
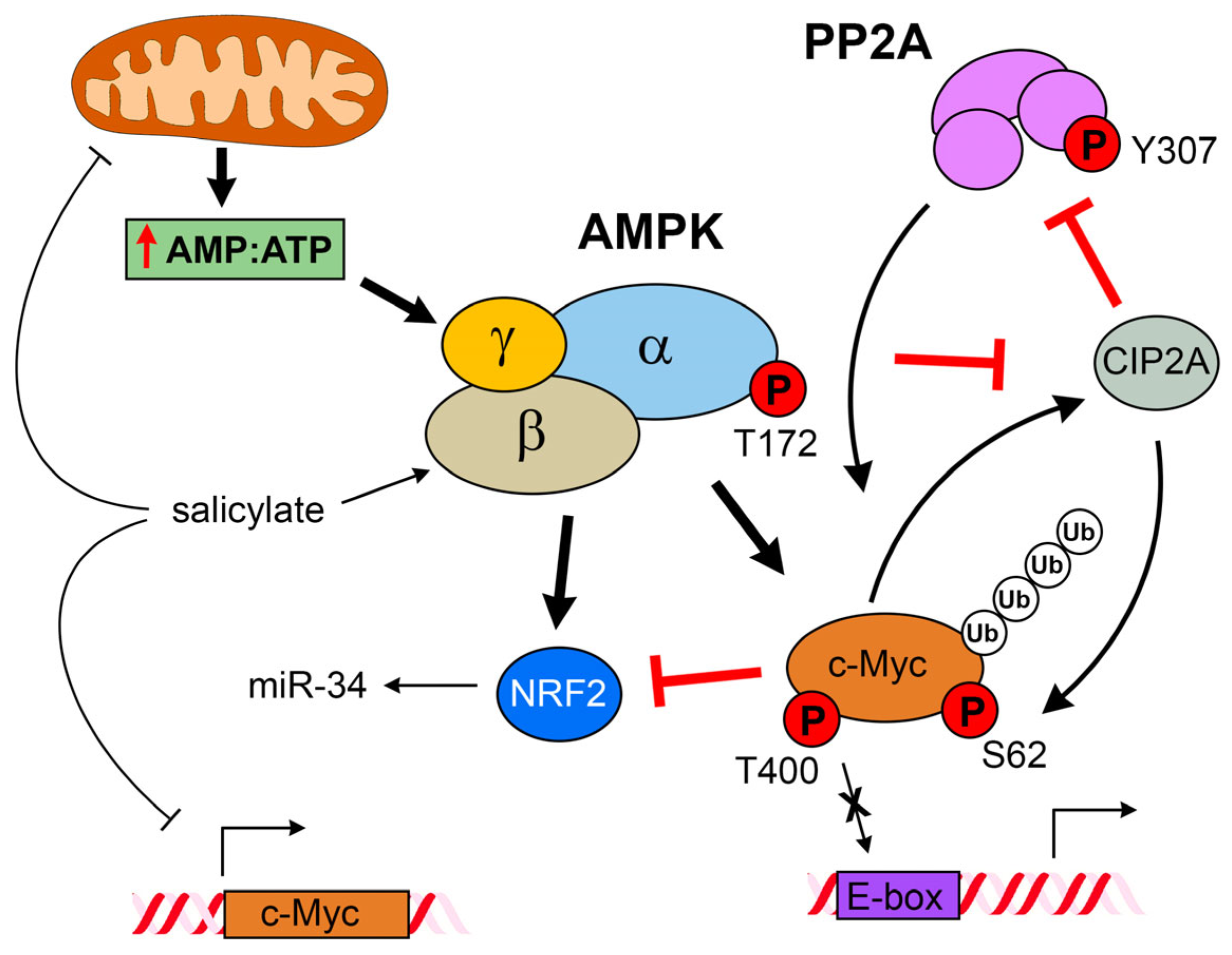
Salicylate-Elicited Activation of AMP-Activated Protein Kinase Directly Triggers Degradation of C-Myc in Colorectal Cancer Cells

 , , ,
, , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culturing
2.2. Animal Studies
2.3. Clinical Samples
2.4. Reagents and Antibodies
2.5. Western Blotting
2.6. Quantitative Real-Time PCR
2.7. In Vitro C-Myc Phosphorylation by AMPK for LC-MS/MS Analysis
2.8. In-Gel Trypsin Digestion of C-Myc and LC-MS/MS Analysis
2.9. Extracellular Flux Analysis
2.10. Lentiviral-Mediated AMPK and MYC Expression and Purification
2.11. Immunofluorescence Staining and Analysis
2.12. Immunohistochemistry (IHC) and Quantitative Assessment of IHC Staining
2.13. Gene Expression Analysis
2.14. Statistical Analysis
3. Results
3.1. Acetylsalicylic Acid Affects the Myc-Dependent Transcriptional Program
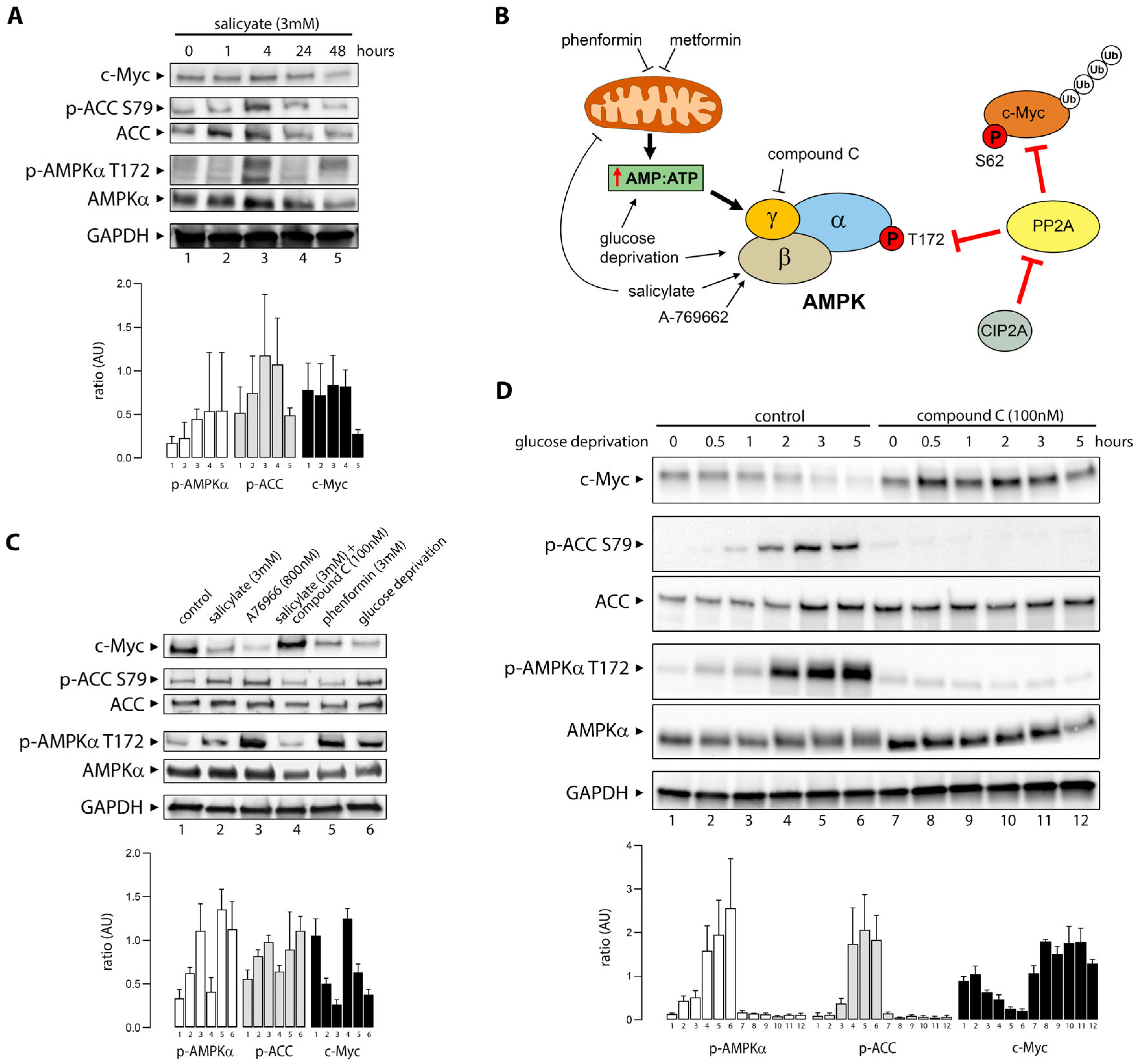
3.2. Salicylate Promotes Activation of AMPK and Degradation of C-Myc
3.3. Salicylate Reduces the Cell Viability of CRC Cells Through an AMPK-Independent Mechanism
3.4. Salicylate Affects the Respiratory Capacity of CRC Cells
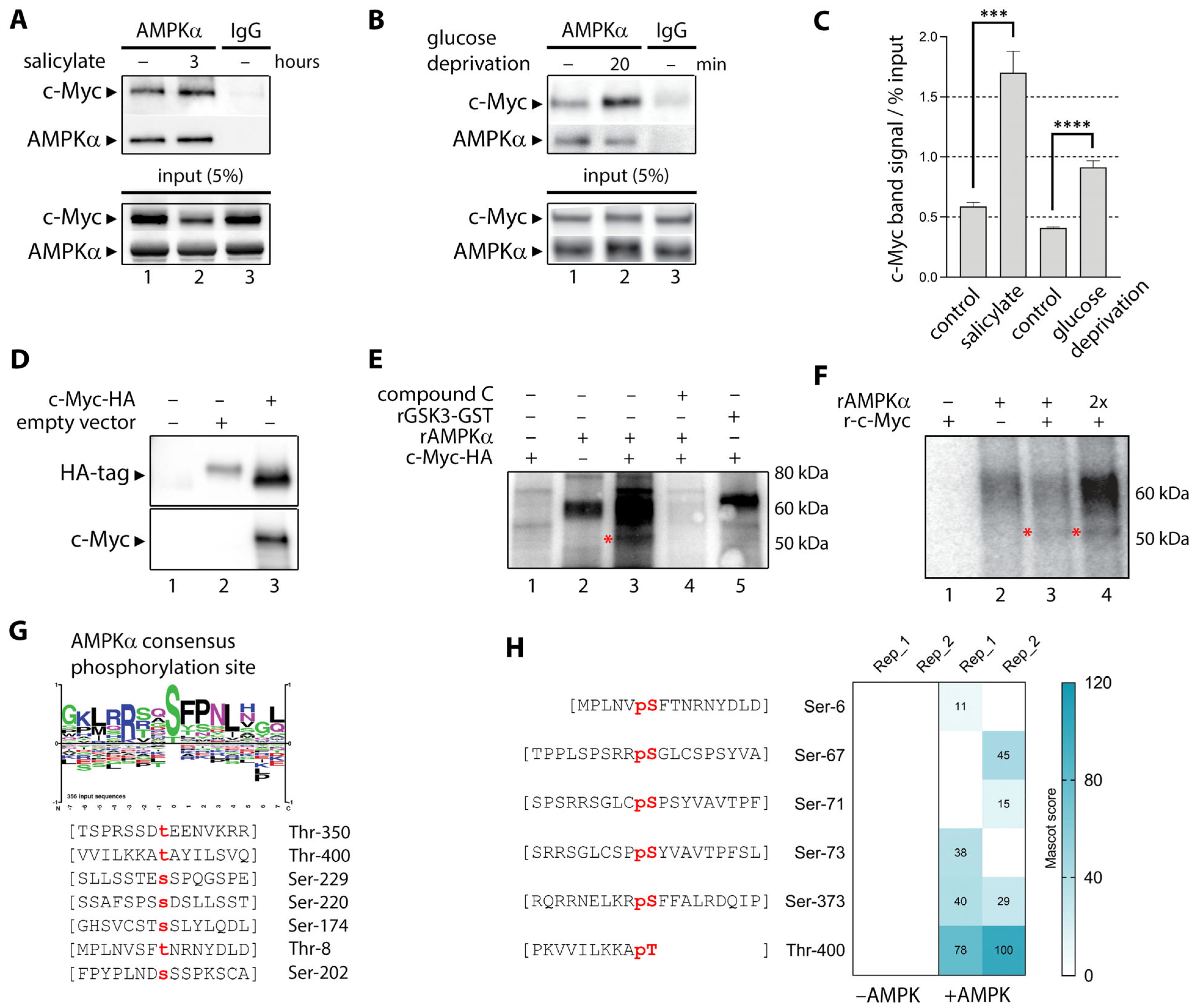
3.5. Activated AMPK Phosphorylates Ser-67, Ser-373, and Thr-400 Residues in C-Myc
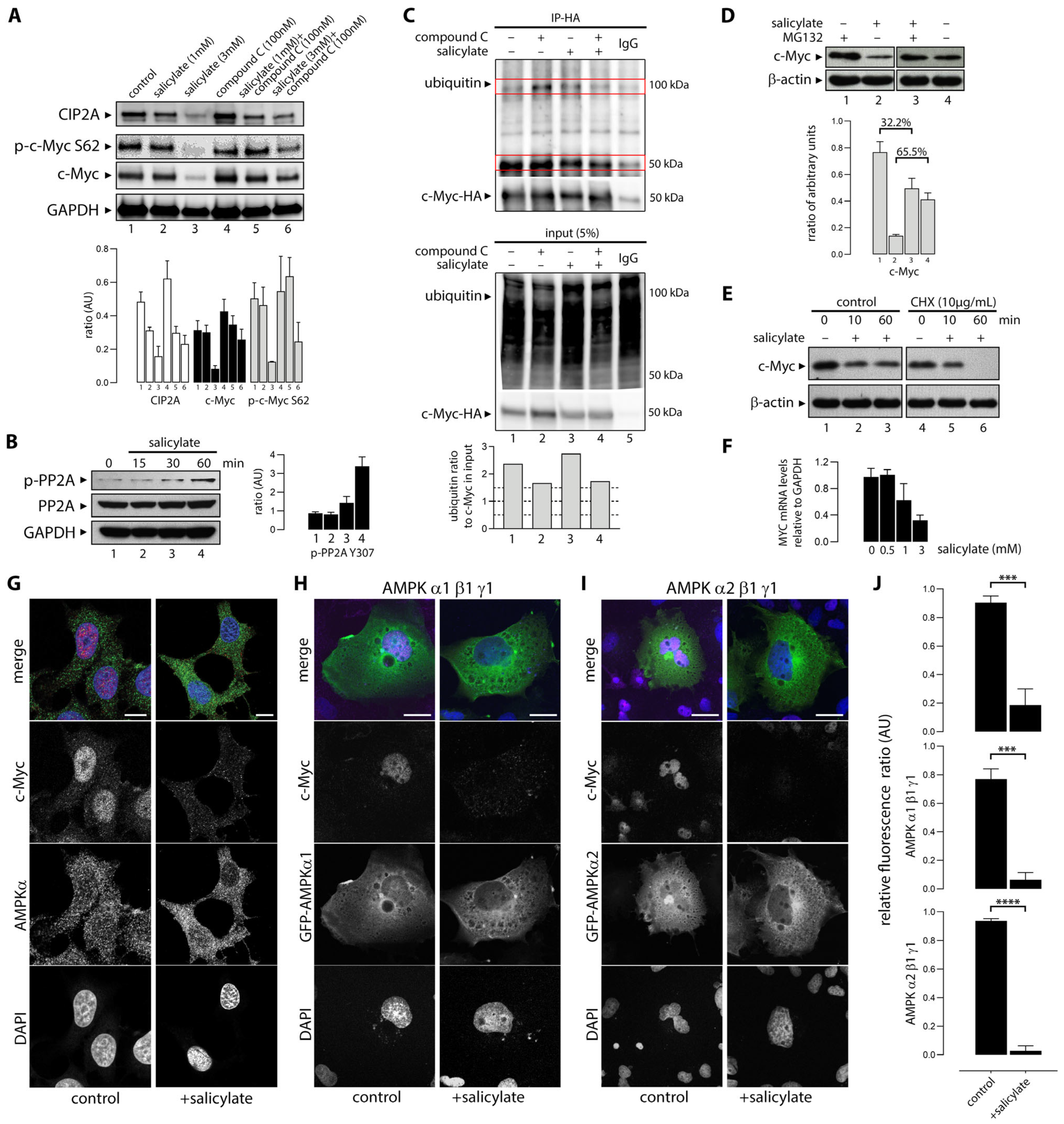
3.6. Salicylate Regulates the Expression of C-Myc Protein in Part Through the Proteosome Pathway
3.7. Sodium Salicylate-Induced AMPK Activation Depleted Expression of Nuclear C-Myc
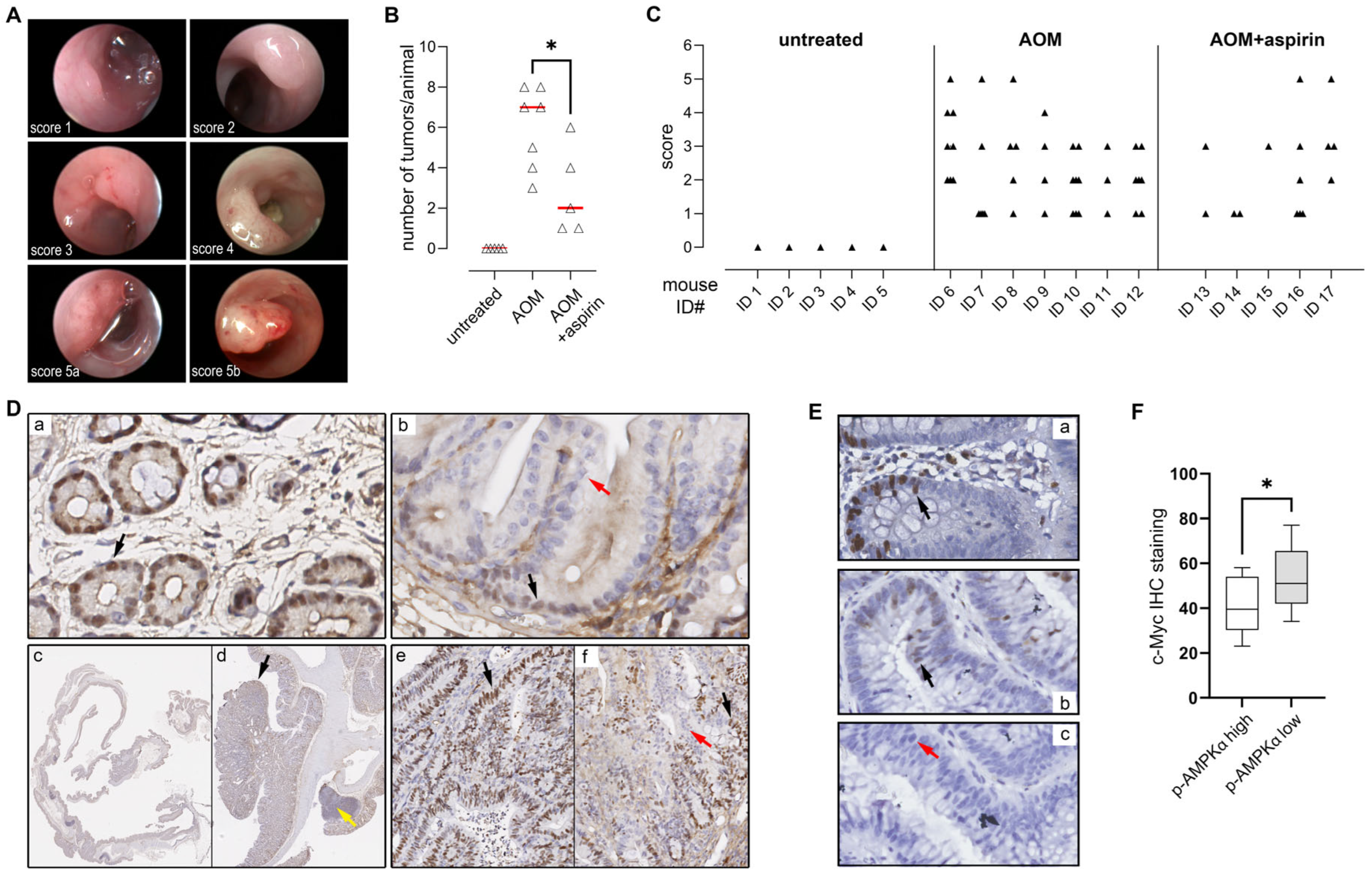
3.8. AMPK Activation Is Negatively Associated with C-Myc Expression in Colorectal Adenomas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Fedirko, V.; Tramacere, I.; Bagnardi, V.; Rota, M.; Scotti, L.; Islami, F.; Negri, E.; Straif, K.; Romieu, I.; La Vecchia, C.; et al. Alcohol drinking and colorectal cancer risk: An overall and dose-response meta-analysis of published studies. Ann. Oncol. 