
Role of PDE4 Family in Cardiomyocyte Physiology and Heart Failure


 , , and
, , and
Abstract
1. Introduction
1.1. PDE Superfamily
1.2. PDE4 Structure and Function
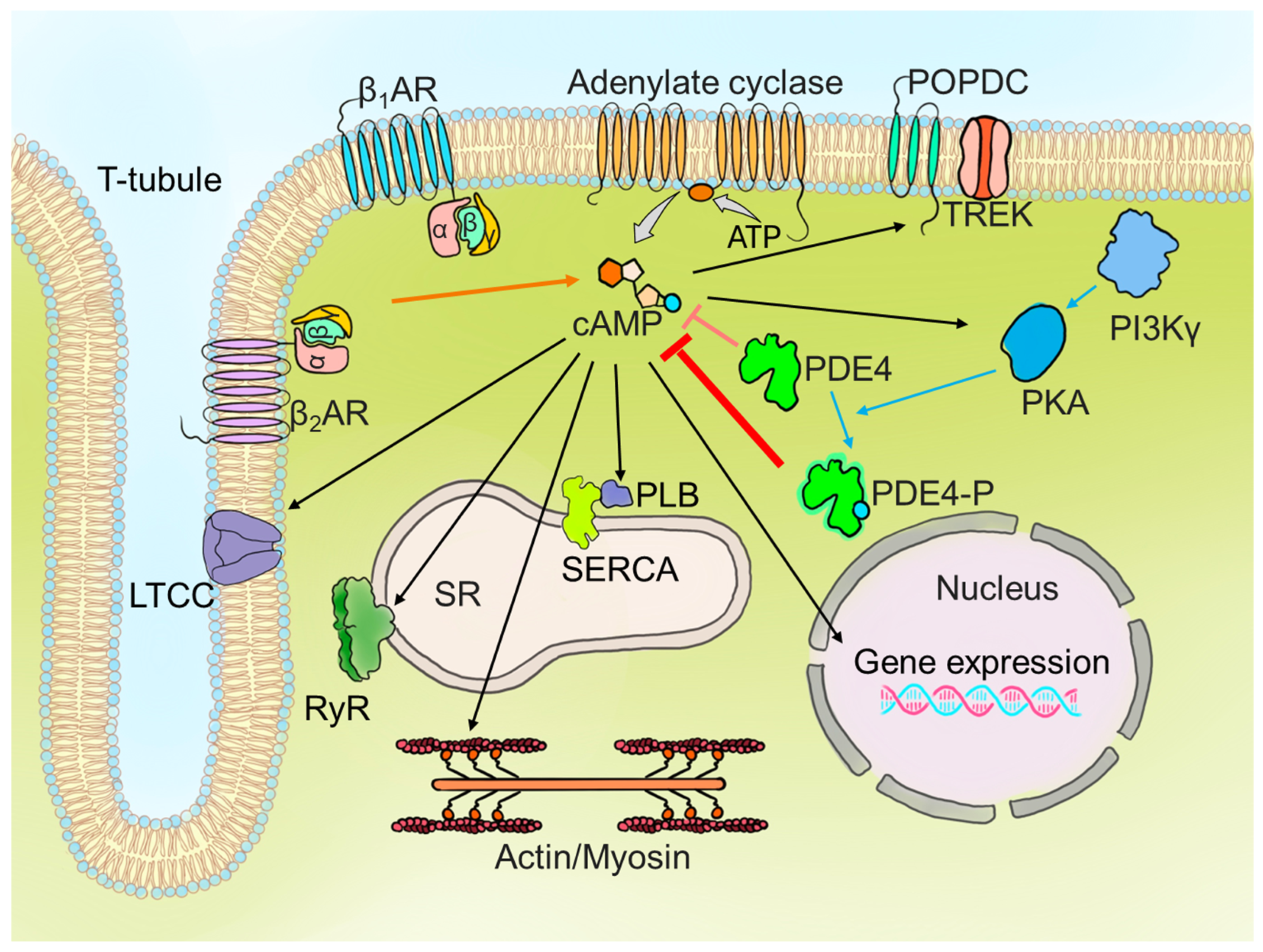
2. PDE4 Expression and Function in the Heart: From Species Variability to Subcellular Dynamics
2.1. PDE4 Expression and Function in Cardiac Tissue Across Different Species
2.2. Regional Differences in PDE4 Activity
2.3. PDE4 Isoform-Specific Functions and Subcellular Localisation
2.3.1. Plasma Membrane: Modulation of βAR Signalling
2.3.2. Calcium Current Regulation by PDE4
2.3.3. Sarcoplasmic Reticulum: Local Regulation of cAMP Microdomains
2.3.4. Nuclear Envelope
3. A-Kinase-Anchoring Proteins in Cardiomyocytes: Regulators of cAMP Compartmentation and Signalling
3.1. Overview of AKAPs: Precision in PKA Signalling
3.2. Non-Conventional AKAPs in Cardiac Signalling
3.3. AKAP-PDE4-PKA Complexes: Masters of cAMP Regulation
4. PDE4 and AKAPs in Heart Failure: Implications for cAMP Signalling and Cardiac Remodelling
4.1. PDE4 and Heart Failure
4.2. AKAPs in Heart Failure: Mechanistic Roles and Pathological Impact
5. Therapeutical Insights and Future Perspectives
6. Conclusions: Can PDE4 Still Be Considered Only a Minor Helper in the Heart?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| βAR | Beta-Adrenergic Receptor |
| β1AR | Beta-1-Adrenergic Receptor |
| β2AR | Beta-2-Adrenergic Receptor |
| AKAP | A-Kinase Anchoring Protein |
| cAMP | Cyclic Adenosine Monophosphate |
| cGMP | Cyclic Guanosine Monophosphate |
| CaMKII | Calcium/Calmodulin-Dependent Protein Kinase II |
| CREB | cAMP Response Element-Binding Protein |
| ERK2 | Extracellular Signal-Regulated Kinase 2 |
| GPCR | G-Protein Coupled Receptor |
| HFpEF | Heart Failure with Preserved Ejection Fraction |
| ICa,L | L-Type Calcium Current |
| LTCC | L-Type Calcium Channel |
| LV | Left Ventricle |
| mAKAP | Muscle A-Kinase Anchoring Protein |
| PDE | Phosphodiesterase |
| PDE4A, PDE4B, PDE4C, PDE4D | Isoforms of Phosphodiesterase 4 |
| PKA | Protein Kinase A |
| PKA-RI/RII | Protein Kinase A Regulatory Subunit I/II |
| PKD | Protein Kinase D |
| PLN | Phospholamban |
| POPDC1 | Popeye Domain Containing 1 |
| RyR2 | Ryanodine Receptor 2 |
| SERCA2a | Sarco/Endoplasmic Reticulum Calcium-ATPase 2a |
| SR | Sarcoplasmic Reticulum |
| UCR | Upstream Conserved Region |
| VASP | Vasodilator-stimulated phosphoprotein |
References
- Puertas-Umbert, L.; Alonso, J.; Hove-Madsen, L.; Martínez-González, J.; Rodríguez, C. PDE4 Phosphodiesterases in Cardiovascular Diseases: Key Pathophysiological Players and Potential Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 17017. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Wang, Y.; Yan, C.; Xiang, Y.K. Phosphodiesterase in Heart and Vessels: From Physiology to Diseases. Physiol. Rev. 2024, 104, 765–834. [Google Scholar] [CrossRef]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic Targeting of 3′,5′-Cyclic Nucleotide Phosphodiesterases: Inhibition and Beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Xiang, Y.K.; Zaccolo, M. Whole-Cell cAMP and PKA Activity Are Epiphenomena, Nanodomain Signaling Matters. Physiology 2019, 34, 240–249. [Google Scholar] [CrossRef]
- Omori, K.; Kotera, J. Overview of PDEs and Their Regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP Signaling Cross-Talk: Role of Phosphodiesterases and Implications for Cardiac Pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs Create Local Domains of cAMP Signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Xuong, N.-H.; Taylor, S.S. Crystal Structure of a Complex Between the Catalytic and Regulatory (RIα) Subunits of PKA. Science 2005, 307, 690–696. [Google Scholar] [CrossRef]
- Viste, K.; Kopperud, R.K.; Christensen, A.E.; Døskeland, S.O. Substrate Enhances the Sensitivity of Type I Protein Kinase A to cAMP. J. Biol. Chem. 2005, 280, 13279–13284. [Google Scholar] [CrossRef]
- Cheadle, C.; Nesterova, M.; Watkins, T.; Barnes, K.C.; Hall, J.C.; Rosen, A.; Becker, K.G.; Cho-Chung, Y.S. Regulatory Subunits of PKA Define an Axis of Cellular Proliferation/Differentiation in Ovarian Cancer Cells. BMC Med. Genom. 2008, 1, 43. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein Kinase A Type I and Type II Define Distinct Intracellular Signaling Compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef]
