
Advancing Glioblastoma Research with Innovative Brain Organoid-Based Models

 , , and
, , and 
Abstract

1. Enduring Challenges in Glioblastoma Research and Therapy
2. Human Stem Cells Used in GBM Models
2.1. Human Embryonic Stem Cells (hESCs) and Induced Pluripotent Stem Cells (hiPSCs)
2.2. Human Expanded Potential Stem Cells (hEPSCs)
2.3. Human Neural Stem Cells (hNSCs)
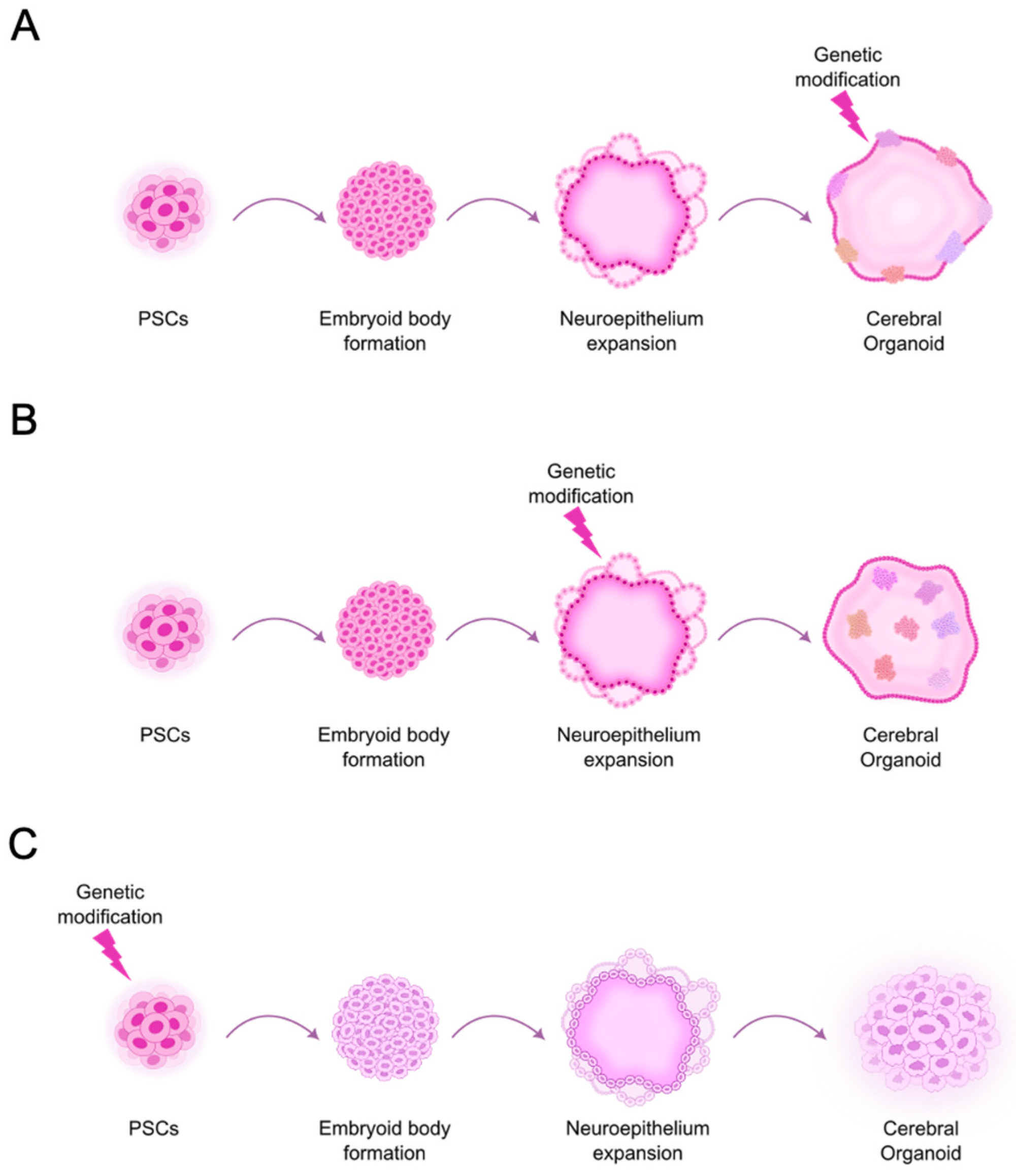
3. Stem Cell-Derived Brain Organoid-Based Models in GBM Research
3.1. Models to Dissect GBM Tumor Initiation and Potential Vulnerabilities
3.1.1. Development of GBM Models Based on Common Genetic Aberrations to Study Tumor Initiation
3.1.2. Impact of Other Molecular Players on GBM Development
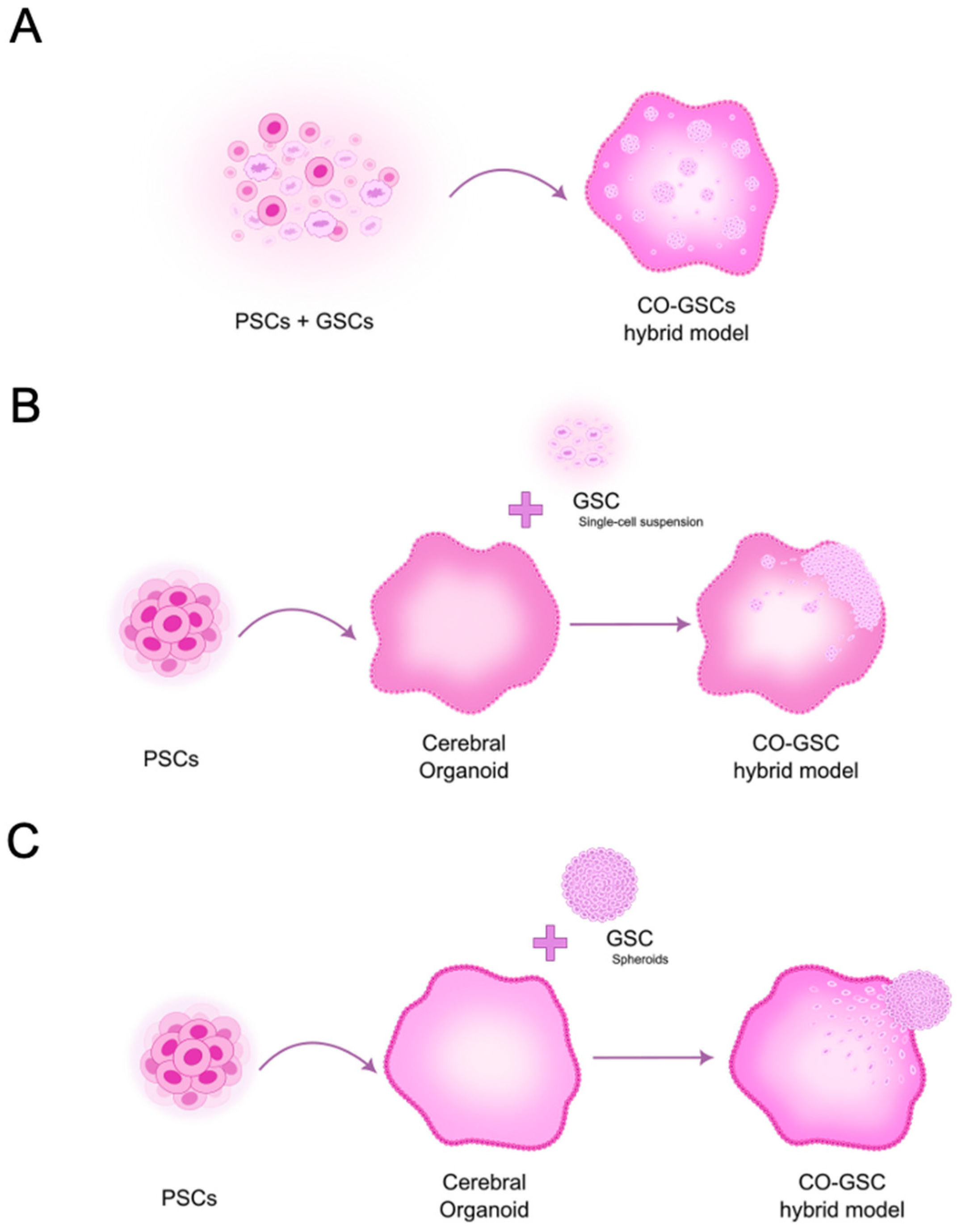
3.2. Models to Study Fully Developed GBM Within the Brain Microenvironment
3.2.1. Pioneering Studies Using 3D GBM-Neural Tissue Hybrid Models
3.2.2. Innovative Studies Using 3D GBM-Brain Organoid Hybrid Models
4. Enabling Technologies for Analyzing Organoid-Based Models of GBM
4.1. Microscopy Technologies
4.2. Omics Technologies
5. Challenges, Opportunities, and Future Perspectives
5.1. Challenges in Finding the Perfect Model to Study GBM
5.2. Harnessing Stem Cell-Derived Organoid Biotechnology to Model Glioblastoma
5.3. Future Perspectives of Stem Cell-Derived Organoids in Glioblastoma Modeling
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| 4D | Four-dimensional |
| ADAM10 | A disintegrin and metalloproteinase domain-containing protein 10 |
| BBB | Blood–brain barrier |
| BCNU | Bis-chloroethylnitrosourea |
| Cas9 | CRISPR-associated nuclease 9 |
| CDKN2A/B | Cyclin-dependent kinase inhibitor 2A/B |
| CO | Cerebral organoid |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| EB | Embryoid body |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| enCOR | Microfilament-engineered cerebral organoid |
| EPSC | Expanded potential stem cell |
| ESC | Embryonic stem cell |
| ERK | Extracellular signal-regulated kinase |
| GBM | Glioblastoma |
| GFP | Green fluorescent protein |
| GLICO | Glioma cerebral organoid |
| GSC | Glioblastoma stem-like cell |
| IDH1/2 | Isocitrate dehydrogenase 1 and 2 genes |
| iPSC | Induced pluripotent stem cell |
| LEGOs | Laboratory-engineered glioblastoma-like organoids |
| LLSM | Lattice-light sheet microscopy |
| LSCM | Laser scanning confocal microscopy |
| LSFM | Light-sheet fluorescent microscopy |
| ltGLICOs | Long-term glioma cerebral organoids |
| MEOX2 | Mesenchyme homeobox 2 |
| METTL7B | Methyltransferase-like 7B |
| NCBI GEO | National Center for Biotechnology Information Gene Expression Omnibus |
| neoCOR | Neoplastic cerebral organoid |
| NF1 | Neurofibromin 1 |
| NLGN3 | Neuroligin-3 |
| NPC | Neural progenitor cell |
| NSC | Neural stem cell |
| oRG-like | Outer radial glia-like cells |
| PSC | Pluripotent stem cell |
| PTEN | Phosphatase and tensin homolog deletions |
| RFP | Red fluorescent protein |
| scATAC-seq | Single-cell assay for transposase-accessible chromatin sequencing |
| scRNA-seq | Single cell RNA sequencing |
| SDCM | Spinning disk confocal microscopy |
| SGZ | Subgranular zone |
| shRNAs | Short hairpins RNAs |
| SVZ | Subventricular zone |
| TEM | Transmission electron microscopy |
| TERT | Telomerase reverse transcriptase |
| TME | Tumor microenvironment |
| TMZ | Temozolomide |
| TP53 | Tumor Protein 53 |
References
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2024, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Ballard, C.; Benedetti, J.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S.; Ostrom, Q.T. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2017–2021. Neuro. Oncol. 2024, 26, vi1–vi85. [Google Scholar] [CrossRef]
- Pellerino, A.; Caccese, M.; Padovan, M.; Cerretti, G.; Lombardi, G. Epidemiology, Risk Factors, and Prognostic Factors of Gliomas. Clin. Transl. Imaging 2022, 10, 467–475. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Lan, Z.; Li, X.; Zhang, X. Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers. Int. J. Mol. Sci. 2024, 25, 3040. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.-V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in Glioblastoma Research: Focus on the Tumor Microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Weller, M.; Le Rhun, E. How Did Lomustine Become Standard of Care in Recurrent Glioblastoma? Cancer Treat. Rev. 2020, 87, 102029. [Google Scholar] [CrossRef]