2011, 22, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Botteri, E.; Iodice, S.; Bagnardi, V.; Raimondi, S.; Lowenfels, A.B.; Maisonneuve, P. Smoking and colorectal cancer: A meta-analysis. JAMA 2008, 300, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.S.; Chen, T.Y.; Giovannucci, E. Cigarette smoking and colorectal cancer incidence and mortality: Systematic review and meta-analysis. Int. J. Cancer 2009, 124, 2406–2415. [Google Scholar] [CrossRef]
- Martinez, M.E.; Giovannucci, E.; Spiegelman, D.; Hunter, D.J.; Willett, W.C.; Colditz, G.A. Leisure-time physical activity, body size, and colon cancer in women. Nurses’ Health Study Research Group. J. Natl. Cancer Inst. 1997, 89, 948–955. [Google Scholar] [CrossRef]
- Lieberman, D.A.; Prindiville, S.; Weiss, D.G.; Willett, W.; Group, V.A.C.S. Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA 2003, 290, 2959–2967. [Google Scholar] [CrossRef]
- Cross, A.J.; Leitzmann, M.F.; Gail, M.H.; Hollenbeck, A.R.; Schatzkin, A.; Sinha, R. A prospective study of red and processed meat intake in relation to cancer risk. PLoS Med. 2007, 4, e325. [Google Scholar] [CrossRef]
- Bray, F.; Moller, B. Predicting the future burden of cancer. Nat. Rev. Cancer 2006, 6, 63–74. [Google Scholar] [CrossRef]
- Brenner, H.; Hoffmeister, M.; Arndt, V.; Stegmaier, C.; Altenhofen, L.; Haug, U. Protection from right- and left-sided colorectal neoplasms after colonoscopy: Population-based study. J. Natl. Cancer Inst. 2010, 102, 89–95. [Google Scholar] [CrossRef]
- Osborne, M.; Boyle, P.; Lipkin, M. Cancer prevention. Lancet 1997, 349 (Suppl. 2), S27–S30. [Google Scholar] [CrossRef]
- Greenwald, P.; Kelloff, G.J. The role of chemoprevention in cancer control. IARC Sci. Publ. 1996, 13–22. [Google Scholar]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Flossmann, E.; Rothwell, P.M. Effect of aspirin on long-term risk of colorectal cancer: Consistent evidence from randomised and observational studies. Lancet 2007, 369, 1603–1613. [Google Scholar] [CrossRef]