- Burgers, P.P.; Bruystens, J.; Burnley, R.J.; Nikolaev, V.O.; Keshwani, M.; Wu, J.; Janssen, B.J.C.; Taylor, S.S.; Heck, A.J.R.; Scholten, A. Structure of Sm AKAP and Its Regulation by PKA -mediated Phosphorylation. FEBS J. 2016, 283, 2132–2148. [Google Scholar] [CrossRef]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. cGMP Signals Modulate cAMP Levels in a Compartment-Specific Manner to Regulate Catecholamine-Dependent Signaling in Cardiac Myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef]
- Manning, C.D.; Burman, M.; Christensen, S.B.; Cieslinski, L.B.; Essayan, D.M.; Grous, M.; Torphy, T.J.; Barnette, M.S. Suppression of Human Inflammatory Cell Function by Subtype-selective PDE4 Inhibitors Correlates with Inhibition of PDE4A and PDE4B. Br. J. Pharmacol. 1999, 128, 1393–1398. [Google Scholar] [CrossRef]
- Wang, P.; Myers, J.G.; Wu, P.; Cheewatrakoolpong, B.; Egan, R.W.; Billah, M.M. Expression, Purification, and Characterization of Human cAMP-Specific Phosphodiesterase (PDE4) Subtypes A, B, C, and D. Biochem. Biophys. Res. Commun. 1997, 234, 320–324. [Google Scholar] [CrossRef]
- Han, P.; Zhu, X.; Michaeli, T. Alternative Splicing of the High Affinity cAMP-Specific Phosphodiesterase (PDE7A) mRNA in Human Skeletal Muscle and Heart. J. Biol. Chem. 1997, 272, 16152–16157. [Google Scholar] [CrossRef]
- Hetman, J.M.; Soderling, S.H.; Glavas, N.A.; Beavo, J.A. Cloning and Characterization of PDE7B, a cAMP-Specific Phosphodiesterase. Proc. Natl. Acad. Sci. USA 2000, 97, 472–476. [Google Scholar] [CrossRef]
- Fisher, D.A.; Smith, J.F.; Pillar, J.S.; St. Denis, S.H.; Cheng, J.B. Isolation and Characterization of PDE8A, a Novel Human cAMP-Specific Phosphodiesterase. Biochem. Biophys. Res. Commun. 1998, 246, 570–577. [Google Scholar] [CrossRef]
- Soderling, S.H.; Bayuga, S.J.; Beavo, J.A. Cloning and Characterization of a cAMP-Specific Cyclic Nucleotide Phosphodiesterase. Proc. Natl. Acad. Sci. USA 1998, 95, 8991–8996. [Google Scholar] [CrossRef] [PubMed]
- Gamanuma, M.; Yuasa, K.; Sasaki, T.; Sakurai, N.; Kotera, J.; Omori, K. Comparison of Enzymatic Characterization and Gene Organization of Cyclic Nucleotide Phosphodiesterase 8 Family in Humans. Cell. Signal. 2003, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Loughney, K.; Hill, T.R.; Florio, V.A.; Uher, L.; Rosman, G.J.; Wolda, S.L.; Jones, B.A.; Howard, M.L.; McAllister-Lucas, L.M.; Sonnenburg, W.K.; et al. Isolation and Characterization of cDNAs Encoding PDE5A, a Human cGMP-Binding, cGMP-Specific 3′,5′-Cyclic Nucleotide Phosphodiesterase. Gene 1998, 216, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Lau, A.; Tu, R.; Lue, T.F. Expression of Three Isoforms of cGMP-Binding cGMP-Specific Phosphodiesterase (PDE5) in Human Penile Cavernosum. Biochem. Biophys. Res. Commun. 2000, 268, 628–635. [Google Scholar] [CrossRef]
- Wang, P.; Wu, P.; Myers, J.G.; Stamford, A.; Egan, R.W.; Billah, M.M. Characterization of Human, Dog and Rabbit Corpus Cavernosum Type 5 Phosphodiesterases. Life Sci. 2001, 68, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, P.G.; Beavo, J.A. Characterization of a Bovine Cone Photoreceptor Phosphodiesterase Purified by Cyclic GMP-Sepharose Chromatography. J. Biol. Chem. 1988, 263, 8133–8141. [Google Scholar]
- Muradov, H.; Boyd, K.K.; Artemyev, N.O. Rod Phosphodiesterase-6 PDE6A and PDE6B Subunits Are Enzymatically Equivalent. J. Biol. Chem. 2010, 285, 39828–39834. [Google Scholar] [CrossRef]
- Fisher, D.A.; Smith, J.F.; Pillar, J.S.; Denis, S.H.S.; Cheng, J.B. Isolation and Characterization of PDE9A, a Novel Human cGMP-Specific Phosphodiesterase. J. Biol. Chem. 1998, 273, 15559–15564. [Google Scholar] [CrossRef] [PubMed]
- Soderling, S.H.; Bayuga, S.J.; Beavo, J.A. Identification and Characterization of a Novel Family of Cyclic Nucleotide Phosphodiesterases. J. Biol. Chem. 1998, 273, 15553–15558. [Google Scholar] [CrossRef]
- Wang, P.; Wu, P.; Egan, R.W.; Billah, M.M. Identification and Characterization of a New Human Type 9 cGMP-Specific Phosphodiesterase Splice Variant (PDE9A5). Gene 2003, 314, 15–27. [Google Scholar] [CrossRef]
- Snyder, P.B.; Florio, V.A.; Ferguson, K.; Loughney, K. Isolation, Expression and Analysis of Splice Variants of a Human Ca2+/Calmodulin-Stimulated Phosphodiesterase (PDE1A). Cell. Signal. 1999, 11, 535–544. [Google Scholar] [CrossRef]
- Sharma, R.K.; Wang, J.H. Calmodulin and Ca2+-Dependent Phosphorylation and Dephosphorylation of 63-kDa Subunit-Containing Bovine Brain Calmodulin-Stimulated Cyclic Nucleotide Phosphodiesterase Isozyme. J. Biol. Chem. 1986, 261, 1322–1328. [Google Scholar] [CrossRef]