- Ma, R.; Taphoorn, M.J.B.; Plaha, P. Advances in the Management of Glioblastoma. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1103–1111. [Google Scholar] [CrossRef]
- Ho, W.-M.; Chen, C.-Y.; Chiang, T.-W.; Chuang, T.-J. A Longer Time to Relapse Is Associated with a Larger Increase in Differences between Paired Primary and Recurrent IDH Wild-Type Glioblastomas at Both the Transcriptomic and Genomic Levels. Acta Neuropathol. Commun. 2024, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Pichol-Thievend, C.; Anezo, O.; Pettiwala, A.M.; Bourmeau, G.; Montagne, R.; Lyne, A.-M.; Guichet, P.-O.; Deshors, P.; Ballestín, A.; Blanchard, B.; et al. VC-Resist Glioblastoma Cell State: Vessel Co-Option as a Key Driver of Chemoradiation Resistance. Nat. Commun. 2024, 15, 3602. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Scully, S.; Francescone, R.; Faibish, M.; Bentley, B.; Taylor, S.L.; Oh, D.; Schapiro, R.; Moral, L.; Yan, W.; Shao, R. Transdifferentiation of Glioblastoma Stem-like Cells into Mural Cells Drives Vasculogenic Mimicry in Glioblastomas. J. Neurosci. 2012, 32, 12950–12960. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour Vascularization via Endothelial Differentiation of Glioblastoma Stem-like Cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma Hijacks Neuronal Mechanisms for Brain Invasion. Cell 2022, 185, 2899–2917.e31. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic Synaptic Input to Glioma Cells Drives Brain Tumour Progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and Synaptic Integration of Glioma into Neural Circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor Stem Cells Derived from Glioblastomas Cultured in bFGF and EGF More Closely Mirror the Phenotype and Genotype of Primary Tumors than Do Serum-Cultured Cell Lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely Hominid Features of Adult Human Astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-Derived Xenografts Undergo Mouse-Specific Tumor Evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef]
- Mariappan, A.; Goranci-Buzhala, G.; Ricci-Vitiani, L.; Pallini, R.; Gopalakrishnan, J. Trends and Challenges in Modeling Glioma Using 3D Human Brain Organoids. Cell Death Differ. 2021, 28, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- HESCRegistry—Public Lines|STEM Cell Information. Available online: https://stemcells.nih.gov/registry/eligible-to-use-lines (accessed on 9 January 2025).
- Koga, T.; Chaim, I.A.; Benitez, J.A.; Markmiller, S.; Parisian, A.D.; Hevner, R.F.; Turner, K.M.; Hessenauer, F.M.; D’Antonio, M.; Nguyen, N.-P.D.; et al. Longitudinal Assessment of Tumor Development Using Cancer Avatars Derived from Genetically Engineered Pluripotent Stem Cells. Nat. Commun. 2020, 11, 550. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically Engineered Cerebral Organoids Model Brain Tumor Formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-M.; Lee, S.-H.; Lim, J.; Yoo, J.; Hwang, D.-Y. The Epidermal Growth Factor Receptor Variant Type III Mutation Frequently Found in Gliomas Induces Astrogenesis in Human Cerebral Organoids. Cell Prolif. 2021, 54, e12965. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, M.; Shao, C.; Schlicker, L.; Zhuo, Y.; Harim, Y.; Peng, T.; Tian, W.; Stöffler, N.; Schneider, M.; et al. A Multidimensional Atlas of Human Glioblastoma-like Organoids Reveals Highly Coordinated Molecular Networks and Effective Drugs. NPJ Precis. Oncol. 2024, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Nowak-Imialek, M.; Chen, X.; Chen, D.; Herrmann, D.; Ruan, D.; Chen, A.C.H.; Eckersley-Maslin, M.A.; Ahmad, S.; Lee, Y.L.; et al. Establishment of Porcine and Human Expanded Potential Stem Cells. Nat. Cell Biol. 2019, 21, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ryan, D.J.; Wang, W.; Tsang, J.C.-H.; Lan, G.; Masaki, H.; Gao, X.; Antunes, L.; Yu, Y.; Zhu, Z.; et al. Establishment of Mouse Expanded Potential Stem Cells. Nature 2017, 550, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ryan, D.J.; Lan, G.; Zou, X.; Liu, P. In Vitro Establishment of Expanded-Potential Stem Cells from Mouse Pre-Implantation Embryos or Embryonic Stem Cells. Nat. Protoc. 2019, 14, 350–378. [Google Scholar] [CrossRef] [PubMed]
