
Mechanosignaling via Integrins: Pivotal Players in Liver Fibrosis Progression and Therapy


Abstract
1. Introduction
2. Overview of Liver Fibrosis
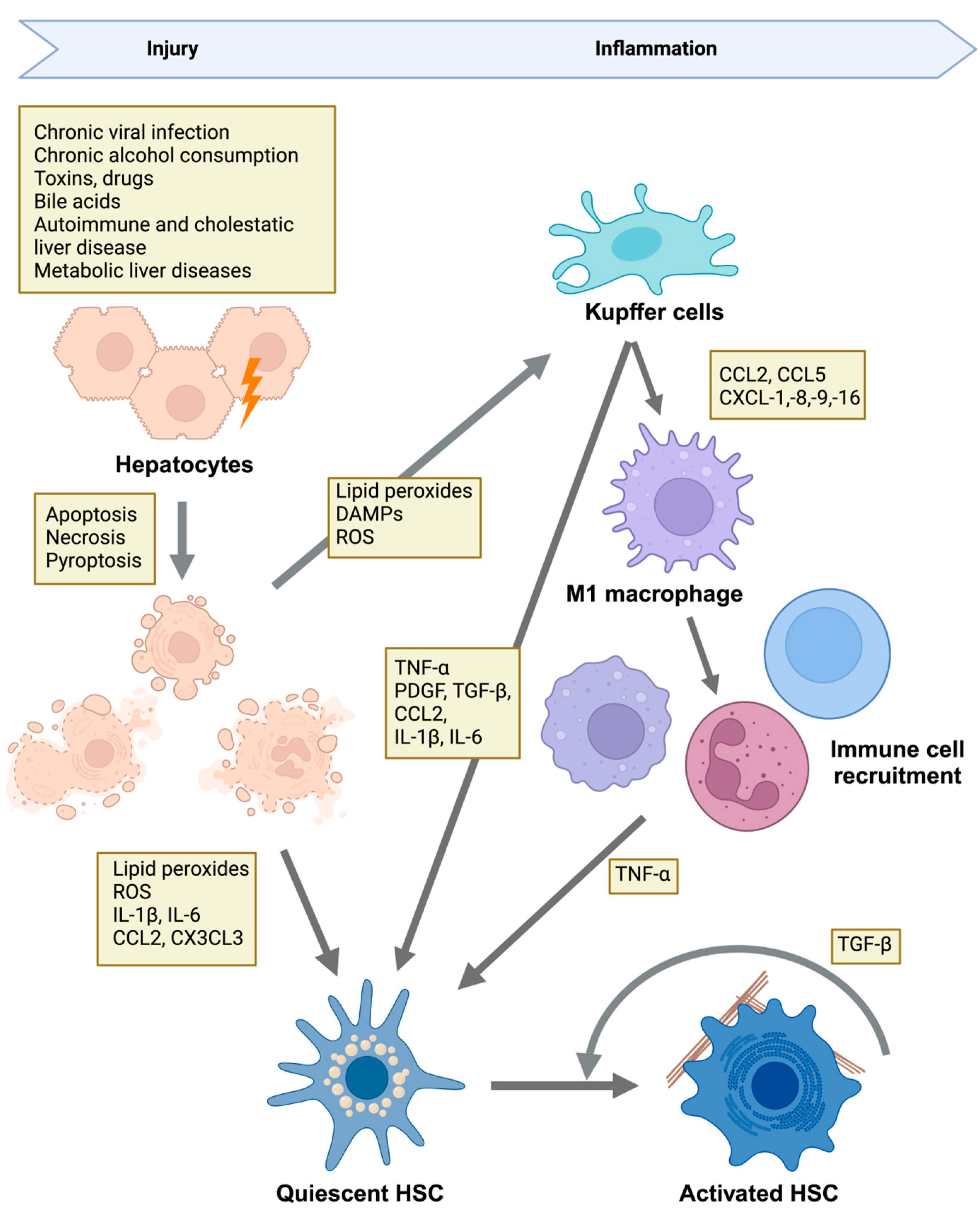
2.1. Etiologies of Liver Injury and Disease Mechanisms
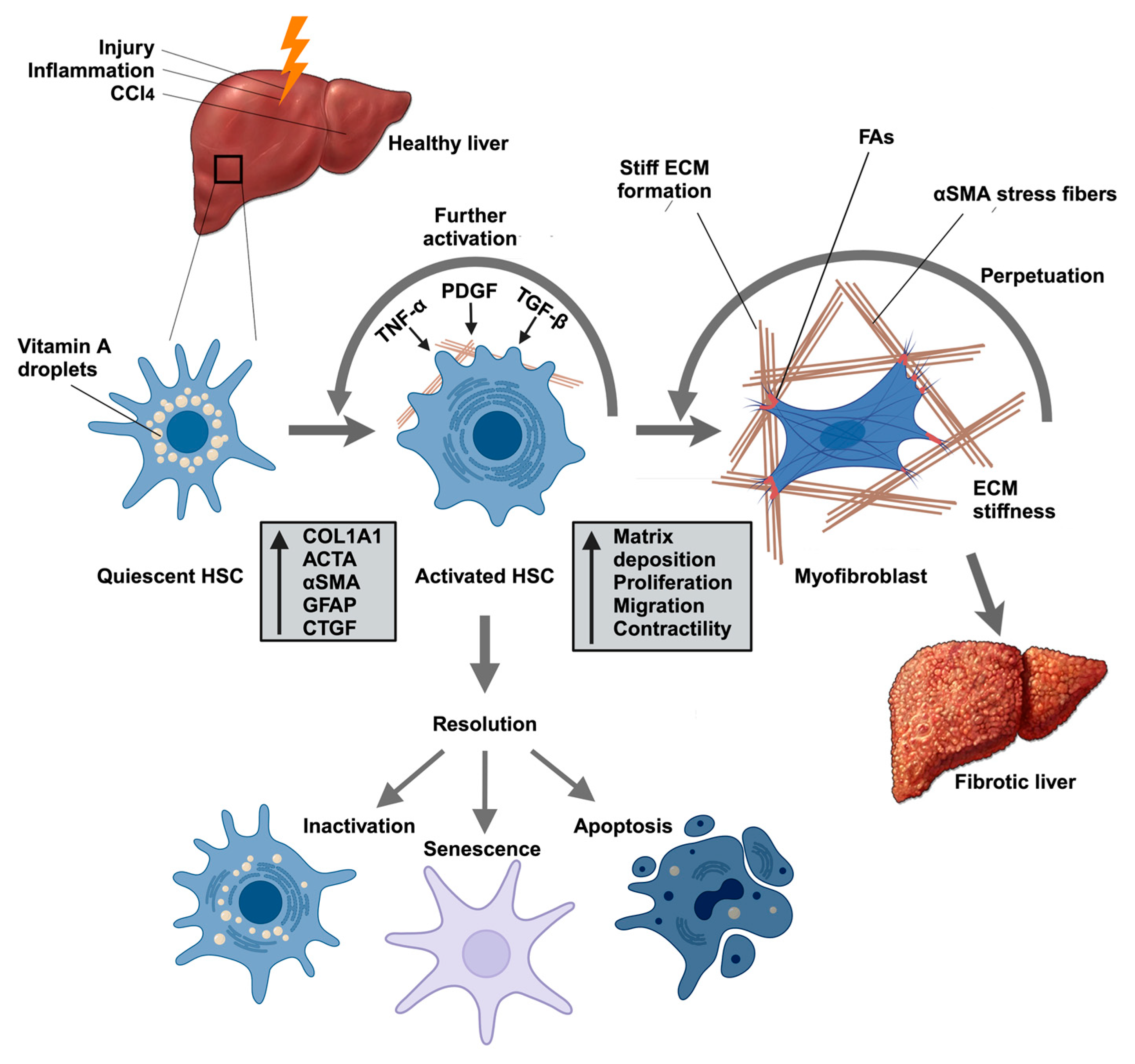
2.2. HSCs Are Responsible for Pathological ECM Deposition
2.3. Targeting HSC Activation to Reversal of Liver Fibrosis
3. Integrin Signaling in Liver Fibrosis
3.1. Overview of Integrin Signaling
3.2. Roles of Integrins in HSC Activation
3.3. Other Disease-Specific Roles of Integrins in Liver Fibrosis
4. Integrin Regulation of Signaling Cascades in Liver Fibrosis
4.1. Integrin-Mediated Mechanotransduction Through Rho/ROCK and MRTF
4.1.1. The Core Rho/ROCK/MRTF Pathway
4.1.2. Experimental Evidence Linking the Rho/ROCK/MRTF/SRF Axis to Fibrosis
4.2. The Hippo/YAP/TAZ Pathway
4.2.1. The Hippo Core Signaling Cascade
4.2.2. Mechanical Regulation of the Hippo Pathway
4.2.3. YAP/TAZ Activation Promotes HSC Activation and Fibrogenesis
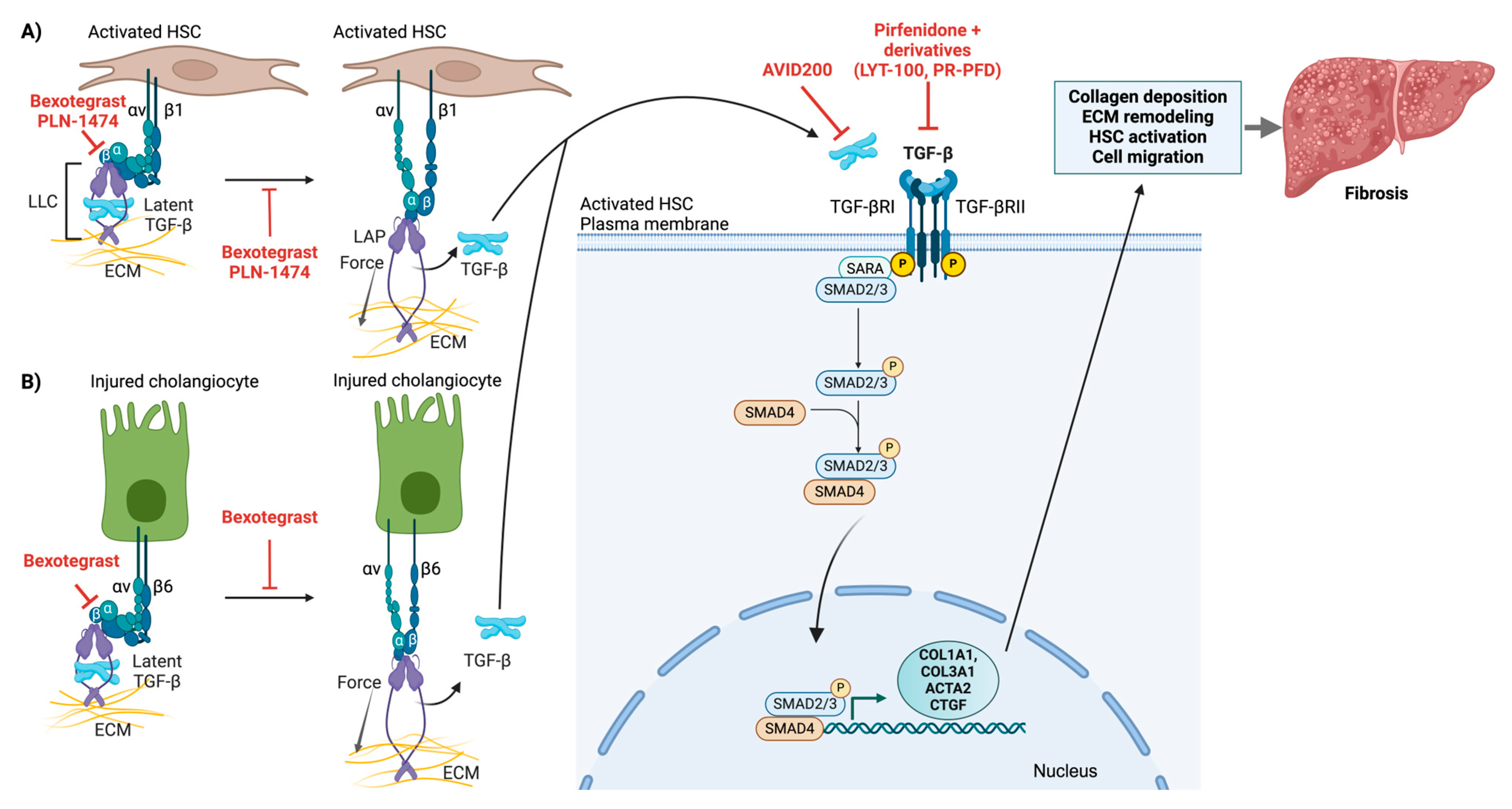
4.3. Integrin αv-Mediated Latent TGF-β Activation
4.3.1. Molecular Mechanisms of Latent TGF-β Activation
4.3.2. Canonical TGF-β Signaling Pathway
4.3.3. Evidence for Integrin-Mediated TGF-β Activation
5. Integration of Mechanosensitive Signaling Pathways in Liver Fibrosis
6. Therapeutic Approaches Targeting Mechanosensitive Signaling in Liver Fibrosis
6.1. Pan-Integrin Inhibitors
6.2. Selective Single and Dual Integrin Inhibitors
6.3. TGF-β Pathway Inhibitors
6.4. Targeting the Rho/ROCK/MRTF Pathway
6.4.1. ROCK Inhibitors
6.4.2. MRTF Inhibitors
6.5. Targeting YAP/TAZ
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinzani, M. Welcome to fibrogenesis & tissue repair. Fibrogenesis Tissue Repair 2008, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat. Clin. Pract. Gastroenterol. Hepatol. 2004, 1, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Dufour, J.F.; Buti, M.; Soriano, V.; Buynak, R.J.; Mantry, P.; Taunk, J.; Stern, J.O.; Vinisko, R.; Gallivan, J.P.; et al. Interferon-free treatment of chronic hepatitis C with faldaprevir, deleobuvir and ribavirin: SOUND-C3, a Phase 2b study. Liver Int. 2015, 35, 417–421. [Google Scholar] [CrossRef]
- Zeuzem, S.; Dusheiko, G.M.; Salupere, R.; Mangia, A.; Flisiak, R.; Hyland, R.H.; Illeperuma, A.; Svarovskaia, E.; Brainard, D.M.; Symonds, W.T.; et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N. Engl. J. Med. 2014, 370, 1993–2001. [Google Scholar] [CrossRef]
- Zeuzem, S.; Jacobson, I.M.; Baykal, T.; Marinho, R.T.; Poordad, F.; Bourliere, M.; Sulkowski, M.S.; Wedemeyer, H.; Tam, E.; Desmond, P.; et al. Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 2014, 370, 1604–1614. [Google Scholar] [CrossRef]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Poordad, F.; Hezode, C.; Trinh, R.; Kowdley, K.V.; Zeuzem, S.; Agarwal, K.; Shiffman, M.L.; Wedemeyer, H.; Berg, T.; Yoshida, E.M.; et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N. Engl. J. Med. 2014, 370, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr161. [Google Scholar] [CrossRef] [PubMed]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Berumen, J.; Baglieri, J.; Kisseleva, T.; Mekeel, K. Liver fibrosis: Pathophysiology and clinical implications. WIREs Mech. Dis. 2021, 13, e1499. [Google Scholar] [CrossRef]
- Torok, N.J.; Dranoff, J.A.; Schuppan, D.; Friedman, S.L. Strategies and endpoints of antifibrotic drug trials: Summary and recommendations from the AASLD Emerging Trends Conference, Chicago, June 2014. Hepatology 2015, 62, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Olsen, A.L.; Bloomer, S.A.; Chan, E.P.; Gaca, M.D.; Georges, P.C.; Sackey, B.; Uemura, M.; Janmey, P.A.; Wells, R.G. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G110–G118. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Alsamman, S.; Christenson, S.A.; Yu, A.; Ayad, N.M.E.; Mooring, M.S.; Segal, J.M.; Hu, J.K.; Schaub, J.R.; Ho, S.S.; Rao, V.; et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Bouvet, M.; Claude, O.; Roux, M.; Skelly, D.; Masurkar, N.; Mougenot, N.; Nadaud, S.; Blanc, C.; Delacroix, C.; Chardonnet, S.; et al. Anti-integrin α(v) therapy improves cardiac fibrosis after myocardial infarction by blunting cardiac PW1(+) stromal cells. Sci. Rep. 2020, 10, 11404. [Google Scholar] [CrossRef] [PubMed]
- Bougueon, M.; Legagneux, V.; Hazard, O.; Bomo, J.; Siegel, A.; Feret, J.; Theret, N. A rule-based multiscale model of hepatic stellate cell plasticity: Critical role of the inactivation loop in fibrosis progression. PLoS Comput. Biol. 2024, 20, e1011858. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.; Kim, D.K.; Yang, W.I.; Shin, D.H.; Jung, I.M.; Park, H.K.; Chang, B.C. Overexpression of transforming growth factor-beta 1 in the valvular fibrosis of chronic rheumatic heart disease. J. Korean Med. Sci. 2008, 23, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; McHutchison, J.; Manns, M.; Trepo, C.; Lindsay, K.; Goodman, Z.; Ling, M.H.; Albrecht, J. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 2002, 122, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Troeger, J.S.; Mederacke, I.; Gwak, G.Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.P.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083.e22. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Calderwood, D.A. Organization, dynamics and mechanoregulation of integrin-mediated cell-ECM adhesions. Nat. Rev. Mol. Cell Biol. 2023, 24, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Chastney, M.R.; Conway, J.R.W.; Ivaska, J. Integrin adhesion complexes. Curr. Biol. 2021, 31, R536–R542. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef]