- Drew, D.A.; Chan, A.T. Aspirin in the Prevention of Colorectal Neoplasia. Annu. Rev. Med. 2021, 72, 415–430. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Roth, G.J.; Stanford, N.; Majerus, P.W. Acetylation of prostaglandin synthase by aspirin. Proc. Natl. Acad. Sci. USA 1975, 72, 3073–3076. [Google Scholar] [CrossRef]
- Stark, L.A.; Din, F.V.; Zwacka, R.M.; Dunlop, M.G. Aspirin-induced activation of the NF-kappaB signaling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [CrossRef]
- Din, F.V.; Dunlop, M.G.; Stark, L.A. Evidence for colorectal cancer cell specificity of aspirin effects on NF kappa B signalling and apoptosis. Br. J. Cancer 2004, 91, 381–388. [Google Scholar] [CrossRef]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef]
- de Souza Almeida Matos, A.L.; Oakhill, J.S.; Moreira, J.; Loh, K.; Galic, S.; Scott, J.W. Allosteric regulation of AMP-activated protein kinase by adenylate nucleotides and small-molecule drugs. Biochem. Soc. Trans. 2019, 47, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem. Soc. Trans. 2011, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef]
- Baba, Y.; Nosho, K.; Shima, K.; Meyerhardt, J.A.; Chan, A.T.; Engelman, J.A.; Cantley, L.C.; Loda, M.; Giovannucci, E.; Fuchs, C.S.; et al. Prognostic significance of AMP-activated protein kinase expression and modifying effect of MAPK3/1 in colorectal cancer. Br. J. Cancer 2010, 103, 1025–1033. [Google Scholar] [CrossRef]
- Petti, C.; Vegetti, C.; Molla, A.; Bersani, I.; Cleris, L.; Mustard, K.J.; Formelli, F.; Hardie, G.D.; Sensi, M.; Anichini, A. AMPK activators inhibit the proliferation of human melanomas bearing the activated MAPK pathway. Melanoma Res. 2012, 22, 341–350. [Google Scholar] [CrossRef]
- Luo, Z.; Saha, A.K.; Xiang, X.; Ruderman, N.B. AMPK, the metabolic syndrome and cancer. Trends Pharmacol. Sci. 2005, 26, 69–76. [Google Scholar] [CrossRef]