- Vinel, C.; Rosser, G.; Guglielmi, L.; Constantinou, M.; Pomella, N.; Zhang, X.; Boot, J.R.; Jones, T.A.; Millner, T.O.; Dumas, A.A.; et al. Comparative Epigenetic Analysis of Tumour Initiating Cells and Syngeneic EPSC-Derived Neural Stem Cells in Glioblastoma. Nat. Commun. 2021, 12, 6130. [Google Scholar] [CrossRef]
- Constantinou, M.; Nicholson, J.; Zhang, X.; Maniati, E.; Lucchini, S.; Rosser, G.; Vinel, C.; Wang, J.; Lim, Y.M.; Brandner, S.; et al. Lineage Specification in Glioblastoma Is Regulated by METTL7B. Cell Rep. 2024, 43, 114309. [Google Scholar] [CrossRef] [PubMed]
- Mora-Bermúdez, F.; Vaid, S.; Huttner, W.B. Neural Stem Cells in Cerebral Cortex Development. In Neuroscience in the 21st Century: From Basic to Clinical; Pfaff, D.W., Volkow, N.D., Rubenstein, J.L., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 161–192. ISBN 978-3-030-88832-9. [Google Scholar]
- Loras, A.; Gonzalez-Bonet, L.G.; Gutierrez-Arroyo, J.L.; Martinez-Cadenas, C.; Marques-Torrejon, M.A. Neural Stem Cells as Potential Glioblastoma Cells of Origin. Life 2023, 13, 905. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, R.; Xiong, Y.; Zhou, L.; Yan, X.; Wang, M.; Li, F.; Xie, C.; Zhang, Y.; Huang, Z.; et al. Sequential Fate-Switches in Stem-like Cells Drive the Tumorigenic Trajectory from Human Neural Stem Cells to Malignant Glioma. Cell Res. 2021, 31, 684–702. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.L.; O’Duibhir, E.; Gangoso, E.; Bressan, R.B.; Bulstrode, H.; Marqués-Torrejón, M.-Á.; Ferguson, K.M.; Blin, C.; Grant, V.; Alfazema, N.; et al. Elevated FOXG1 in Glioblastoma Stem Cells Cooperates with Wnt/β-Catenin to Induce Exit from Quiescence. Cell Rep. 2023, 42, 112561. [Google Scholar] [CrossRef]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct Isolation of Human Central Nervous System Stem Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and Expansion Using Only Small Molecules of Human Neural Progenitors for Neurodegenerative Disease Modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, D.; Zarnowska, E.D.; Du, Z.; Werbel, B.; Valliere, C.; Pearce, R.A.; Thomson, J.A.; Zhang, S.-C. Directed Differentiation of Dopaminergic Neuronal Subtypes from Human Embryonic Stem Cells. Stem Cells 2005, 23, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Pollard, S.; Conti, L.; Toselli, M.; Biella, G.; Parkin, G.; Willatt, L.; Falk, A.; Cattaneo, E.; Smith, A. Long-Term Tripotent Differentiation Capacity of Human Neural Stem (NS) Cells in Adherent Culture. Mol. Cell Neurosci. 2008, 38, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef]
- Pine, A.R.; Cirigliano, S.M.; Singhania, R.; Nicholson, J.; da Silva, B.; Leslie, C.S.; Fine, H.A. Microenvironment-Driven Dynamic Chromatin Changes in Glioblastoma Recapitulate Early Neural Development at Single-Cell Resolution. Cancer Res. 2023, 83, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, V.; Pospisilova, V.; Vanova, T.; Amruz Cerna, K.; Abaffy, P.; Sedmik, J.; Raska, J.; Vochyanova, S.; Matusova, Z.; Houserova, J.; et al. Glioblastoma and Cerebral Organoids: Development and Analysis of an in Vitro Model for Glioblastoma Migration. Mol. Oncol. 2023, 17, 647–663. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Liu, P.; Griffiths, S.; Veljanoski, D.; Vaughn-Beaucaire, P.; Speirs, V.; Brüning-Richardson, A. Preclinical Models of Glioblastoma: Limitations of Current Models and the Promise of New Developments. Expert Rev. Mol. Med. 2021, 23, e20. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome Editing. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Taubenschmid-Stowers, J.; Orthofer, M.; Laemmerer, A.; Krauditsch, C.; Rózsová, M.; Studer, C.; Lötsch, D.; Gojo, J.; Gabler, L.; Dyczynski, M.; et al. A Whole-Genome Scan for Artemisinin Cytotoxicity Reveals a Novel Therapy for Human Brain Tumors. EMBO Mol. Med. 2023, 15, e16959. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Wang, Y.; Habib, A.; Priyadarshini, M.; Kodavali, C.V.; Chen, A.; Ma, W.; Wang, J.; Hameed, N.U.F.; Hu, B.; et al. TP53-PTEN-NF1 Depletion in Human Brain Organoids Produces a Glioma Phenotype in Vitro. Front. Oncol. 2023, 13, 1279806. [Google Scholar] [CrossRef] [PubMed]