- Berrier, A.L.; Yamada, K.M. Cell-matrix adhesion. J. Cell. Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Miranti, C.K.; Brugge, J.S. Sensing the environment: A historical perspective on integrin signal transduction. Nat. Cell Biol. 2002, 4, E83–E90. [Google Scholar] [CrossRef] [PubMed]
- Singer, I.I.; Kawka, D.W.; Kazazis, D.M.; Clark, R.A. In vivo co-distribution of fibronectin and actin fibers in granulation tissue: Immunofluorescence and electron microscope studies of the fibronexus at the myofibroblast surface. J. Cell Biol. 1984, 98, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Dugina, V.; Fontao, L.; Chaponnier, C.; Vasiliev, J.; Gabbiani, G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J. Cell Sci. 2001, 114, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, M.B.; Howard, E.W.; Tomasek, J.J. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 2000, 257, 180–189. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Conroy, K.P.; Kitto, L.J.; Henderson, N.C. alphav integrins: Key regulators of tissue fibrosis. Cell Tissue Res. 2016, 365, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Schiller, H.B.; Hermann, M.R.; Polleux, J.; Vignaud, T.; Zanivan, S.; Friedel, C.C.; Sun, Z.; Raducanu, A.; Gottschalk, K.E.; Thery, M.; et al. beta1- and alphav-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat. Cell Biol. 2013, 15, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, E.; Christen, U. The Many Roles of Cell Adhesion Molecules in Hepatic Fibrosis. Cells 2019, 8, 1503. [Google Scholar] [CrossRef]
- Zhubanchaliyev, A.; Temirbekuly, A.; Kongrtay, K.; Wanshura, L.C.; Kunz, J. Targeting Mechanotransduction at the Transcriptional Level: YAP and BRD4 Are Novel Therapeutic Targets for the Reversal of Liver Fibrosis. Front. Pharmacol. 2016, 7, 462. [Google Scholar] [CrossRef]
- Shi, Z.; Ren, M.; Rockey, D.C. Myocardin and myocardin-related transcription factor-A synergistically mediate actin cytoskeletal-dependent inhibition of liver fibrogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G504–G517. [Google Scholar] [CrossRef] [PubMed]
- Kanta, J. Collagen matrix as a tool in studying fibroblastic cell behavior. Cell Adhes. Migr. 2015, 9, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef]
- Tang, Y.; Rowe, R.G.; Botvinick, E.L.; Kurup, A.; Putnam, A.J.; Seiki, M.; Weaver, V.M.; Keller, E.T.; Goldstein, S.; Dai, J.; et al. MT1-MMP-dependent control of skeletal stem cell commitment via a beta1-integrin/YAP/TAZ signaling axis. Dev. Cell 2013, 25, 402–416. [Google Scholar] [CrossRef]
- Venugopal, S.; Dan, Q.; Sri Theivakadadcham, V.S.; Wu, B.; Kofler, M.; Layne, M.D.; Connelly, K.A.; Rzepka, M.F.; Friedberg, M.K.; Kapus, A.; et al. Regulation of the RhoA exchange factor GEF-H1 by profibrotic stimuli through a positive feedback loop involving RhoA, MRTF, and Sp1. Am. J. Physiol. Cell Physiol. 2024, 327, C387–C402. [Google Scholar] [CrossRef] [PubMed]
- Cavin, S.; Maric, D.; Diviani, D. A-kinase anchoring protein-Lbc promotes pro-fibrotic signaling in cardiac fibroblasts. Biochim. Biophys. Acta 2014, 1843, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Colo, G.P.; Seiwert, A.; Haga, R.B. Lfc subcellular localization and activity is controlled by alphav-class integrin. J. Cell Sci. 2023, 136. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Friedl, A. Syndecan-1-Induced ECM Fiber Alignment Requires Integrin alphavbeta3 and Syndecan-1 Ectodomain and Heparan Sulfate Chains. PLoS ONE 2016, 11, e0150132. [Google Scholar] [CrossRef]
- Park, S.; Kim, J.; Yang, S.; Kang, S.H.; Kang, W.; Paik, Y.H. Exogenous S1P via S1P receptor 2 induces CTGF expression through Src-RhoA-ROCK-YAP pathway in hepatic stellate cells. Mol. Biol. Rep. 2024, 51, 950. [Google Scholar] [CrossRef] [PubMed]
- Kanno, K.; Tazuma, S.; Nishioka, T.; Hyogo, H.; Chayama, K. Angiotensin II participates in hepatic inflammation and fibrosis through MCP-1 expression. Dig. Dis. Sci. 2005, 50, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Khalifa, M.O.; Li, P.; Huang, Y.; Gu, W.; Li, T.S. Angiotensin receptor blocker alleviates liver fibrosis by altering the mechanotransduction properties of hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G446–G456. [Google Scholar] [CrossRef] [PubMed]