- Puustinen, P.; Keldsbo, A.; Corcelle-Termeau, E.; Ngoei, K.; Sonder, S.L.; Farkas, T.; Kaae Andersen, K.; Oakhill, J.S.; Jaattela, M. DNA-dependent protein kinase regulates lysosomal AMP-dependent protein kinase activation and autophagy. Autophagy 2020, 16, 1871–1888. [Google Scholar] [CrossRef]
- Dachineni, R.; Kumar, D.R.; Callegari, E.; Kesharwani, S.S.; Sankaranarayanan, R.; Seefeldt, T.; Tummala, H.; Bhat, G.J. Salicylic acid metabolites and derivatives inhibit CDK activity: Novel insights into aspirin’s chemopreventive effects against colorectal cancer. Int. J. Oncol. 2017, 51, 1661–1673. [Google Scholar] [CrossRef]
- Becker, C.; Fantini, M.C.; Neurath, M.F. High resolution colonoscopy in live mice. Nat. Protoc. 2006, 1, 2900–2904. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Wisidagama, D.R.; Setoguchi, K.; Hong, J.S.; Van Horn, C.M.; Imam, S.S.; Vergnes, L.; Malone, C.S.; Koehler, C.M.; et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012, 7, 1068–1085. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc deletion rescues Apc deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Ai, G.; Dachineni, R.; Muley, P.; Tummala, H.; Bhat, G.J. Aspirin and salicylic acid decrease c-Myc expression in cancer cells: A potential role in chemoprevention. Tumor Biol. 2016, 37, 1727–1738. [Google Scholar] [CrossRef]
- Mitrugno, A.; Sylman, J.L.; Ngo, A.T.; Pang, J.; Sears, R.C.; Williams, C.D.; McCarty, O.J. Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: Implications for the oncoprotein c-MYC. Am. J. Physiol. Cell Physiol. 2017, 312, C176–C189. [Google Scholar] [CrossRef]
- Bettess, M.D.; Dubois, N.; Murphy, M.J.; Dubey, C.; Roger, C.; Robine, S.; Trumpp, A. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol. Cell. Biol. 2005, 25, 7868–7878. [Google Scholar] [CrossRef]
- Haikala, H.M.; Anttila, J.M.; Klefstrom, J. MYC and AMPK-Save Energy or Die! Front. Cell Dev. Biol. 2017, 5, 38. [Google Scholar] [CrossRef]