- Kincaid, R.L.; Stith-Coleman, I.E.; Vaughan, M. Proteolytic Activation of Calmodulin-Dependent Cyclic Nucleotide Phosphodiesterase. J. Biol. Chem. 1985, 260, 9009–9015. [Google Scholar] [PubMed]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and Characterization of Human cDNAs Encoding a cGMP-Stimulated 3′,5′-Cyclic Nucleotide Phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Reifsnyder, D.H.; Gallis, B.; Cadd, G.G.; Beavo, J.A. Isolation and Characterization of Bovine Cardiac Muscle cGMP-Inhibited Phosphodiesterase: A Receptor for New Cardiotonic Drugs. Mol. Pharmacol. 1986, 29, 506–514. [Google Scholar] [CrossRef]
- Grant, P.G.; Colman, R.W. Purification and Characterization of a Human Platelet Cyclic Nucleotide Phosphodiesterase. Biochemistry 1984, 23, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Meacci, E.; Taira, M.; Moos, M.; Smith, C.J.; Movsesian, M.A.; Degerman, E.; Belfrage, P.; Manganiello, V. Molecular Cloning and Expression of Human Myocardial cGMP-Inhibited cAMP Phosphodiesterase. Proc. Natl. Acad. Sci. USA 1992, 89, 3721–3725. [Google Scholar] [CrossRef]
- Degerman, E.; Belfrage, P.; Newman, A.H.; Rice, K.C.; Manganiello, V.C. Purification of the Putative Hormone-Sensitive Cyclic AMP Phosphodiesterase from Rat Adipose Tissue Using a Derivative of Cilostamide as a Novel Affinity Ligand. J. Biol. Chem. 1987, 262, 5797–5807. [Google Scholar] [CrossRef]
- Fujishige, K.; Kotera, J.; Michibata, H.; Yuasa, K.; Takebayashi, S.; Okumura, K.; Omori, K. Cloning and Characterization of a Novel Human Phosphodiesterase That Hydrolyzes Both cAMP and cGMP (PDE10A). J. Biol. Chem. 1999, 274, 18438–18445. [Google Scholar] [CrossRef]
- Loughney, K.; Snyder, P.B.; Uher, L.; Rosman, G.J.; Ferguson, K.; Florio, V.A. Isolation and Characterization of PDE10A, a Novel Human 3′, 5′-Cyclic Nucleotide Phosphodiesterase. Gene 1999, 234, 109–117. [Google Scholar] [CrossRef]
- Fawcett, L.; Baxendale, R.; Stacey, P.; McGrouther, C.; Harrow, I.; Soderling, S.; Hetman, J.; Beavo, J.A.; Phillips, S.C. Molecular Cloning and Characterization of a Distinct Human Phosphodiesterase Gene Family: PDE11A. Proc. Natl. Acad. Sci. USA 2000, 97, 3702–3707. [Google Scholar] [CrossRef]
- Hetman, J.M.; Robas, N.; Baxendale, R.; Fidock, M.; Phillips, S.C.; Soderling, S.H.; Beavo, J.A. Cloning and Characterization of Two Splice Variants of Human Phosphodiesterase 11A. Proc. Natl. Acad. Sci. USA 2000, 97, 12891–12895. [Google Scholar] [CrossRef]
- Loughney, K.; Martins, T.J.; Harris, E.A.S.; Sadhu, K.; Hicks, J.B.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K. Isolation and Characterization of cDNAs Corresponding to Two Human Calcium, Calmodulin-Regulated, 3′,5′-Cyclic Nucleotide Phosphodiesterases. J. Biol. Chem. 1996, 271, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.M.; Erdogan, S.; Rena, G.; Borchert, G.; Hoch, B.; Bartel, S.; Scotland, G.; Huston, E.; Houslay, M.D.; Krause, E.-G. Altered Expression of PDE1 and PDE4 Cyclic Nucleotide Phosphodiesterase Isoforms in 7-Oxo-Prostacyclin-Preconditioned Rat Heart. J. Mol. Cell. Cardiol. 1997, 29, 3135–3146. [Google Scholar] [CrossRef] [PubMed]
- Senzaki, H.; Smith, C.J.; Juang, G.J.; Isoda, T.; Mayer, S.P.; Ohler, A.; Paolocci, N.; Tomaselli, G.F.; Hare, J.M.; Kass, D.A. Cardiac Phosphodiesterase 5 (cGMP-specific) Modulates Β-adrenergic Signaling in Vivo and Is Down-regulated in Heart Failure. FASEB J. 2001, 15, 1718–1726. [Google Scholar] [CrossRef]
- Ónody, A.; Zvara, Á.; Hackler, L.; Vígh, L.; Ferdinandy, P.; Puskás, L.G. Effect of Classic Preconditioning on the Gene Expression Pattern of Rat Hearts: A DNA Microarray Study. FEBS Lett. 2003, 536, 35–40. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Y.; Lighthouse, J.K.; Mickelsen, D.M.; Wu, J.; Yao, P.; Small, E.M.; Yan, C. A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction. Circulation 2020, 141, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Kim, G.E.; Tunin, R.S.; Adesiyun, T.; Hsu, S.; Nakagawa, R.; Zhu, G.; O’Brien, J.J.; Hendrick, J.P.; Davis, R.E.; et al. Acute Enhancement of Cardiac Function by Phosphodiesterase Type 1 Inhibition: Translational Study in the Dog and Rabbit. Circulation 2018, 138, 1974–1987. [Google Scholar] [CrossRef]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved Expression and Functions of PDE4 in Rodent and Human Heart. Basic. Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.V.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P.; et al. Phosphodiesterase 4B in the Cardiac L-Type Ca2+ Channel Complex Regulates Ca2+ Current and Protects against Ventricular Arrhythmias in Mice. J. Clin. Investig. 2011, 121, 2651–2661. [Google Scholar] [CrossRef]