- Yanase, M.; Ikeda, H.; Matsui, A.; Maekawa, H.; Noiri, E.; Tomiya, T.; Arai, M.; Yano, T.; Shibata, M.; Ikebe, M.; et al. Lysophosphatidic acid enhances collagen gel contraction by hepatic stellate cells: Association with rho-kinase. Biochem. Biophys. Res. Commun. 2000, 277, 72–78. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Z.; Wang, S.; Xiu, A.; Zhang, C. Carvedilol Inhibits Angiotensin II-Induced Proliferation and Contraction in Hepatic Stellate Cells through the RhoA/Rho-Kinase Pathway. Biomed. Res. Int. 2019, 2019, 7932046. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Meng, Y.; Ji, H.L.; Pan, C.Q.; Huang, S.; Yu, C.H.; Xiao, L.M.; Cui, K.; Ni, S.Y.; Zhang, Z.S.; et al. Spironolactone lowers portal hypertension by inhibiting liver fibrosis, ROCK-2 activity and activating NO/PKG pathway in the bile-duct-ligated rat. PLoS ONE 2012, 7, e34230. [Google Scholar] [CrossRef]
- Ji, H.; Meng, Y.; Zhang, X.; Luo, W.; Wu, P.; Xiao, B.; Zhang, Z.; Li, X. Aldosterone induction of hepatic stellate cell contraction through activation of RhoA/ROCK-2 signaling pathway. Regul. Pept. 2011, 169, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Akahoshi, T.; Kawanaka, H.; Hashimoto, N.; Nagao, Y.; Tomikawa, M.; Taketomi, A.; Shirabe, K.; Hashizume, M.; Maehara, Y. Endothelin-1 derived from spleen-activated Rho-kinase pathway in rats with secondary biliary cirrhosis. Hepatol. Res. 2012, 42, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Kwok, W.; Clemens, M.G. Rho-kinase activation contributes to Lps-induced impairment of endothelial nitric oxide synthase activation by endothelin-1 in cultured hepatic sinusoidal endothelial cells. Shock 2014, 42, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef]
- Chaturvedi, L.S.; Marsh, H.M.; Basson, M.D. Role of RhoA and its effectors ROCK and mDia1 in the modulation of deformation-induced FAK, ERK, p38, and MLC motogenic signals in human Caco-2 intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2011, 301, C1224–C1238. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Khalifa, M.O.; Gu, W.; Li, T.S. Hydrostatic pressure induces profibrotic properties in hepatic stellate cells via the RhoA/ROCK signaling pathway. FEBS Open Bio 2022, 12, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Totsukawa, G.; Yamakita, Y.; Yamashiro, S.; Hartshorne, D.J.; Sasaki, Y.; Matsumura, F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 2000, 150, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Wong, C.M.; Ko, F.C.; Chan, L.K.; Ching, Y.P.; Yam, J.W.; Ng, I.O. Deleted in liver cancer 1 (DLC1) negatively regulates Rho/ROCK/MLC pathway in hepatocellular carcinoma. PLoS ONE 2008, 3, e2779. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Wang, D.; Chang, P.S.; Wang, Z.; Sutherland, L.; Richardson, J.A.; Small, E.; Krieg, P.A.; Olson, E.N. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 2001, 105, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, Y.; Sakai, N.; Iwata, Y.; Lagares, D.; Hara, A.; Kitajima, S.; Toyama, T.; Miyagawa, T.; Ogura, H.; Sato, K.; et al. Myocardin-related transcription factor contributes to renal fibrosis through the regulation of extracellular microenvironment surrounding fibroblasts. FASEB J. 2023, 37, e23005. [Google Scholar] [CrossRef] [PubMed]
- Gualdrini, F.; Esnault, C.; Horswell, S.; Stewart, A.; Matthews, N.; Treisman, R. SRF Co-factors Control the Balance between Cell Proliferation and Contractility. Mol. Cell 2016, 64, 1048–1061. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.Z.; Bialik, J.F.; Speight, P.; Dan, Q.; Yeung, T.; Szaszi, K.; Pedersen, S.F.; Kapus, A. TGF-beta1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017, 292, 14902–14920. [Google Scholar] [CrossRef]
- Speight, P.; Kofler, M.; Szaszi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFbeta-regulated Smad3. Nat. Commun. 2016, 7, 11642. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.R.; Jakobson, M.; Colo, G.P.; Rognoni, E.; Jakobson, M.; Kupatt, C.; Posern, G.; Fassler, R. Integrins synergise to induce expression of the MRTF-A-SRF target gene ISG15 for promoting cancer cell invasion. J. Cell Sci. 2016, 129, 1391–1403. [Google Scholar] [CrossRef]
- Small, E.M. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J. Cardiovasc. Transl. Res. 2012, 5, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.Z.; Lichner, Z.; Szaszi, K.; Kapus, A. MRTF: Basic Biology and Role in Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6040. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Sladojevic, N.; Blair, J.E.; Liao, J.K. Targeting Rho-associated coiled-coil forming protein kinase (ROCK) in cardiovascular fibrosis and stiffening. Expert. Opin. Ther. Targets 2020, 24, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fang, C.; Zhang, L.; Deng, Y.; Wang, M.; Meng, F. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, ameliorates hepatic fibrosis in rats with type 2 diabetes. Chin. Med. J. 2014, 127, 225–231. [Google Scholar] [CrossRef]
- Iwamoto, H.; Nakamuta, M.; Tada, S.; Sugimoto, R.; Enjoji, M.; Nawata, H. A p160ROCK-specific inhibitor, Y-27632, attenuates rat hepatic stellate cell growth. J. Hepatol. 2000, 32, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.J.; Mu, Y.L.; Zhao, H.J.; Zhao, R.R.; Guo, Q.J.; Su, Y.H.; Zhang, J. Fasudil prevents liver fibrosis via activating natural killer cells and suppressing hepatic stellate cells. World J. Gastroenterol. 2021, 27, 3581–3594. [Google Scholar] [CrossRef]