- Brody, T.M. Action of sodium salicylate and related compounds on tissue metabolism in vitro. J. Pharmacol. Exp. Ther. 1956, 117, 39–51. [Google Scholar] [CrossRef]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mayer, J.A.; Mazumdar, A.; Fertuck, K.; Kim, H.; Brown, M.; Brown, P.H. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol. Endocrinol. 2011, 25, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Dikshit, P.; Chatterjee, M.; Goswami, A.; Mishra, A.; Jana, N.R. Aspirin induces apoptosis through the inhibition of proteasome function. J. Biol. Chem. 2006, 281, 29228–29235. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Ming, L.; Yan, Y.; Zhang, Y.; Xue, H. Beclin 1 acetylation impairs the anticancer effect of aspirin in colorectal cancer cells. Oncotarget 2017, 8, 74781–74790. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, L.; Misirkic, M.; Janjetovic, K.; Vilimanovich, U.; Sudar, E.; Isenovic, E.; Prica, M.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Bumbasirevic, V.; et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011, 7, 40–50. [Google Scholar] [CrossRef]
- Wu, Y.; Yan, B.; Xu, W.; Guo, L.; Wang, Z.; Li, G.; Hou, N.; Zhang, J.; Ling, R. Compound C enhances the anticancer effect of aspirin in HER-2-positive breast cancer by regulating lipid metabolism in an AMPK-independent pathway. Int. J. Biol. Sci. 2020, 16, 583–597. [Google Scholar] [CrossRef]
- Norman, C.; Howell, K.A.; Millar, A.H.; Whelan, J.M.; Day, D.A. Salicylic acid is an uncoupler and inhibitor of mitochondrial electron transport. Plant Physiol. 2004, 134, 492–501. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Vervoorts, J.; Luscher-Firzlaff, J.; Luscher, B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006, 281, 34725–34729. [Google Scholar] [CrossRef]
- Schaffer, B.E.; Levin, R.S.; Hertz, N.T.; Maures, T.J.; Schoof, M.L.; Hollstein, P.E.; Benayoun, B.A.; Banko, M.R.; Shaw, R.J.; Shokat, K.M.; et al. Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction. Cell Metab. 2015, 22, 907–921. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Lewis, B.A.; Levens, D. MYC: A complex problem. Trends Cell Biol. 2023, 33, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.W.; Buchan, H.L.; Civin, C.I.; Kastan, M.B. Altered cytoplasmic/nuclear distribution of the c-myc protein in differentiating ML-1 human myeloid leukemia cells. Cell Growth Differ. 1993, 4, 349–357. [Google Scholar] [PubMed]
- Sanchez-Arevalo Lobo, V.J.; Doni, M.; Verrecchia, A.; Sanulli, S.; Faga, G.; Piontini, A.; Bianchi, M.; Conacci-Sorrell, M.; Mazzarol, G.; Peg, V.; et al. Dual regulation of Myc by Abl. Oncogene 2013, 32, 5261–5271. [Google Scholar] [CrossRef]
- Stapleton, D.; Mitchelhill, K.I.; Gao, G.; Widmer, J.; Michell, B.J.; Teh, T.; House, C.M.; Fernandez, C.S.; Cox, T.; Witters, L.A.; et al. Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem. 1996, 271, 611–614. [Google Scholar] [CrossRef]
- Qiu, S.; Liu, T.; Piao, C.; Wang, Y.; Wang, K.; Zhou, Y.; Cai, L.; Zheng, S.; Lan, F.; Du, J. AMPKalpha2 knockout enhances tumour inflammation through exacerbated liver injury and energy deprivation-associated AMPKalpha1 activation. J. Cell. Mol. Med. 2019, 23, 1687–1697. [Google Scholar] [CrossRef]
- Baron, J.A.; Cole, B.F.; Sandler, R.S.; Haile, R.W.; Ahnen, D.; Bresalier, R.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.; Burke, C.A.; et al. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med. 2003, 348, 891–899. [Google Scholar] [CrossRef]
- Cole, B.F.; Logan, R.F.; Halabi, S.; Benamouzig, R.; Sandler, R.S.; Grainge, M.J.; Chaussade, S.; Baron, J.A. Aspirin for the chemoprevention of colorectal adenomas: Meta-analysis of the randomized trials. J. Natl. Cancer Inst. 2009, 101, 256–266. [Google Scholar] [CrossRef]
- Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013, 191, 39–65. [Google Scholar] [CrossRef]
- Shimura, T.; Toden, S.; Komarova, N.L.; Boland, C.; Wodarz, D.; Goel, A. A comprehensive in vivo and mathematic modeling-based kinetic characterization for aspirin-induced chemoprevention in colorectal cancer. Carcinogenesis 2020, 41, 751–760. [Google Scholar] [CrossRef]