- Marumoto, T.; Tashiro, A.; Friedmann-Morvinski, D.; Scadeng, M.; Soda, Y.; Gage, F.H.; Verma, I.M. Development of a Novel Mouse Glioma Model Using Lentiviral Vectors. Nat. Med. 2009, 15, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Pașca, A.M.; Park, J.-Y.; Shin, H.-W.; Qi, Q.; Revah, O.; Krasnoff, R.; O’Hara, R.; Willsey, A.J.; Palmer, T.D.; Pașca, S.P. Human 3D Cellular Model of Hypoxic Brain Injury of Prematurity. Nat. Med. 2019, 25, 784–791. [Google Scholar] [CrossRef]
- Schnalzger, T.E.; de Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D Model for CAR-Mediated Cytotoxicity Using Patient-Derived Colorectal Cancer Organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef] [PubMed]
- Tachon, G.; Masliantsev, K.; Rivet, P.; Petropoulos, C.; Godet, J.; Milin, S.; Wager, M.; Guichet, P.-O.; Karayan-Tapon, L. Prognostic Significance of MEOX2 in Gliomas. Mod. Pathol. 2019, 32, 774–786. [Google Scholar] [CrossRef]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-Dependent Chromatin State Reprogramming, Reversibility, and Persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Schönrock, A.; Heinzelmann, E.; Steffl, B.; Demirdizen, E.; Narayanan, A.; Krunic, D.; Bähr, M.; Park, J.-W.; Schmidt, C.; Özduman, K.; et al. MEOX2 Homeobox Gene Promotes Growth of Malignant Gliomas. Neuro Oncol. 2022, 24, 1911–1924. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Song, H.; Ming, G.-L. Brain Organoids: Advances, Applications and Challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [PubMed]
- Nayernia, Z.; Turchi, L.; Cosset, E.; Peterson, H.; Dutoit, V.; Dietrich, P.-Y.; Tirefort, D.; Chneiweiss, H.; Lobrinus, J.-A.; Krause, K.-H.; et al. The Relationship between Brain Tumor Cell Invasion of Engineered Neural Tissues and in Vivo Features of Glioblastoma. Biomaterials 2013, 34, 8279–8290. [Google Scholar] [CrossRef] [PubMed]
- Preynat-Seauve, O.; Suter, D.M.; Tirefort, D.; Turchi, L.; Virolle, T.; Chneiweiss, H.; Foti, M.; Lobrinus, J.-A.; Stoppini, L.; Feki, A.; et al. Development of Human Nervous Tissue upon Differentiation of Embryonic Stem Cells in Three-Dimensional Culture. Stem Cells 2009, 27, 509–520. [Google Scholar] [CrossRef]
- Cosset, E.; Petty, T.; Dutoit, V.; Tirefort, D.; Otten-Hernandez, P.; Farinelli, L.; Dietrich, P.-Y.; Preynat-Seauve, O. Human Tissue Engineering Allows the Identification of Active miRNA Regulators of Glioblastoma Aggressiveness. Biomaterials 2016, 107, 74–87. [Google Scholar] [CrossRef]
- Bassot, A.; Dragic, H.; Haddad, S.A.; Moindrot, L.; Odouard, S.; Corlazzoli, F.; Marinari, E.; Bomane, A.; Brassens, A.; Marteyn, A.; et al. Identification of a miRNA Multi-Targeting Therapeutic Strategy in Glioblastoma. Cell Death Dis. 2023, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Plummer, S.; Wallace, S.; Ball, G.; Lloyd, R.; Schiapparelli, P.; Quiñones-Hinojosa, A.; Hartung, T.; Pamies, D. A Human iPSC-Derived 3D Platform Using Primary Brain Cancer Cells to Study Drug Development and Personalized Medicine. Sci. Rep. 2019, 9, 1407. [Google Scholar] [CrossRef] [PubMed]
- Linkous, A.; Fine, H.A. Generating Patient-Derived Gliomas within Cerebral Organoids. STAR Protoc. 2020, 1, 100008. [Google Scholar] [CrossRef]
- Pine, A.R.; Cirigliano, S.M.; Nicholson, J.G.; Hu, Y.; Linkous, A.; Miyaguchi, K.; Edwards, L.; Singhania, R.; Schwartz, T.H.; Ramakrishna, R.; et al. Tumor Microenvironment Is Critical for the Maintenance of Cellular States Found in Primary Glioblastomas. Cancer Discov. 2020, 10, 964–979. [Google Scholar] [CrossRef]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63.e6. [Google Scholar] [CrossRef] [PubMed]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-Organization of Axial Polarity, inside-out Layer Pattern, and Species-Specific Progenitor Dynamics in Human ES Cell-Derived Neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [PubMed]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Jechow, K.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling Glioblastoma Invasion Using Human Brain Organoids and Single-Cell Transcriptomics. Neuro Oncol. 2020, 22, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain Tumour Cells Interconnect to a Functional and Resistant Network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Goranci-Buzhala, G.; Mariappan, A.; Gabriel, E.; Ramani, A.; Ricci-Vitiani, L.; Buccarelli, M.; D’Alessandris, Q.G.; Pallini, R.; Gopalakrishnan, J. Rapid and Efficient Invasion Assay of Glioblastoma in Human Brain Organoids. Cell Rep. 2020, 31, 107738. [Google Scholar] [CrossRef]