- Bhat, A.; Ray, B.; Mahalakshmi, A.M.; Tuladhar, S.; Nandakumar, D.; Srinivasan, M.; Essa, M.M.; Chidambaram, S.B.; Guillemin, G.J.; Sakharkar, M.K. Phosphodiesterase-4 Enzyme as a Therapeutic Target in Neurological Disorders. Pharmacol. Res. 2020, 160, 105078. [Google Scholar] [CrossRef]
- Zhang, K.; Farooqui, S.M.; O’Donnell, J.M. Ontogeny of Rolipram-Sensitive, Low-Km, Cyclic AMP-Specific Phosphodiesterase in Rat Brain. Dev. Brain Res. 1999, 112, 11–19. [Google Scholar] [CrossRef]
- Rybalkin, S.D.; Hinds, T.R.; Beavo, J.A. Enzyme Assays for cGMP Hydrolyzing Phosphodiesterases. In Guanylate Cyclase and Cyclic GMP; Krieg, T., Lukowski, R., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1020, pp. 51–62. ISBN 978-1-62703-458-6. [Google Scholar]
- Xu, R.; Fu, J.; Hu, Y.; Yang, X.; Tao, X.; Chen, L.; Huang, K.; Fu, Q. Roflumilast-Mediated Phosphodiesterase 4D Inhibition Reverses Diabetes-Associated Cardiac Dysfunction and Remodeling: Effects Beyond Glucose Lowering. Diabetes 2022, 71, 1660–1678. [Google Scholar] [CrossRef] [PubMed]
- Qasim, H.; Rajaei, M.; Xu, Y.; Reyes-Alcaraz, A.; Abdelnasser, H.Y.; Stewart, M.D.; Lahiri, S.K.; Wehrens, X.H.T.; McConnell, B.K. AKAP12 Upregulation Associates with PDE8A to Accelerate Cardiac Dysfunction. Circ. Res. 2024, 134, 1006–1022. [Google Scholar] [CrossRef] [PubMed]
- Grammatika Pavlidou, N.; Dobrev, S.; Beneke, K.; Reinhardt, F.; Pecha, S.; Jacquet, E.; Abu-Taha, I.H.; Schmidt, C.; Voigt, N.; Kamler, M.; et al. Phosphodiesterase 8 Governs cAMP/PKA-Dependent Reduction of L-Type Calcium Current in Human Atrial Fibrillation: A Novel Arrhythmogenic Mechanism. Eur. Heart J. 2023, 44, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, D.C.; Anderson, S.G.; Caldwell, J.L.; Trafford, A.W. Phosphodiesterase-5 Inhibitors and the Heart: Compound Cardioprotection? Heart 2018, 104, 1244–1250. [Google Scholar] [CrossRef]
- Gong, W.; Yan, M.; Chen, J.; Chaugai, S.; Chen, C.; Wang, D. Chronic Inhibition of Cyclic Guanosine Monophosphate-Specific Phosphodiesterase 5 Prevented Cardiac Fibrosis through Inhibition of Transforming Growth Factor β-Induced Smad Signaling. Front. Med. 2014, 8, 445–455. [Google Scholar] [CrossRef]
- Wang, P.; Li, Z.; Cai, S.; Li, J.; He, P.; Huang, Y.; Feng, G.; Luo, H.; Chen, S.; Liu, P. C33(S), a Novel PDE9A Inhibitor, Protects against Rat Cardiac Hypertrophy through Upregulating cGMP Signaling. Acta Pharmacol. Sin. 2017, 38, 1257–1268. [Google Scholar] [CrossRef]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.-S.; Hamdani, N.; Holewinski, R.; Jo, S.-H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A Controls Nitric-Oxide-Independent cGMP and Hypertrophic Heart Disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef]
- Vandeput, F.; Wolda, S.L.; Krall, J.; Hambleton, R.; Uher, L.; McCaw, K.N.; Radwanski, P.B.; Florio, V.; Movsesian, M.A. Cyclic Nucleotide Phosphodiesterase PDE1C1 in Human Cardiac Myocytes. J. Biol. Chem. 2007, 282, 32749–32757. [Google Scholar] [CrossRef]
- Miller, C.L.; Cai, Y.; Oikawa, M.; Thomas, T.; Dostmann, W.R.; Zaccolo, M.; Fujiwara, K.; Yan, C. Cyclic Nucleotide Phosphodiesterase 1A: A Key Regulator of Cardiac Fibroblast Activation and Extracellular Matrix Remodeling in the Heart. Basic Res. Cardiol. 2011, 106, 1023–1039. [Google Scholar] [CrossRef]
- Monterisi, S.; Lobo, M.J.; Livie, C.; Castle, J.C.; Weinberger, M.; Baillie, G.; Surdo, N.C.; Musheshe, N.; Stangherlin, A.; Gottlieb, E.; et al. PDE2A2 Regulates Mitochondria Morphology and Apoptotic Cell Death via Local Modulation of cAMP/PKA Signalling. eLife 2017, 6, e21374. [Google Scholar] [CrossRef]
- Sadek, M.S.; Cachorro, E.; El-Armouche, A.; Kämmerer, S. Therapeutic Implications for PDE2 and cGMP/cAMP Mediated Crosstalk in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 7462. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M.; Ahmad, F.; Hirsch, E. Functions of PDE3 Isoforms in Cardiac Muscle. J. Cardiovasc. Dev. Dis. 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsesian, M.; et al. Phosphodiesterase Type 3A Regulates Basal Myocardial Contractility Through Interacting with Sarcoplasmic Reticulum Calcium ATPase Type 2a Signaling Complexes in Mouse Heart. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef]
- Kamel, R.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. Cyclic Nucleotide Phosphodiesterases as Therapeutic Targets in Cardiac Hypertrophy and Heart Failure. Nat. Rev. Cardiol. 2023, 20, 90–108. [Google Scholar] [CrossRef]