- Xie, Y.; Song, T.; Huo, M.; Zhang, Y.; Zhang, Y.Y.; Ma, Z.H.; Wang, N.; Zhang, J.P.; Chu, L. Fasudil alleviates hepatic fibrosis in type 1 diabetic rats: Involvement of the inflammation and RhoA/ROCK pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5665–5677. [Google Scholar] [CrossRef]
- Tian, W.; Fan, Z.; Li, J.; Hao, C.; Li, M.; Xu, H.; Wu, X.; Zhou, B.; Zhang, L.; Fang, M.; et al. Myocardin-related transcription factor A (MRTF-A) plays an essential role in hepatic stellate cell activation by epigenetically modulating TGF-beta signaling. Int. J. Biochem. Cell Biol. 2016, 71, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Hao, C.; Fan, Z.; Weng, X.; Qin, H.; Wu, X.; Fang, M.; Chen, Q.; Shen, A.; Xu, Y. Myocardin related transcription factor A programs epigenetic activation of hepatic stellate cells. J. Hepatol. 2015, 62, 165–174. [Google Scholar] [CrossRef]
- Kong, M.; Hong, W.; Shao, Y.; Lv, F.; Fan, Z.; Li, P.; Xu, Y.; Guo, J. Ablation of serum response factor in hepatic stellate cells attenuates liver fibrosis. J. Mol. Med. 2019, 97, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Sasaki, H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 2008, 135, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.O.; Camargo, F.D. Hippo signalling in the liver: Role in development, regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Scott, K.E.; Fraley, S.I.; Rangamani, P. A spatial model of YAP/TAZ signaling reveals how stiffness, dimensionality, and shape contribute to emergent outcomes. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Oliver-De La Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skladal, P.; Pesl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Ito, M.; Naoe, Y.; Lacy-Hulbert, A.; Ikeda, K. Integrin alphav in the mechanical response of osteoblast lineage cells. Biochem. Biophys. Res. Commun. 2014, 447, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Weiler, S.M.E.; Lutz, T.; Bissinger, M.; Sticht, C.; Knaub, M.; Gretz, N.; Schirmacher, P.; Breuhahn, K. TAZ target gene ITGAV regulates invasion and feeds back positively on YAP and TAZ in liver cancer cells. Cancer Lett. 2020, 473, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Kitsugi, K.; Noritake, H.; Matsumoto, M.; Hanaoka, T.; Umemura, M.; Yamashita, M.; Takatori, S.; Ito, J.; Ohta, K.; Chida, T.; et al. Arg-Gly-Asp-binding integrins activate hepatic stellate cells via the hippo signaling pathway. Cell Signal. 2022, 99, 110437. [Google Scholar] [CrossRef]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e13. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.X.; Yao, Y.; Bu, F.T.; Chen, Y.; Wu, Y.T.; Yang, Y.; Chen, X.; Zhu, Y.; Wang, Q.; Pan, X.Y.; et al. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol. Immunol. 2019, 107, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhang, N.; Xu, Y.; Chen, Q.; Khan, M.; Potter, J.J.; Nayar, S.K.; Cornish, T.; Alpini, G.; Bronk, S.; et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology 2012, 56, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, J.; Keski-Oja, J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell 2000, 11, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-beta-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Heldin, C.H. Latent forms of TGF-beta: Molecular structure and mechanisms of activation. Ciba Found. Symp. 1991, 157, 81–89; discussion 89–92. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Chia, Z.J.; Cao, Y.N.; Little, P.J.; Kamato, D. Transforming growth factor-beta receptors: Versatile mechanisms of ligand activation. Acta Pharmacol. Sin. 2024, 45, 1337–1348. [Google Scholar] [CrossRef]
- Lu, M.; Munger, J.S.; Steadele, M.; Busald, C.; Tellier, M.; Schnapp, L.M. Integrin alpha8beta1 mediates adhesion to LAP-TGFbeta1. J. Cell Sci. 2002, 115, 4641–4648. [Google Scholar] [CrossRef]
- Thomas, G.J.; Hart, I.R.; Speight, P.M.; Marshall, J.F. Binding of TGF-beta1 latency-associated peptide (LAP) to alpha(v)beta6 integrin modulates behaviour of squamous carcinoma cells. Br. J. Cancer 2002, 87, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef]
- Gyorfi, A.H.; Matei, A.E.; Distler, J.H.W. Targeting TGF-beta signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Felli, E.; Selicean, S.; Guixe-Muntet, S.; Wang, C.; Bosch, J.; Berzigotti, A.; Gracia-Sancho, J. Mechanobiology of portal hypertension. JHEP Rep. 2023, 5, 100869. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Hinz, B. TGF-beta1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef]
- Roberts, A.B.; Kim, S.J.; Noma, T.; Glick, A.B.; Lafyatis, R.; Lechleider, R.; Jakowlew, S.B.; Geiser, A.; O’Reilly, M.A.; Danielpour, D.; et al. Multiple forms of TGF-beta: Distinct promoters and differential expression. Ciba Found. Symp. 1991, 157, 7–15; discussion 15–28. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Sheppard, D. Integrin-mediated activation of transforming growth factor-beta(1) in pulmonary fibrosis. Chest 2001, 120, 49S–53S. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. Roles of alphav integrins in vascular biology and pulmonary pathology. Curr. Opin. Cell Biol. 2004, 16, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005, 24, 395–402. [Google Scholar] [CrossRef]