- Gabriel, E.; Gopalakrishnan, J. Generation of iPSC-Derived Human Brain Organoids to Model Early Neurodevelopmental Disorders. J. Vis. Exp. 2017, 122, 55372. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting Neuronal Activity-Regulated Neuroligin-3 Dependency in High-Grade Glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Azzarelli, R.; Ori, M.; Philpott, A.; Simons, B.D. Three-Dimensional Model of Glioblastoma by Co-Culturing Tumor Stem Cells with Human Brain Organoids. Biol. Open 2021, 10, bio056416. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Phillips, A.W.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Knoblich, J.A. Guided Self-Organization and Cortical Plate Formation in Human Brain Organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef]
- Nicholson, J.G.; Cirigliano, S.; Singhania, R.; Haywood, C.; Shahidi Dadras, M.; Yoshimura, M.; Vanderbilt, D.; Liechty, B.; Fine, H.A. Chronic Hypoxia Remodels the Tumor Microenvironment to Support Glioma Stem Cell Growth. Acta Neuropathol. Commun. 2024, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Wang, H.; Gu, T.; Liu, H.; Deng, P.; Li, B.; Yang, H.; Mao, Y.; Shao, Z. Modeling the Precise Interaction of Glioblastoma with Human Brain Region-Specific Organoids. iScience 2024, 27, 109111. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Noh, H.; Bin Kim, W.; Ni, P.; Nguyen, C.; Cote, S.E.; Noyes, E.; Zhao, J.; Parsons, T.; Park, J.M.; et al. Dysregulated Protocadherin-Pathway Activity as an Intrinsic Defect in Induced Pluripotent Stem Cell-Derived Cortical Interneurons from Subjects with Schizophrenia. Nat. Neurosci. 2019, 22, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of Human Cortical Organoid Generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Mangena, V.; Chanoch-Myers, R.; Sartore, R.; Paulsen, B.; Gritsch, S.; Weisman, H.; Hara, T.; Breakefield, X.O.; Breyne, K.; Regev, A.; et al. Glioblastoma-Cortical Organoids Recapitulate Cell State Heterogeneity and Intercellular Transfer. Cancer Discov. 2024, 15, 299–315. [Google Scholar] [CrossRef]
- Ramani, A.; Pasquini, G.; Gerkau, N.J.; Jadhav, V.; Vinchure, O.S.; Altinisik, N.; Windoffer, H.; Muller, S.; Rothenaigner, I.; Lin, S.; et al. Reliability of High-Quantity Human Brain Organoids for Modeling Microcephaly, Glioma Invasion and Drug Screening. Nat. Commun. 2024, 15, 10703. [Google Scholar] [CrossRef] [PubMed]
- Goranci-Buzhala, G.; Mariappan, A.; Ricci-Vitiani, L.; Josipovic, N.; Pacioni, S.; Gottardo, M.; Ptok, J.; Schaal, H.; Callaini, G.; Rajalingam, K.; et al. Cilium Induction Triggers Differentiation of Glioma Stem Cells. Cell Rep. 2021, 36, 109656. [Google Scholar] [CrossRef]
- Francois, L.; Boskovic, P.; Knerr, J.; He, W.; Sigismondo, G.; Schwan, C.; More, T.H.; Schlotter, M.; Conway, M.E.; Krijgsveld, J.; et al. BCAT1 Redox Function Maintains Mitotic Fidelity. Cell Rep. 2022, 41, 111524. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin Avβ5 Axis. Cell Stem Cell 2020, 26, 187–204.e10. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.S.; Jandrey, E.H.F.; Granha, I.; Endo, A.K.; Ferreira, R.O.; Araujo, B.H.S.; Zatz, M.; Okamoto, O.K. Differential Replication and Oncolytic Effects of Zika Virus in Aggressive CNS Tumor Cells: Insights from Organoid and Tumoroid Models. Viruses 2024, 16, 1764. [Google Scholar] [CrossRef]
- Fawal, M.-A.; Jungas, T.; Davy, A. Inhibition of DHFR Targets the Self-Renewing Potential of Brain Tumor Initiating Cells. Cancer Lett. 2021, 503, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Jermakowicz, A.M.; Rybin, M.J.; Suter, R.K.; Sarkaria, J.N.; Zeier, Z.; Feng, Y.; Ayad, N.G. The Novel BET Inhibitor UM-002 Reduces Glioblastoma Cell Proliferation and Invasion. Sci. Rep. 2021, 11, 23370. [Google Scholar] [CrossRef]