- Jäger, R.; Russwurm, C.; Schwede, F.; Genieser, H.-G.; Koesling, D.; Russwurm, M. Activation of PDE10 and PDE11 Phosphodiesterases. J. Biol. Chem. 2012, 287, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Richter, W.; Mehats, C.; Livera, G.; Park, J.-Y.; Jin, C. Cyclic AMP-Specific PDE4 Phosphodiesterases as Critical Components of Cyclic AMP Signaling. J. Biol. Chem. 2003, 278, 5493–5496. [Google Scholar] [CrossRef]
- Shi, Q.; Li, M.; Mika, D.; Fu, Q.; Kim, S.; Phan, J.; Shen, A.; Vandecasteele, G.; Xiang, Y.K. Heterologous Desensitization of Cardiac β-Adrenergic Signal via Hormone-Induced βAR/Arrestin/PDE4 Complexes. Cardiovasc. Res. 2017, 113, 656–670. [Google Scholar] [CrossRef]
- De Arcangelis, V.; Liu, R.; Soto, D.; Xiang, Y. Differential Association of Phosphodiesterase 4D Isoforms with Β2-Adrenoceptor in Cardiac Myocytes. J. Biol. Chem. 2009, 284, 33824–33832. [Google Scholar] [CrossRef]
- Richter, W.; Day, P.; Agrawal, R.; Bruss, M.D.; Granier, S.; Wang, Y.L.; Rasmussen, S.G.F.; Horner, K.; Wang, P.; Lei, T.; et al. Signaling from Β1- and Β2-Adrenergic Receptors Is Defined by Differential Interactions with PDE4. EMBO J. 2008, 27, 384–393. [Google Scholar] [CrossRef]
- Kajimoto, K.; Hagiwara, N.; Kasanuki, H.; Hosoda, S. Contribution of Phosphodiesterase Isozymes to the Regulation of the L-type Calcium Current in Human Cardiac Myocytes. Br. J. Pharmacol. 1997, 121, 1549–1556. [Google Scholar] [CrossRef]
- Molina, C.E.; Leroy, J.; Richter, W.; Xie, M.; Scheitrum, C.; Lee, I.-O.; Maack, C.; Rucker-Martin, C.; Donzeau-Gouge, P.; Verde, I.; et al. Cyclic Adenosine Monophosphate Phosphodiesterase Type 4 Protects Against Atrial Arrhythmias. J. Am. Coll. Cardiol. 2012, 59, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Berisha, F.; Götz, K.R.; Wegener, J.W.; Brandenburg, S.; Subramanian, H.; Molina, C.E.; Rüffer, A.; Petersen, J.; Bernhardt, A.; Girdauskas, E.; et al. cAMP Imaging at Ryanodine Receptors Reveals β2 -Adrenoceptor Driven Arrhythmias. Circ. Res. 2021, 129, 81–94. [Google Scholar] [CrossRef]
- Sin, Y.Y.; Edwards, H.V.; Li, X.; Day, J.P.; Christian, F.; Dunlop, A.J.; Adams, D.R.; Zaccolo, M.; Houslay, M.D.; Baillie, G.S. Disruption of the Cyclic AMP Phosphodiesterase-4 (PDE4)–HSP20 Complex Attenuates the β-Agonist Induced Hypertrophic Response in Cardiac Myocytes. J. Mol. Cell. Cardiol. 2011, 50, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, P.; Aulabaugh, A.; Geoghegan, K.F.; McLellan, T.J.; Pandit, J. Engineered Stabilization and Structural Analysis of the Autoinhibited Conformation of PDE4. Proc. Natl. Acad. Sci. USA 2015, 112, E1414–E1422. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Conti, M. Dimerization of the Type 4 cAMP-Specific Phosphodiesterases Is Mediated by the Upstream Conserved Regions (UCRs). J. Biol. Chem. 2002, 277, 40212–40221. [Google Scholar] [CrossRef]
- MacKenzie, S.J.; Baillie, G.S.; McPhee, I.; MacKenzie, C.; Seamons, R.; McSorley, T.; Millen, J.; Beard, M.B.; Van Heeke, G.; Houslay, M.D. Long PDE4 cAMP Specific Phosphodiesterases Are Activated by Protein Kinase A-mediated Phosphorylation of a Single Serine Residue in Upstream Conserved Region 1 (UCR1). Br. J. Pharmacol. 2002, 136, 421–433. [Google Scholar] [CrossRef]
- Sette, C.; Vicini, E.; Conti, M. The ratPDE3/IVd Phosphodiesterase Gene Codes for Multiple Proteins Differentially Activated by cAMP-Dependent Protein Kinase. J. Biol. Chem. 1994, 269, 18271–18274. [Google Scholar] [CrossRef]
- Sette, C.; Conti, M. Phosphorylation and Activation of a cAMP-Specific Phosphodiesterase by the cAMP-Dependent Protein Kinase. J. Biol. Chem. 1996, 271, 16526–16534. [Google Scholar] [CrossRef]
- Alvarez, R.; Sette, C.; Yang, D.; Eglen, R.M.; Wilhelm, R.; Shelton, E.R.; Conti, M. Activation and Selective Inhibition of a Cyclic AMP-Specific Phosphodiesterase, PDE-4D3. Mol. Pharmacol. 1995, 48, 616–622. [Google Scholar]
- Hoffmann, R.; Wilkinson, I.R.; McCallum, J.F.; Engels, P.; Houslay, M.D. cAMP-Specific Phosphodiesterase HSPDE4D3 Mutants Which Mimic Activation and Changes in Rolipram Inhibition Triggered by Protein Kinase A Phosphorylation of Ser-54: Generation of a Molecular Model. Biochem. J. 1998, 333 Pt 1, 139–149. [Google Scholar] [CrossRef]
- Beard, M.B.; Olsen, A.E.; Jones, R.E.; Erdogan, S.; Houslay, M.D.; Bolger, G.B. UCR1 and UCR2 Domains Unique to the cAMP-Specific Phosphodiesterase Family Form a Discrete Module via Electrostatic Interactions. J. Biol. Chem. 2000, 275, 10349–10358. [Google Scholar] [CrossRef]