- Nishimichi, N.; Tsujino, K.; Kanno, K.; Sentani, K.; Kobayashi, T.; Chayama, K.; Sheppard, D.; Yokosaki, Y. Induced hepatic stellate cell integrin, alpha8beta1, enhances cellular contractility and TGFbeta activity in liver fibrosis. J. Pathol. 2021, 253, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Nejjari, M.; Couvelard, A.; Mosnier, J.F.; Moreau, A.; Feldmann, G.; Degott, C.; Marcellin, P.; Scoazec, J.Y. Integrin up-regulation in chronic liver disease: Relationship with inflammation and fibrosis in chronic hepatitis C. J. Pathol. 2001, 195, 473–481. [Google Scholar] [CrossRef]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef]
- Patsenker, E.; Popov, Y.; Stickel, F.; Jonczyk, A.; Goodman, S.L.; Schuppan, D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology 2008, 135, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Dolinski, B.M.; Kikuchi, N.; Leone, D.R.; Peters, M.G.; Weinreb, P.H.; Violette, S.M.; Bissell, D.M. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology 2007, 46, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Jalan-Sakrikar, N.; Guicciardi, M.E.; O’Hara, S.P.; Azad, A.; LaRusso, N.F.; Gores, G.J.; Huebert, R.C. Central role for cholangiocyte pathobiology in cholestatic liver diseases. Hepatology 2024. [Google Scholar] [CrossRef] [PubMed]
- Guillot, A.; Guerri, L.; Feng, D.; Kim, S.J.; Ahmed, Y.A.; Paloczi, J.; He, Y.; Schuebel, K.; Dai, S.; Liu, F.; et al. Bile acid-activated macrophages promote biliary epithelial cell proliferation through integrin alphavbeta6 upregulation following liver injury. J. Clin. Investig. 2021, 131, e132305. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Chan, S.W.; Guo, F.; Toloczko, A.; Cui, L.; Hong, W. MRTF/SRF dependent transcriptional regulation of TAZ in breast cancer cells. Oncotarget 2016, 7, 13706–13716. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Hwang, D.; Lee, D.; Kim, J.H.; Kim, S.Y.; Lim, D.S. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J. 2017, 36, 520–535. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.M.; Benitez, J.A.; Plouffe, S.W.; Ryback, D.; Klein, A.; Smith, J.; Greenbaum, J.; Delatte, B.; Rao, A.; Guan, K.L.; et al. YAP and MRTF-A, transcriptional co-activators of RhoA-mediated gene expression, are critical for glioblastoma tumorigenicity. Oncogene 2018, 37, 5492–5507. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; He, Y.; Wei, Z.; Lu, Y.; Du, F.; Ou, G.; Wang, N.; Luo, X.G.; Ma, W.; Zhang, T.C.; et al. MRTF-A mediates the activation of COL1A1 expression stimulated by multiple signaling pathways in human breast cancer cells. Biomed. Pharmacother. 2018, 104, 718–728. [Google Scholar] [CrossRef]
- Kim, J.; Kang, W.; Kang, S.H.; Park, S.H.; Kim, J.Y.; Yang, S.; Ha, S.Y.; Paik, Y.H. Proline-rich tyrosine kinase 2 mediates transforming growth factor-beta-induced hepatic stellate cell activation and liver fibrosis. Sci. Rep. 2020, 10, 21018. [Google Scholar] [CrossRef] [PubMed]
- Jirouskova, M.; Harant, K.; Cejnar, P.; Ojha, S.; Korelova, K.; Sarnova, L.; Sticova, E.; Mayr, C.; Schiller, H.; Gregor, M. Dynamics of compartment-specific proteomic landscapes of hepatotoxic and cholestatic models of liver fibrosis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lin, P.; Yan, X.; Jing, S.; Wu, Y.; Shan, Y.; Guo, W.; Gu, J.; Li, Y.; Zhang, H.; Li, H. Single-cell and spatially resolved transcriptomics for liver biology. Hepatology 2024, 80, 698–720. [Google Scholar] [CrossRef]
- Ramachandran, P.; Matchett, K.P.; Dobie, R.; Wilson-Kanamori, J.R.; Henderson, N.C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.R.; Roper, J.A.; Grove, J.I.; Aithal, G.P.; Pun, K.T.; Bennett, A.J. Integrins as a drug target in liver fibrosis. Liver Int. 2022, 42, 507–521. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Mouded, M.; Chambers, D.C.; Martinez, F.J.; Richeldi, L.; Lancaster, L.H.; Hamblin, M.J.; Gibson, K.F.; Rosas, I.O.; Prasse, A.; et al. A Phase IIb Randomized Clinical Study of an Anti-alpha(v)beta(6) Monoclonal Antibody in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 206, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Mouded, M.; Prasse, A.; Stebbins, C.; Zhao, G.; Song, G.; Arefayene, M.; Violette, S.M.; Gallagher, D.; Gibson, K.F. Randomized Phase IIa Clinical Study of an Anti-alpha(v)beta(6) Monoclonal Antibody in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 206, 1166–1168. [Google Scholar] [CrossRef]
- Tampe, D.; Zeisberg, M. Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Maden, C.H.; Fairman, D.; Chalker, M.; Costa, M.J.; Fahy, W.A.; Garman, N.; Lukey, P.T.; Mant, T.; Parry, S.; Simpson, J.K.; et al. Safety, tolerability and pharmacokinetics of GSK3008348, a novel integrin alphavbeta6 inhibitor, in healthy participants. Eur. J. Clin. Pharmacol. 2018, 74, 701–709. [Google Scholar] [CrossRef] [PubMed]