- Akinyeke, T.; Matsumura, S.; Wang, X.; Wu, Y.; Schalfer, E.D.; Saxena, A.; Yan, W.; Logan, S.K.; Li, X. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis 2013, 34, 2823–2832. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Yang, C.S.; DiPaola, R.S.; Tan, X.L. Repurposing of metformin and aspirin by targeting AMPK-mTOR and inflammation for pancreatic cancer prevention and treatment. Cancer Prev. Res. 2014, 7, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Macek, P.; Cliff, M.J.; Embrey, K.J.; Holdgate, G.A.; Nissink, J.W.M.; Panova, S.; Waltho, J.P.; Davies, R.A. Myc phosphorylation in its basic helix-loop-helix region destabilizes transient alpha-helical structures, disrupting Max and DNA binding. J. Biol. Chem. 2018, 293, 9301–9310. [Google Scholar] [CrossRef] [PubMed]
- von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef]
- Liu, C.; Rokavec, M.; Huang, Z.; Hermeking, H. Salicylate induces AMPK and inhibits c-MYC to activate a NRF2/ARE/miR-34a/b/c cascade resulting in suppression of colorectal cancer metastasis. Cell Death Dis. 2023, 14, 707. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef]
- Nieminen, A.I.; Eskelinen, V.M.; Haikala, H.M.; Tervonen, T.A.; Yan, Y.; Partanen, J.I.; Klefstrom, J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, E1839–E1848. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef]
- Pathi, S.; Jutooru, I.; Chadalapaka, G.; Nair, V.; Lee, S.O.; Safe, S. Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (Sp) transcription factors. PLoS ONE 2012, 7, e48208. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, S.; Hardie, D.G. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim. Biophys. Acta 2010, 1804, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Rivas-San Vicente, M.; Plasencia, J. Salicylic acid beyond defence: Its role in plant growth and development. J. Exp. Bot. 2011, 62, 3321–3338. [Google Scholar] [CrossRef]