- MacKenzie, S.J.; Baillie, G.S.; McPhee, I.; Bolger, G.B.; Houslay, M.D. ERK2 Mitogen-Activated Protein Kinase Binding, Phosphorylation, and Regulation of the PDE4D cAMP-Specific Phosphodiesterases. J. Biol. Chem. 2000, 275, 16609–16617. [Google Scholar] [CrossRef] [PubMed]
- Lenhard, J.M.; Kassel, D.B.; Rocque, W.J.; Hamacher, L.; Holmes, W.D.; Patel, I.; Hoffman, C.; Luther, M. Phosphorylation of a cAMP-Specific Phosphodiesterase (HSPDE4B2B) by Mitogen-Activated Protein Kinase. Biochem. J. 1996, 316, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Baillie, G.S.; MacKenzie, S.J.; Yarwood, S.J.; Houslay, M.D. The MAP Kinase ERK2 Inhibits the Cyclic AMP-Specific Phosphodiesterase HSPDE4D3 by Phosphorylating It at Ser579. EMBO J. 1999, 18, 893–903. [Google Scholar] [CrossRef]
- Baillie, G.S.; MacKenzie, S.J.; McPhee, I.; Houslay, M.D. Sub-family Selective Actions in the Ability of Erk2 MAP Kinase to Phosphorylate and Regulate the Activity of PDE4 Cyclic AMP-specific Phosphodiesterases. Br. J. Pharmacol. 2000, 131, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.; Burgin, A.B.; Gurney, M.E. Structural Basis for the Design of Selective Phosphodiesterase 4B Inhibitors. Cell. Signal. 2014, 26, 657–663. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing Structure Coverage for over 214 Million Protein Sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Leroy, J.; Abi-Gerges, A.; Nikolaev, V.O.; Richter, W.; Lechêne, P.; Mazet, J.-L.; Conti, M.; Fischmeister, R.; Vandecasteele, G. Spatiotemporal Dynamics of β-Adrenergic cAMP Signals and L-Type Ca2+ Channel Regulation in Adult Rat Ventricular Myocytes: Role of Phosphodiesterases. Circ. Res. 2008, 102, 1091–1100. [Google Scholar] [CrossRef]
- Kerfant, B.-G.; Zhao, D.; Lorenzen-Schmidt, I.; Wilson, L.S.; Cai, S.; Chen, S.R.W.; Maurice, D.H.; Backx, P.H. PI3Kγ Is Required for PDE4, Not PDE3, Activity in Subcellular Microdomains Containing the Sarcoplasmic Reticular Calcium ATPase in Cardiomyocytes. Circ. Res. 2007, 101, 400–408. [Google Scholar] [CrossRef]
- Lehnart, S.E.; Wehrens, X.H.T.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.-L.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D Deficiency in the Ryanodine-Receptor Complex Promotes Heart Failure and Arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.B.; Katugampola, S.; Able, S.; Napier, C.; Harding, S.E. Profiling of cAMP and cGMP Phosphodiesterases in Isolated Ventricular Cardiomyocytes from Human Hearts: Comparison with Rat and Guinea Pig. Life Sci. 2012, 90, 328–336. [Google Scholar] [CrossRef]
- Reinhardt, R.R.; Chin, E.; Zhou, J.; Taira, M.; Murata, T.; Manganiello, V.C.; Bondy, C.A. Distinctive Anatomical Patterns of Gene Expression for cGMP-Inhibited Cyclic Nucleotide Phosphodiesterases. J. Clin. Investig. 1995, 95, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, J.; Choi, Y.-H.; Krall, J.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of Cyclic Nucleotide Phosphodiesterase PDE3A in Cardiac Myocytes. J. Biol. Chem. 2002, 277, 38072–38078. [Google Scholar] [CrossRef]
- Reeves, M.L.; Leigh, B.K.; England, P.J. The Identification of a New Cyclic Nucleotide Phosphodiesterase Activity in Human and Guinea-Pig Cardiac Ventricle. Implications for the Mechanism of Action of Selective Phosphodiesterase Inhibitors. Biochem. J. 1987, 241, 535–541. [Google Scholar] [CrossRef]
- Muller, B.; Stoclet, J.-C.; Lugnier, C. Cytosolic and Membrane-Bound Cyclic Nucleotide Phosphodiesterases from Guinea Pig Cardiac Ventricles. Eur. J. Pharmacol. Mol. Pharmacol. 1992, 225, 263–272. [Google Scholar] [CrossRef]
- Patrucco, E.; Albergine, M.S.; Santana, L.F.; Beavo, J.A. Phosphodiesterase 8A (PDE8A) Regulates Excitation–Contraction Coupling in Ventricular Myocytes. J. Mol. Cell. Cardiol. 2010, 49, 330–333. [Google Scholar] [CrossRef]
- Subramanian, H.; Nikolaev, V.O. AKAP12 Overexpression Affects Cardiac Function via PDE8. Circ. Res. 2024, 134, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Ismaili, D.; Petersen, J.; Schulz, C.; Eschenhagen, T.; Koivumäki, J.T.; Christ, T. PDE8 Inhibition and Its Impact on I Ca,L in Persistent Atrial Fibrillation: Evaluation of PDE8 as a Potential Drug Target. J. Cardiovasc. Pharmacol. 2024, 84, 606–612. [Google Scholar] [CrossRef]
- Willoughby, D.; Baillie, G.S.; Lynch, M.J.; Ciruela, A.; Houslay, M.D.; Cooper, D.M.F. Dynamic Regulation, Desensitization, and Cross-Talk in Discrete Subcellular Microdomains during Β2-Adrenoceptor and Prostanoid Receptor cAMP Signaling. J. Biol. Chem. 2007, 282, 34235–34249. [Google Scholar] [CrossRef]