- John, A.E.; Graves, R.H.; Pun, K.T.; Vitulli, G.; Forty, E.J.; Mercer, P.F.; Morrell, J.L.; Barrett, J.W.; Rogers, R.F.; Hafeji, M.; et al. Translational pharmacology of an inhaled small molecule alphavbeta6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat. Commun. 2020, 11, 4659. [Google Scholar] [CrossRef] [PubMed]
- Hryczanek, H.F.; Barrett, J.; Barrett, T.N.; Burley, G.A.; Cookson, R.E.; Hatley, R.J.D.; Measom, N.D.; Roper, J.A.; Rowedder, J.E.; Slack, R.J.; et al. Core Modifications of GSK3335103 toward Orally Bioavailable alpha(v)beta(6) Inhibitors with Improved Synthetic Tractability. J. Med. Chem. 2024, 67, 19689–19715. [Google Scholar] [CrossRef]
- Lancaster, L.; Cottin, V.; Ramaswamy, M.; Wuyts, W.A.; Jenkins, R.G.; Scholand, M.B.; Kreuter, M.; Valenzuela, C.; Ryerson, C.J.; Goldin, J.; et al. Bexotegrast in Patients with Idiopathic Pulmonary Fibrosis: The INTEGRIS-IPF Clinical Trial. Am. J. Respir. Crit. Care Med. 2024, 210, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Mukhatayev, Z.; Adilbayeva, A.; Kunz, J. CTHRC1: An Emerging Hallmark of Pathogenic Fibroblasts in Lung Fibrosis. Cells 2024, 13, 946. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Liu, J.; Wu, Q.; Wang, B.; Hu, T.; Li, Y.; Yan, X.; Ma, L.; Tan, Z. A dual alphavbeta1/alphavbeta6 integrin inhibitor Bexotegrast (PLN-74809) ameliorates organ injury and fibrogenesis in fibrotic kidney disease. Eur. J. Pharmacol. 2024, 983, 176983. [Google Scholar] [CrossRef]
- Wim, A.W.; Claudia, V.; Gisli, J.; Jonathan, G.G.; Grace Hyun, J.K.; Marzena, J.; Scott, T.; Martin, D.; Chris, N.B.; Éric, L.; et al. Late Breaking Abstract—Safety, tolerability and antifibrotic activity of bexotegrast: Phase 2a INTEGRIS-IPF study (NCT04396756). Eur. Respir. J. 2023, 62, OA1423. [Google Scholar] [CrossRef]
- Bellani, S.; Molyneaux, P.L.; Maher, T.M.; Spagnolo, P. Potential of alphavbeta6 and alphavbeta1 integrin inhibition for treatment of idiopathic pulmonary fibrosis. Expert Opin. Ther. Targets 2024, 28, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular Mechanisms and Potential Clinical Applications in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef]
- Poo, J.L.; Torre, A.; Aguilar-Ramirez, J.R.; Cruz, M.; Mejia-Cuan, L.; Cerda, E.; Velazquez, A.; Patino, A.; Ramirez-Castillo, C.; Cisneros, L.; et al. Benefits of prolonged-release pirfenidone plus standard of care treatment in patients with advanced liver fibrosis: PROMETEO study. Hepatol. Int. 2020, 14, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Sansores, R.H.; Ramirez-Venegas, A.; Montiel-Lopez, F.; Dominguez-Arellano, S.; Alva-Lopez, L.F.; Falfan-Valencia, R.; Perez-Rubio, G.; Olaya-Lopez, E.; Zavaleta-Martinez, E.O.; Aguilar-Medina, S.; et al. Prolonged-release pirfenidone in patients with pulmonary fibrosis as a phenotype of post-acute sequelae of COVID-19 pneumonia. Safety and efficacy. Respir. Med. 2023, 217, 107362. [Google Scholar] [CrossRef] [PubMed]
- Hahmann, C.; Schroeter, T. Rho-kinase inhibitors as therapeutics: From pan inhibition to isoform selectivity. Cell. Mol. Life Sci. 2010, 67, 171–177. [Google Scholar] [CrossRef]
- Koch, J.C.; Kuttler, J.; Maass, F.; Lengenfeld, T.; Zielke, E.; Bahr, M.; Lingor, P. Compassionate Use of the ROCK Inhibitor Fasudil in Three Patients With Amyotrophic Lateral Sclerosis. Front. Neurol. 2020, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Collu, R.; Yin, Z.; Giunti, E.; Daley, S.; Chen, M.; Morin, P.; Killick, R.; Wong, S.T.C.; Xia, W. Effect of the ROCK inhibitor fasudil on the brain proteomic profile in the tau transgenic mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2024, 16, 1323563. [Google Scholar] [CrossRef] [PubMed]
- Zanin-Zhorov, A.; Chen, W.; Moretti, J.; Nyuydzefe, M.S.; Zhorov, I.; Munshi, R.; Ghosh, M.; Serdjebi, C.; MacDonald, K.; Blazar, B.R.; et al. Selectivity matters: Selective ROCK2 inhibitor ameliorates established liver fibrosis via targeting inflammation, fibrosis, and metabolism. Commun. Biol. 2023, 6, 1176. [Google Scholar] [CrossRef] [PubMed]
- Zanin-Zhorov, A.; Blazar, B.R. ROCK2, a critical regulator of immune modulation and fibrosis has emerged as a therapeutic target in chronic graft-versus-host disease. Clin. Immunol. 2021, 230, 108823. [Google Scholar] [CrossRef] [PubMed]
- Przepiorka, D.; Le, R.Q.; Ionan, A.; Li, R.J.; Wang, Y.H.; Gudi, R.; Mitra, S.; Vallejo, J.; Okusanya, O.O.; Ma, L.; et al. FDA Approval Summary: Belumosudil for Adult and Pediatric Patients 12 Years and Older with Chronic GvHD after Two or More Prior Lines of Systemic Therapy. Clin. Cancer Res. 2022, 28, 2488–2492. [Google Scholar] [CrossRef] [PubMed]