- Rybin, M.J.; Laverde-Paz, M.J.; Suter, R.K.; Affer, M.; Ayad, N.G.; Feng, Y.; Zeier, Z. A Dual Aurora and Lim Kinase Inhibitor Reduces Glioblastoma Proliferation and Invasion. Bioorg. Med. Chem. Lett. 2022, 61, 128614. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, A.; Reed, M.R.; Heflin, B.; Gaydos, J.; Piña-Oviedo, S.; Jędrzejczyk, M.; Klejborowska, G.; Stępczyńska, N.; Chambers, T.C.; Tackett, A.J.; et al. Anti-Glioblastoma Activity of Monensin and Its Analogs in an Organoid Model of Cancer. Biomed. Pharmacother. 2022, 153, 113440. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, L.; Bedia, C.; Diao, D.; Mosteiro, A.; Ferrés, A.; Stanzani, E.; Martínez-Soler, F.; Tortosa, A.; Pineda, E.; Aldecoa, I.; et al. Preclinical Studies with Glioblastoma Brain Organoid Co-Cultures Show Efficient 5-ALA Photodynamic Therapy. Cells 2023, 12, 1125. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, D.; Hoffmann, D.C.; Venkataramani, V.; Jung, E.; Horschitz, S.; Tetzlaff, S.K.; Jabali, A.; Hai, L.; Kessler, T.; Azoŕin, D.D.; et al. Autonomous Rhythmic Activity in Glioma Networks Drives Brain Tumour Growth. Nature 2023, 613, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Schlieper-Scherf, S.; Hebach, N.; Hausmann, D.; Azorín, D.D.; Hoffmann, D.C.; Horschitz, S.; Maier, E.; Koch, P.; Karreman, M.A.; Etminan, N.; et al. Disrupting Glioblastoma Networks with Tumor Treating Fields (TTFields) in in Vitro Models. J. Neurooncol. 2024, 170, 139–151. [Google Scholar] [CrossRef]
- Kural, M.H.; Djakbarova, U.; Cakir, B.; Tanaka, Y.; Chan, E.T.; Arteaga Muniz, V.I.; Madraki, Y.; Qian, H.; Park, J.; Sewanan, L.R.; et al. Mechano-Inhibition of Endocytosis Sensitizes Cancer Cells to Fas-Induced Apoptosis. Cell Death Dis. 2024, 15, 440. [Google Scholar] [CrossRef] [PubMed]
- Fujii, E.; Yamazaki, M.; Kawai, S.; Ohtani, Y.; Watanabe, T.; Kato, A.; Suzuki, M. A Simple Method for Histopathological Evaluation of Organoids. J. Toxicol. Pathol. 2018, 31, 81–85. [Google Scholar] [CrossRef]
- Antonica, F.; Kasprzyk, D.F.; Opitz, R.; Iacovino, M.; Liao, X.-H.; Dumitrescu, A.M.; Refetoff, S.; Peremans, K.; Manto, M.; Kyba, M.; et al. Generation of Functional Thyroid from Embryonic Stem Cells. Nature 2012, 491, 66–71. [Google Scholar] [CrossRef]
- Linnemann, J.R.; Miura, H.; Meixner, L.K.; Irmler, M.; Kloos, U.J.; Hirschi, B.; Bartsch, H.S.; Sass, S.; Beckers, J.; Theis, F.J.; et al. Quantification of Regenerative Potential in Primary Human Mammary Epithelial Cells. Development 2015, 142, 3239–3251. [Google Scholar] [CrossRef]
- Bock, N.; Röhl, J. Real-Time and 3D Quantification of Cancer Cell Dynamics: Exploiting a Bioengineered Human Bone Metastatic Microtissue. Methods Mol. Biol. 2019, 2054, 59–77. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-Organizing Optic-Cup Morphogenesis in Three-Dimensional Culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Okkelman, I.A.; Foley, T.; Papkovsky, D.B.; Dmitriev, R.I. Live Cell Imaging of Mouse Intestinal Organoids Reveals Heterogeneity in Their Oxygenation. Biomaterials 2017, 146, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D Human Triculture System Modeling Neurodegeneration and Neuroinflammation in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Völkner, M.; Zschätzsch, M.; Rostovskaya, M.; Overall, R.W.; Busskamp, V.; Anastassiadis, K.; Karl, M.O. Retinal Organoids from Pluripotent Stem Cells Efficiently Recapitulate Retinogenesis. Stem Cell Rep. 2016, 6, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Lukonin, I.; Serra, D.; Challet Meylan, L.; Volkmann, K.; Baaten, J.; Zhao, R.; Meeusen, S.; Colman, K.; Maurer, F.; Stadler, M.B.; et al. Phenotypic Landscape of Intestinal Organoid Regeneration. Nature 2020, 586, 275–280. [Google Scholar] [CrossRef]
- Li, Y.; Chen, M.; Hu, J.; Sheng, R.; Lin, Q.; He, X.; Guo, M. Volumetric Compression Induces Intracellular Crowding to Control Intestinal Organoid Growth via Wnt/β-Catenin Signaling. Cell Stem Cell 2021, 28, 63–78.e7. [Google Scholar] [CrossRef] [PubMed]
- Sloan, S.A.; Andersen, J.; Pașca, A.M.; Birey, F.; Pașca, S.P. Generation and Assembly of Human Brain Region-Specific Three-Dimensional Cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef]
- McEvoy, E.; Han, Y.L.; Guo, M.; Shenoy, V.B. Gap Junctions Amplify Spatial Variations in Cell Volume in Proliferating Tumor Spheroids. Nat. Commun. 2020, 11, 6148. [Google Scholar] [CrossRef]
- Beghin, A.; Grenci, G.; Sahni, G.; Guo, S.; Rajendiran, H.; Delaire, T.; Mohamad Raffi, S.B.; Blanc, D.; de Mets, R.; Ong, H.T.; et al. Automated High-Speed 3D Imaging of Organoid Cultures with Multi-Scale Phenotypic Quantification. Nat. Methods 2022, 19, 881–892. [Google Scholar] [CrossRef]