- Barnes, A.P.; Livera, G.; Huang, P.; Sun, C.; O’Neal, W.K.; Conti, M.; Stutts, M.J.; Milgram, S.L. Phosphodiesterase 4D Forms a cAMP Diffusion Barrier at the Apical Membrane of the Airway Epithelium. J. Biol. Chem. 2005, 280, 7997–8003. [Google Scholar] [CrossRef]
- McCahill, A.; Campbell, L.; McSorley, T.; Sood, A.; Lynch, M.J.; Li, X.; Yan, C.; Baillie, G.S.; Houslay, M.D. In Cardiac Myocytes, cAMP Elevation Triggers the down-Regulation of Transcripts and Promoter Activity for Cyclic AMP Phosphodiesterase-4A10 (PDE4A10). Cell. Signal. 2008, 20, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Richter, W.; Westenbroek, R.E.; Catterall, W.A.; Conti, M. PDE4B Mediates Local Feedback Regulation of Β1-Adrenergic cAMP Signaling in a Sarcolemmal Compartment of Cardiac Myocytes. J. Cell Sci. 2014, 127, 1033–1042. [Google Scholar] [CrossRef]
- Willoughby, D.; Wong, W.; Schaack, J.; Scott, J.D.; Cooper, D.M.F. An Anchored PKA and PDE4 Complex Regulates Subplasmalemmal cAMP Dynamics. EMBO J. 2006, 25, 2051–2061. [Google Scholar] [CrossRef]
- Houslay, M.D.; Baillie, G.S.; Maurice, D.H. cAMP-Specific Phosphodiesterase-4 Enzymes in the Cardiovascular System: A Molecular Toolbox for Generating Compartmentalized cAMP Signaling. Circ. Res. 2007, 100, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.J.; Baillie, G.S.; Houslay, M.D. cAMP-Specific Phosphodiesterase-4D5 (PDE4D5) Provides a Paradigm for Understanding the Unique Non-Redundant Roles That PDE4 Isoforms Play in Shaping Compartmentalized cAMP Cell Signalling. Biochem. Soc. Trans. 2007, 35, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Kyurkchieva, E.; Baillie, G.S. Short PDE4 Isoforms as Drug Targets in Disease. Front. Biosci. (Landmark Ed) 2023, 28, 133. [Google Scholar] [CrossRef]
- Karam, S.; Margaria, J.P.; Bourcier, A.; Mika, D.; Varin, A.; Bedioune, I.; Lindner, M.; Bouadjel, K.; Dessillons, M.; Gaudin, F.; et al. Cardiac Overexpression of PDE4B Blunts β-Adrenergic Response and Maladaptive Remodeling in Heart Failure. Circulation 2020, 142, 161–174. [Google Scholar] [CrossRef]
- Mika, D.; Bobin, P.; Pomérance, M.; Lechêne, P.; Westenbroek, R.E.; Catterall, W.A.; Vandecasteele, G.; Leroy, J.; Fischmeister, R. Differential Regulation of Cardiac Excitation–Contraction Coupling by cAMP Phosphodiesterase Subtypes. Cardiovasc. Res. 2013, 100, 336–346. [Google Scholar] [CrossRef]
- Molenaar, P.; Christ, T.; Hussain, R.I.; Engel, A.; Berk, E.; Gillette, K.T.; Chen, L.; Galindo-Tovar, A.; Krobert, K.A.; Ravens, U.; et al. PDE3, but Not PDE4, Reduces β1 - and β2 -adrenoceptor-mediated Inotropic and Lusitropic Effects in Failing Ventricle from Metoprolol-treated Patients. Br. J. Pharmacol. 2013, 169, 528–538. [Google Scholar] [CrossRef]
- Bobin, P.; Varin, A.; Lefebvre, F.; Fischmeister, R.; Vandecasteele, G.; Leroy, J. Calmodulin Kinase II Inhibition Limits the Pro-Arrhythmic Ca2+ Waves Induced by cAMP-Phosphodiesterase Inhibitors. Cardiovasc. Res. 2016, 110, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.T.; Bhogal, N.K.; Diakonov, I.; Pannell, L.M.K.; Perera, R.K.; Bork, N.I.; Schobesberger, S.; Lucarelli, C.; Faggian, G.; Alvarez-Laviada, A.; et al. Cardiomyocyte Membrane Structure and cAMP Compartmentation Produce Anatomical Variation in β2AR-cAMP Responsiveness in Murine Hearts. Cell Rep. 2018, 23, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.E.; Johnson, D.M.; Mehel, H.; Spätjens, R.L.H.M.G.; Mika, D.; Algalarrondo, V.; Slimane, Z.H.; Lechêne, P.; Abi-Gerges, N.; Van Der Linde, H.J.; et al. Interventricular Differences in β-Adrenergic Responses in the Canine Heart: Role of Phosphodiesterases. JAHA 2014, 3, e000858. [Google Scholar] [CrossRef]
- La Gerche, A.; Heidbüchel, H.; Burns, A.T.; Mooney, D.J.; Taylor, A.J.; Pfluger, H.B.; Inder, W.J.; Macisaac, A.I.; Prior, D.L. Disproportionate Exercise Load and Remodeling of the Athlete’s Right Ventricle. Med. Sci. Sports Exerc. 2011, 43, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.V.; Balycheva, M.; Sanchez-Alonso, J.L.; Ilkan, Z.; Alvarez-Laviada, A.; Bhogal, N.; Diakonov, I.; Schobesberger, S.; Sikkel, M.B.; Bhargava, A.; et al. Direct Evidence for Microdomain-Specific Localization and Remodeling of Functional L-Type Calcium Channels in Rat and Human Atrial Myocytes. Circulation 2015, 132, 2372–2384. [Google Scholar] [CrossRef]
- Judina, A.; Niglas, M.; Leonov, V.; Kirkby, N.S.; Diakonov, I.; Wright, P.T.; Zhao, L.; Mitchell, J.A.; Gorelik, J. Pulmonary Hypertension-Associated Right Ventricular Cardiomyocyte Remodelling Reduces Treprostinil Function. Cells 2023, 12, 2764. [Google Scholar] [CrossRef]