- Nalkurthi, C.; Schroder, W.A.; Melino, M.; Irvine, K.M.; Nyuydzefe, M.; Chen, W.; Liu, J.; Teng, M.W.L.; Hill, G.R.; Bertolino, P.; et al. ROCK2 inhibition attenuates profibrogenic immune cell function to reverse thioacetamide-induced liver fibrosis. JHEP Rep. 2022, 4, 100386. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, S.; Egger-Heidrich, K.; Halter, J.P.; Jost, L.; Stolzel, F.; Perl, M.; Denk, A.; Edinger, M.; Herr, W.; Kroger, N.; et al. Safety and efficacy of the ROCK-2-inhibitor Belumosudil in cGvHD treatment—A retrospective, German-Swiss multicenter real-world data analysis. Bone Marrow Transplant. 2025. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Watanabe, B.; Nakagawa, Y.; Minami, S.; Morita, T. RPEL proteins are the molecular targets for CCG-1423, an inhibitor of Rho signaling. PLoS ONE 2014, 9, e89016. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Rodansky, E.S.; Haak, A.J.; Larsen, S.D.; Neubig, R.R.; Higgins, P.D. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis. 2014, 20, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Qin, J.; Sun, L.; Gui, L.; Zhang, C.; Huang, Y.; Deng, W.; Huang, A.; Sun, D.; Luo, M. Intrahepatic upregulation of MRTF-A signaling contributes to increased hepatic vascular resistance in cirrhotic rats with portal hypertension. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Zhang, Y.; Sun, Y.; Kong, M.; Han, S.; Wang, C.; Wang, Y.; Xu, D.; Tu, Q.; Zhu, K.; et al. Inhibition of RhoA/MRTF-A signaling alleviates nucleus pulposus fibrosis induced by mechanical stress overload. Connect. Tissue Res. 2022, 63, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Knipe, R.S.; Nurunnabi, M.; Probst, C.K.; Spinney, J.J.; Abe, E.; Bose, R.J.C.; Ha, K.; Logue, A.; Nguyen, T.; Servis, R.; et al. Myofibroblast-specific inhibition of the Rho kinase-MRTF-SRF pathway using nanotechnology for the prevention of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2023, 324, L190–L198. [Google Scholar] [CrossRef]
- Rzepka, M.F.; Raschzok, S.; Lee, X.A.; Yazaki, K.; Dauz, J.; Sun, M.; Meister, T.; Nghiem, L.; Kabir, G.; Desjardins, J.F.; et al. Inhibition of MRTF-A Ameliorates Pathological Remodeling of the Pressure-loaded Right Ventricle. Am. J. Respir. Cell Mol. Biol. 2024. [Google Scholar] [CrossRef]
- Appleton, K.M.; Palsuledesai, C.C.; Misek, S.A.; Blake, M.; Zagorski, J.; Gallo, K.A.; Dexheimer, T.S.; Neubig, R.R. Inhibition of the Myocardin-Related Transcription Factor Pathway Increases Efficacy of Trametinib in NRAS-Mutant Melanoma Cell Lines. Cancers 2021, 13, 2012. [Google Scholar] [CrossRef]
- Leal, A.S.; Misek, S.A.; Lisabeth, E.M.; Neubig, R.R.; Liby, K.T. The Rho/MRTF pathway inhibitor CCG-222740 reduces stellate cell activation and modulates immune cell populations in Kras(G12D); Pdx1-Cre (KC) mice. Sci. Rep. 2019, 9, 7072. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, K.M.; Varnum, M.; Harkema, J.R.; Auerbach, B.; Larsen, S.D.; Neubig, R.R. Prevention of bleomycin-induced lung fibrosis via inhibition of the MRTF/SRF transcription pathway. Pharmacol. Res. Perspect. 2022, 10, e01028. [Google Scholar] [CrossRef]
- Hutchings, K.M.; Lisabeth, E.M.; Rajeswaran, W.; Wilson, M.W.; Sorenson, R.J.; Campbell, P.L.; Ruth, J.H.; Amin, A.; Tsou, P.S.; Leipprandt, J.R.; et al. Pharmacokinetic optimitzation of CCG-203971: Novel inhibitors of the Rho/MRTF/SRF transcriptional pathway as potential antifibrotic therapeutics for systemic scleroderma. Bioorg. Med. Chem. Lett. 2017, 27, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Hashimoto, Y.; Majima, R.; Nakao, E.; Aoki, H.; Nishihara, M.; Ohno-Urabe, S.; Furusho, A.; Hirakata, S.; Nishida, N.; et al. MRTF-A promotes angiotensin II-induced inflammatory response and aortic dissection in mice. PLoS ONE 2020, 15, e0229888. [Google Scholar] [CrossRef]
- Yu-Wai-Man, C.; Spencer-Dene, B.; Lee, R.M.H.; Hutchings, K.; Lisabeth, E.M.; Treisman, R.; Bailly, M.; Larsen, S.D.; Neubig, R.R.; Khaw, P.T. Local delivery of novel MRTF/SRF inhibitors prevents scar tissue formation in a preclinical model of fibrosis. Sci. Rep. 2017, 7, 518. [Google Scholar] [CrossRef] [PubMed]
- Chapeau, E.A.; Sansregret, L.; Galli, G.G.; Chene, P.; Wartmann, M.; Mourikis, T.P.; Jaaks, P.; Baltschukat, S.; Barbosa, I.A.M.; Bauer, D.; et al. Direct and selective pharmacological disruption of the YAP-TEAD interface by IAG933 inhibits Hippo-dependent and RAS-MAPK-altered cancers. Nat. Cancer 2024, 5, 1102–1120. [Google Scholar] [CrossRef]
- Hagenbeek, T.J.; Zbieg, J.R.; Hafner, M.; Mroue, R.; Lacap, J.A.; Sodir, N.M.; Noland, C.L.; Afghani, S.; Kishore, A.; Bhat, K.P.; et al. An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ signaling and overcomes KRAS G12C inhibitor resistance. Nat. Cancer 2023, 4, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Akao, K.; Sato, T.; Mishiro-Sato, E.; Mukai, S.; Ghani, F.I.; Kondo-Ida, L.; Imaizumi, K.; Sekido, Y. TEAD-independent cell growth of Hippo-inactive mesothelioma cells: Unveiling resistance to TEAD inhibitor K-975 through MYC signaling activation. Mol. Cancer Ther. 2024. [Google Scholar] [CrossRef]
- Otsuki, H.; Uemori, T.; Inai, Y.; Suzuki, Y.; Araki, T.; Nan-Ya, K.I.; Yoshinari, K. Reversible and monitorable nephrotoxicity in rats by the novel potent transcriptional enhanced associate domain (TEAD) inhibitor, K-975. J. Toxicol. Sci. 2024, 49, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, A.; Seike, T.; Danjo, T.; Nakajima, T.; Otsubo, N.; Yamaguchi, D.; Tsuji, Y.; Hamaguchi, K.; Yasunaga, M.; Nishiya, Y.; et al. The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am. J. Cancer Res. 2020, 10, 4399–4415. [Google Scholar]