- Tan, S.; Abas, M.; Verstraeten, I.; Glanc, M.; Molnar, G.; Hajny, J.; Lasak, P.; Petrik, I.; Russinova, E.; Petrasek, J.; et al. Salicylic Acid Targets Protein Phosphatase 2A to Attenuate Growth in Plants. Curr. Biol. 2020, 30, 381–395.e388. [Google Scholar] [CrossRef]
- Nomoto, M.; Skelly, M.J.; Itaya, T.; Mori, T.; Suzuki, T.; Matsushita, T.; Tokizawa, M.; Kuwata, K.; Mori, H.; Yamamoto, Y.Y.; et al. Suppression of MYC transcription activators by the immune cofactor NPR1 fine-tunes plant immune responses. Cell Rep. 2021, 37, 110125. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matos, A.L.S.A.; Ovens, A.J.; Jakobsen, E.; Iglesias-Gato, D.; Bech, J.M.; Friis, S.; Bak, L.K.; Madsen, G.I.; Oakhill, J.S.; Puustinen, P.; et al. Salicylate-Elicited Activation of AMP-Activated Protein Kinase Directly Triggers Degradation of C-Myc in Colorectal Cancer Cells. Cells 2025, 14, 294. https://doi.org/10.3390/cells14040294
Matos ALSA, Ovens AJ, Jakobsen E, Iglesias-Gato D, Bech JM, Friis S, Bak LK, Madsen GI, Oakhill JS, Puustinen P, et al. Salicylate-Elicited Activation of AMP-Activated Protein Kinase Directly Triggers Degradation of C-Myc in Colorectal Cancer Cells. Cells. 2025; 14(4):294. https://doi.org/10.3390/cells14040294
Chicago/Turabian StyleMatos, Ana Laura S. A., Ashley J. Ovens, Emil Jakobsen, Diego Iglesias-Gato, Jacob M. Bech, Stine Friis, Lasse Kristoffer Bak, Gunvor I. Madsen, Jonathan S. Oakhill, Pietri Puustinen, and et al. 2025. "Salicylate-Elicited Activation of AMP-Activated Protein Kinase Directly Triggers Degradation of C-Myc in Colorectal Cancer Cells" Cells 14, no. 4: 294. https://doi.org/10.3390/cells14040294
APA StyleMatos, A. L. S. A., Ovens, A. J., Jakobsen, E., Iglesias-Gato, D., Bech, J. M., Friis, S., Bak, L. K., Madsen, G. I., Oakhill, J. S., Puustinen, P., & Moreira, J. M. A. (2025). Salicylate-Elicited Activation of AMP-Activated Protein Kinase Directly Triggers Degradation of C-Myc in Colorectal Cancer Cells. Cells, 14(4), 294. https://doi.org/10.3390/cells14040294