, p-ERK↓, p-MAPK↓, Aβ1–42↓, p-tau↓ | [123] |
| p38/NLRP3 inflammasome | 3xTg AD mouse model, BV-2 cell line | Aβ plaque↓, PSD95↑, synaptophysin↑, TNF-α↓, IL-1β↓, IL-6↓, IL-4↑, IL-10↑, Iba-1↓, CD16↓, Arg1↑, NLRP3↓, ASC↓, caspase-1↓, IL-18↓, FIS1↓, DRP1↓, OPA1↑, MFN2↑, TFAM↑, NRF1↑, p-p38↓, JC-1↓ | [124] |
| MAPK/p38 | LPS-treated BV-2 cell line, 5XFAD AD mouse model | IL-6↓, IL-1β↓, IL-12↓, TNF-α↓, p-p38↓, Aβ↓, Iba-1↓, GFAP↓, p-tau↓ | [125] |
| PI3K/AKT | APP/PS1 AD mouse model | iNOS↓, CD11b↓, TREM2↑, TMEM119↑, Iba-1↓, Aβ↓, p-AKT↑, p-PI3K↓, p-tau↓, IL-1β↓, NF-κB↓, iNOS↓ | [126] |
| PI3K/AKT/mTOR | AlCl3-triggered AD rat model | Aβ↓, PI3K↑, AKT↑, mTOR↑, GSK3β↑, BACE-1 ↓, p-tau↓, iNOS↓, NF-κB↓ | [127] |
| PI3K/AKT | LPS-ICV-induced mouse | iNOS ↓, pro-IL-1β↓, NO↓, GSK3β↑, SOD↑, GPX1↑, CREB↑ | [128] |
| PI3K/AKT | 5xFAD AD mouse model | Thioflavin↓, Iba-1↓, GFAP↓, NF-κB↓, p-IκBα↓, p-AKT↓ | [129] |
| PI3K/AKT/GSK3β | APP/PS1 AD mouse model | Iiba-1↓, Aβ↓, p-AKT↑, p-PI3K↑, p-tau↓, GSK3β↑ | [130] |
| PDE4 | Rotenone, oral injection, rat model | DA level↑, PDE4 activity↓, pCREB↑, PKA↑, PP-1↓, BDNF↑, NGF↑, NF-κB↓, GSH↑, IL-6↓, TNF-α↓ | [131] |
| PDE3 | C57BL/6 (23 months) | NOR↓, GFAP↓, Iba-1↓ | [132] |
| PDE2 | MCAO C57BL/6 | PDE2↓, cerebral edema↓, TNF-α↓, IL-6↓, IL-1β↓, MCP-1↓, NF-κB↓, PKA↑, Iba-1↓ | [133] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.; Chang, Y. Modulating Neuroinflammation as a Prospective Therapeutic Target in Alzheimer’s Disease. Cells 2025, 14, 168. https://doi.org/10.3390/cells14030168
Lee E, Chang Y. Modulating Neuroinflammation as a Prospective Therapeutic Target in Alzheimer’s Disease. Cells. 2025; 14(3):168. https://doi.org/10.3390/cells14030168
Chicago/Turabian StyleLee, Eunshil, and Yongmin Chang. 2025. "Modulating Neuroinflammation as a Prospective Therapeutic Target in Alzheimer’s Disease" Cells 14, no. 3: 168. https://doi.org/10.3390/cells14030168
APA StyleLee, E., & Chang, Y. (2025). Modulating Neuroinflammation as a Prospective Therapeutic Target in Alzheimer’s Disease. Cells, 14(3), 168. https://doi.org/10.3390/cells14030168