- Favreau, P.F.; He, J.; Gil, D.A.; Deming, D.A.; Huisken, J.; Skala, M.C. Label-Free Redox Imaging of Patient-Derived Organoids Using Selective Plane Illumination Microscopy. Biomed. Opt. Express 2020, 11, 2591–2606. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.S.; Guan, W.; Matsumoto, K.; Pan, C.; Chung, K.; Ertürk, A.; Ueda, H.R.; Lichtman, J.W. TISSUE CLEARING. Nat. Rev. Methods Primers 2021, 1, 84. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice Light-Sheet Microscopy: Imaging Molecules to Embryos at High Spatiotemporal Resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef]
- Ma, Q.; Tao, H.; Li, Q.; Zhai, Z.; Zhang, X.; Lin, Z.; Kuang, N.; Pan, J. OrganoidDB: A Comprehensive Organoid Database for the Multi-Perspective Exploration of Bulk and Single-Cell Transcriptomic Profiles of Organoids. Nucleic Acids Res. 2023, 51, D1086–D1093. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Li, X.; Xiong, H.; Fan, H.; Gao, X.; Vemireddy, V.; Margolis, R.; Li, J.; Ge, X.; Giannotta, M.; et al. Optical Blood-Brain-Tumor Barrier Modulation Expands Therapeutic Options for Glioblastoma Treatment. Nat. Commun. 2023, 14, 4934. [Google Scholar] [CrossRef] [PubMed]
- Haque, W.; Thong, Y.; Verma, V.; Rostomily, R.; Brian Butler, E.; Teh, B.S. Patterns of Management and Outcomes of Unifocal versus Multifocal Glioblastoma. J. Clin. Neurosci. 2020, 74, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Miki, S.; Koga, T.; Mckinney, A.M.; Parisian, A.D.; Tadokoro, T.; Vadla, R.; Marsala, M.; Hevner, R.F.; Costello, J.F.; Furnari, F. TERT Promoter C228T Mutation in Neural Progenitors Confers Growth Advantage Following Telomere Shortening in Vivo. Neuro. Oncol. 2022, 24, 2063–2075. [Google Scholar] [CrossRef]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.-D.; Göke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.-P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M.; Takahashi, J.; Eiraku, M.; Sasai, Y. Generation of Functional Hippocampal Neurons from Self-Organizing Human Embryonic Stem Cell-Derived Dorsomedial Telencephalic Tissue. Nat. Commun. 2015, 6, 8896. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-Organization of Polarized Cerebellar Tissue in 3D Culture of Human Pluripotent Stem Cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human Pluripotent Stem Cells Recurrently Acquire and Expand Dominant Negative P53 Mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Hirst, A.J.; Gokhale, P.J.; Juarez, M.A.; Williams, S.; Wheeler, M.; Bean, K.; Allison, T.F.; Moore, H.D.; Andrews, P.W.; et al. Detecting Genetic Mosaicism in Cultures of Human Pluripotent Stem Cells. Stem Cell Rep. 2016, 7, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Cakir, B.; Xiang, Y.; Sullivan, G.J.; Park, I.-H. Synthetic Analyses of Single-Cell Transcriptomes from Multiple Brain Organoids and Fetal Brain. Cell Rep. 2020, 30, 1682–1689.e3. [Google Scholar] [CrossRef] [PubMed]
- Marotta, D.; Ijaz, L.; Barbar, L.; Nijsure, M.; Stein, J.; Pirjanian, N.; Kruglikov, I.; Clements, T.; Stoudemire, J.; Grisanti, P.; et al. Effects of Microgravity on Human iPSC-Derived Neural Organoids on the International Space Station. Stem Cells Transl. Med. 2024, 13, 1186–1197. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Genetic Alterations Introduced | Reporter | hPSCs Used | Ref. |
|---|---|---|---|---|
| Ogawa’s | Insertion of the HRasG12V transgene into the TP53 locus, leading to TP53 disruption. | tdTomato | H9-hESCs | [29] |
| Bian’s neoCOR | CDKN2A–/CDKN2B–/EGFROE/EGFRvIIIOE PTEN–/TP53–/NF1– CDKN2A–/EGFRvIIIOE/PTEN– | GFP | H9-hESCs | [30] |
| Kim’s | Introduction of the EGFRvIII genetic variant | N/A | H9-hESCs | [31] |
| Wang’s LEGO | PTEN−/−; TP53−/− PTEN−/−; TP53−/−; CDKN2A−/−; CDKN2B−/− PTEN−/−; TP53−/−; NF1−/− | GFP | iPSC lines | [32] |
| Taubenschmid-Stowers neoCOR | PTEN–/TP53–/NF1– | GFP | H9-hESCs | [57] |
| Singh’s | Expression of specific shRNAs using the Tet-On to knckdown PTEN, TP53, and NF1 knockdown | GFP | H1-hESCs H9-hESCs | [58] |
| Application | Model | Main Findings 1 | In Vivo Study | Ref. |
|---|---|---|---|---|
| Study Mechanisms of Disease | ||||
| Ciliogenesis and its impact on GSC invasiveness | 10-day old COs + mCherry-tagged patient-derived GSCs (genetically modified NEK2 conditional KD vs. control) | GSCs induced with cilia (NEK2 KD) failed to infiltrate COs. | Brain xenografts in ID mice | [89] |