- Hillen, H.; Candi, A.; Vanderhoydonck, B.; Kowalczyk, W.; Sansores-Garcia, L.; Kesikiadou, E.C.; Van Huffel, L.; Spiessens, L.; Nijs, M.; Soons, E.; et al. A Novel Irreversible TEAD Inhibitor, SWTX-143, Blocks Hippo Pathway Transcriptional Output and Causes Tumor Regression in Preclinical Mesothelioma Models. Mol. Cancer Ther. 2024, 23, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Yang, L.; Xiao, M.X.; Li, N.; Huang, X.; Ye, L.H.; Zhang, H.C.; Liu, Z.Q.; Li, J.Q.; Liu, Y.Y.; et al. Spatial and Single-Cell Transcriptomics Reveals the Regional Division of the Spatial Structure of MASH Fibrosis. Liver Int. 2024, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Hundertmark, J.; Ritz, T.P.; Weiskirchen, R.; Tacke, F. Single Cell RNA Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis. Cells 2019, 8, 503. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Huang, C.; Shi, X.; Wu, M.; Li, H.; Liu, Q.; Zhang, X.; Zhao, Y.; Li, X. Single-cell transcriptomics of hepatic stellate cells uncover crucial pathways and key regulators involved in non-alcoholic steatohepatitis. Endocr. Connect. 2023, 12, e220502. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.; Gentile, S.D.; Ben Saad, A.; Rahman, R.U.; Habboub, T.; Pratt, D.S.; Mullen, A.C. Single-cell transcriptomics stratifies organoid models of metabolic dysfunction-associated steatotic liver disease. EMBO J. 2023, 42, e113898. [Google Scholar] [CrossRef] [PubMed]
- Merens, V.; Knetemann, E.; Gurbuz, E.; De Smet, V.; Messaoudi, N.; Reynaert, H.; Verhulst, S.; van Grunsven, L.A. Hepatic stellate cell single cell atlas reveals a highly similar activation process across liver disease aetiologies. JHEP Rep. 2025, 7, 101223. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Stage of Evaluation | ClinicalTrials.gov Identifier | |
|---|---|---|---|
| PLN-74809 (Bexotegrast) | PLN-74809 is an oral small-molecule dual inhibitor of αvβ1 and αvβ6 integrins. Developed by Pliant Therapeutics. By inhibiting these integrins, it aims to prevent the activation of latent TGF-β1 and TGF-β3, reducing fibrosis. | PLN-74809 is in two phase II clinical trials targeting IPF and primary sclerosing cholangitis (PSC). Clinical data suggest it is well-tolerated and reduces biomarkers of fibrosis. Bexotegrast has been granted fast track designation and orphan drug designation by the FDA for IPF. Additionally, it received Orphan Drug Designation from both the FDA and EMA for PSC. | 04480840 04072315 05621252 04396796 |
| PLN-1474 | PLN-1474 is a selective inhibitor of αvβ1 integrin, targeting the activation of TGF-β1 in the liver to reduce fibrosis. Developed by Pliant Therapeutics, in collaboration with Novartis | PLN-1474 is has completed phase I clinical trials for liver fibrosis associated with NASH. | Not reported |
| Pirfenidone (PFD) | Inhibition of TGFβ synthesis and activation of downstream signaling. Anti-inflammatory effects through downregulation of TNFα, IL-1 and IL-6, IFNγ along with NFκB inactivation among other effects. | Approved by FDA for IPF. | |
| PR-PFD | Prolonged-release pirfenidone, administered via nebulizer | Approved by FDA for IPF and in Mexico approved by COFEPRIS for advanced liver fibrosis. | 05542615 |
| LYT-100 | Deuterated form of pirfenidone (PureTech Health plc) | Successful Phase 2b Trial for IPF concluded. | 05321420 |
| AVID200 | AVID200 is a TGF-β ligand trap engineered to selectively inhibit TGF-β1 and TGF-β3 isoforms while sparing TGF-β2, aiming to reduce fibrosis with fewer side effects. | In early clinical phase I trials for fibrotic diseases, including systemic sclerosis (SSC) and myelofibrosis (MF). Studies have shown promising results warranting potential applications in other fibrotic conditions. | 03831438, 03895112 |
| Zelasudil (RXC007) | Zelasudil, developed by Redx Pharma, is a potent and highly selective ROCK2 inhibitor in clinical development for IPF. | Preclinical studies have shown promising results for treating other fibrotic indications, including liver fibrosis. Completed a Phase 2a study in IPF as of Q4 2024. | Not reported |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharip, A.; Kunz, J. Mechanosignaling via Integrins: Pivotal Players in Liver Fibrosis Progression and Therapy. Cells 2025, 14, 266. https://doi.org/10.3390/cells14040266
Sharip A, Kunz J. Mechanosignaling via Integrins: Pivotal Players in Liver Fibrosis Progression and Therapy. Cells. 2025; 14(4):266. https://doi.org/10.3390/cells14040266
Chicago/Turabian StyleSharip, Aigul, and Jeannette Kunz. 2025. "Mechanosignaling via Integrins: Pivotal Players in Liver Fibrosis Progression and Therapy" Cells 14, no. 4: 266. https://doi.org/10.3390/cells14040266
APA StyleSharip, A., & Kunz, J. (2025). Mechanosignaling via Integrins: Pivotal Players in Liver Fibrosis Progression and Therapy. Cells, 14(4), 266. https://doi.org/10.3390/cells14040266