| The role of BCAT1 in GBM proliferation | >25 days old COs + GFP-tagged LN229 established GBM cell line (genetically modified BCAT1 conditional KO vs. control) | BCAT1 is crucial for tumor growth. | Syngeneic mouse model (orthotopic implantation of GBM cells) | [90] |
| Test therapeutic approaches | ||||
| Study the therapeutic potential of ZIKV in GBM | 6-month-old COs + GFP-tagged GSCs from 2 patient-derived xenografts | ZIKV preferentially infected and presented oncolytic activity against GSCs that expressed high αvβ5 integrin levels. No signs of toxicity to non-cancerous brain cells. | Brain xenografts in ID mice | [91] |
| Test ZIKV oncolytic effect in CNS malignant tumors | >40 days old COs + GFP-tagged LN18 or U343-MG established GBM cell lines | ZIKV infection led to a significant reduction in tumor cell proportion in COs with GBM. No signs of toxicity to non-cancerous brain cells. | N/D | [92] |
| Target DHFR to eliminate GSCs | 30-day-old COs + 4 patient-derived GFP-tagged GSC lines (i.e., 12M, 25M, 50EF, 53M) | DHFR inhibition in GSCs by pretreatment with methotrexate reduced GSC growth and invasion in COs. No signs of toxicity to non-cancerous brain cells. | N/D | [93] |
| Test novel BET protein inhibitors for GBM treatment | >35 days old Cos 2 + GFP-labeled GBM22 patient-derived xenograft GBM cell line | BET inhibitor UM-002 reduced proliferation and invasion in COs. No signs of toxicity to non-cancerous brain cells. | Brain xenografts in ID mice | [94] |
| Test a dual Aurora and LIM kinase inhibitor for GBM treatment | >35 days old Cos 2 + GFP-labeled GBM22 patient-derived xenograft GBM cell line | Dual Aurora and LIM kinase inhibitor F114 reduced the total number of GFP+ cells and invasion in COs. Potential toxicity to non-cancerous brain cells. | N/D | [95] |
| Test Monensin and derivatives for GBM treatment | >51-day-old Cos 2 + RFP-tagged U87MG established GBM cell line | Compound 1 and monensin reduced tumor size and the expression of PARP. | N/D | [96] |
| Test 5-ALA-mediated photodynamic therapy (PDT) in GBM | 41-day-old COs + 2 GFP-tagged GSCs (i.e., GIC7 and PG88) | 5-ALA/PDT decreased proliferation and increased apoptosis in cancer cells. No signs of toxicity to non-cancerous brain cells. | N/D | [97] |
| Target autonomous rhythmic activity of GBM cells | >27-day-old Cos 2 + GFP-tagged primary human GSCs (i.e., S24 and BG5) | KCa3.1 inhibitors (TRAM-34 and senicapoc) reduced both global Ca2+ activity and tumor cell proliferation. No signs of toxicity to non-cancerous brain cells. | Brain xenografts in ID mice | [98] |
| Test Tumor Treating Fields (TTFields) in GBM | >27-day-old Cos 2 + GFP-tagged primary human GSCs (S24) | TTFields alter tumor cell proliferation and infiltration. Significant reduction in the size of younger (86 days) GBM organoids but not older (159 days). | No | [99] |
| Test a dual treatment with Rho-kinase inhibitor fasudil and soluble FasL in GBM | hESCs mixed with RFP-tagged U87MG cells and differentiated in COs (used after 75 days) | Decrease in the volume occupied by RFP-labeled U87 cells due to increased apoptosis. No signs of toxicity to non-cancerous brain cells. | Subcutaneous xenografts in ID mice | [100] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, C.D.; Calado, S.M.; Matos, A.; Esteves, F.; De Sousa-Coelho, A.L.; Campinho, M.A.; Fernandes, M.T. Advancing Glioblastoma Research with Innovative Brain Organoid-Based Models. Cells 2025, 14, 292. https://doi.org/10.3390/cells14040292
Correia CD, Calado SM, Matos A, Esteves F, De Sousa-Coelho AL, Campinho MA, Fernandes MT. Advancing Glioblastoma Research with Innovative Brain Organoid-Based Models. Cells. 2025; 14(4):292. https://doi.org/10.3390/cells14040292
Chicago/Turabian StyleCorreia, Cátia D., Sofia M. Calado, Alexandra Matos, Filipa Esteves, Ana Luísa De Sousa-Coelho, Marco A. Campinho, and Mónica T. Fernandes. 2025. "Advancing Glioblastoma Research with Innovative Brain Organoid-Based Models" Cells 14, no. 4: 292. https://doi.org/10.3390/cells14040292
APA StyleCorreia, C. D., Calado, S. M., Matos, A., Esteves, F., De Sousa-Coelho, A. L., Campinho, M. A., & Fernandes, M. T. (2025). Advancing Glioblastoma Research with Innovative Brain Organoid-Based Models. Cells, 14(4), 292. https://doi.org/10.3390/cells14040292