- Fu, J.; Mansfield, C.; Diakonov, I.; Judina, A.; Delahaye, M.; Bhogal, N.; Sanchez-Alonso, J.L.; Kamp, T.; Gorelik, J. Stretch Regulation of Β2-Adrenoceptor Signalling in Cardiomyocytes Requires Caveolae. Cardiovasc. Res. 2025, cvae265. [Google Scholar] [CrossRef]
- Wright, P.T.; Nikolaev, V.O.; O’Hara, T.; Diakonov, I.; Bhargava, A.; Tokar, S.; Schobesberger, S.; Shevchuk, A.I.; Sikkel, M.B.; Wilkinson, R.; et al. Caveolin-3 Regulates Compartmentation of Cardiomyocyte Beta2-Adrenergic Receptor-Mediated cAMP Signaling. J. Mol. Cell. Cardiol. 2014, 67, 38–48. [Google Scholar] [CrossRef]
- Bedioune, I.; Lefebvre, F.; Lechêne, P.; Varin, A.; Domergue, V.; Kapiloff, M.S.; Fischmeister, R.; Vandecasteele, G. PDE4 and mAKAPβ Are Nodal Organizers of Β2-ARs Nuclear PKA Signalling in Cardiac Myocytes. Cardiovasc. Res. 2018, 114, 1499–1511. [Google Scholar] [CrossRef]
- Xiang, Y.; Naro, F.; Zoudilova, M.; Jin, S.-L.C.; Conti, M.; Kobilka, B. Phosphodiesterase 4D Is Required for β2 Adrenoceptor Subtype-Specific Signaling in Cardiac Myocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 909–914. [Google Scholar] [CrossRef]
- Epstein, P.M.; Andrenyak, D.M.; Smith, C.J.; Pappano, A.J. Ontogenetic Changes in Adenylate Cyclase, Cyclic AMP Phosphodiesterase and Calmodulin in Chick Ventricular Myocardium. Biochem. J. 1987, 243, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.; Pappano, A.J. Developmental Changes in the Sensitivity of the Chick Embryo Ventricle to Beta-Adrenergic Agonist during Adrenergic Innervation. Circ. Res. 1981, 48, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Berthouze-Duquesnes, M.; Lucas, A.; Saulière, A.; Sin, Y.Y.; Laurent, A.-C.; Galés, C.; Baillie, G.; Lezoualc’h, F. Specific Interactions between Epac1, β-Arrestin2 and PDE4D5 Regulate β-Adrenergic Receptor Subtype Differential Effects on Cardiac Hypertrophic Signaling. Cell. Signal. 2013, 25, 970–980. [Google Scholar] [CrossRef]
- Kraft, A.E.; Bork, N.I.; Subramanian, H.; Pavlaki, N.; Failla, A.V.; Zobiak, B.; Conti, M.; Nikolaev, V.O. Phosphodiesterases 4B and 4D Differentially Regulate cAMP Signaling in Calcium Handling Microdomains of Mouse Hearts. Cells 2024, 13, 476. [Google Scholar] [CrossRef]
- Wright, P.T.; Sanchez-Alonso, J.L.; Lucarelli, C.; Alvarez-Laviada, A.; Poulet, C.E.; Bello, S.O.; Faggian, G.; Terracciano, C.M.; Gorelik, J. Partial Mechanical Unloading of the Heart Disrupts L-Type Calcium Channel and Beta-Adrenoceptor Signaling Microdomains. Front. Physiol. 2018, 9, 1302. [Google Scholar] [CrossRef]
- Rochais, F.; Abi-Gerges, A.; Horner, K.; Lefebvre, F.; Cooper, D.M.F.; Conti, M.; Fischmeister, R.; Vandecasteele, G. A Specific Pattern of Phosphodiesterases Controls the cAMP Signals Generated by Different Gs-Coupled Receptors in Adult Rat Ventricular Myocytes. Circ. Res. 2006, 98, 1081–1088. [Google Scholar] [CrossRef]
- Verde, I.; Vandecasteele, G.; Lezoualc’h, F.; Fischmeister, R. Characterization of the Cyclic Nucleotide Phosphodiesterase Subtypes Involved in the Regulation of the L-type Ca2+ Current in Rat Ventricular Myocytes. Br. J. Pharmacol. 1999, 127, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Helli, P.B.; Simpson, J.A.; Zhao, D.; Farman, G.P.; Jones, P.P.; Tian, X.; Wilson, L.S.; Ahmad, F.; Chen, S.R.W.; et al. Phosphodiesterase 4D Regulates Baseline Sarcoplasmic Reticulum Ca2+ Release and Cardiac Contractility, Independently of L-Type Ca2+ Current. Circ. Res. 2011, 109, 1024–1030. [Google Scholar] [CrossRef]
- Schwinger, R.H.G.; Münch, G.; Bölck, B.; Karczewski, P.; Krause, E.-G.; Erdmann, E. Reduced Ca2+-Sensitivity of SERCA 2a in Failing Human Myocardium Due to Reduced Serin-16 Phospholamban Phoshorylation. J. Mol. Cell. Cardiol. 1999, 31, 479–491. [Google Scholar] [CrossRef]
- Sande, J.B.; Sjaastad, I.; Hoen, I.B.; Bøkenes, J.; Tønnessen, T.; Holt, E.; Lunde, P.K.; Christensen, G. Reduced Level of Serine16 Phosphorylated Phospholamban in the Failing Rat Myocardium: A Major Contributor to Reduced SERCA2 Activity. Cardiovasc. Res. 2002, 53, 382–391. [Google Scholar] [CrossRef]
- Minamisawa, S.; Hoshijima, M.; Chu, G.; Ward, C.A.; Frank, K.; Gu, Y.; Martone, M.E.; Wang, Y.; Ross, J.; Kranias, E.G.; et al. Chronic Phospholamban–Sarcoplasmic Reticulum Calcium ATPase Interaction Is the Critical Calcium Cycling Defect in Dilated Cardiomyopathy. Cell 1999, 99, 313–322. [Google Scholar] [CrossRef]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In Vivo Model with Targeted cAMP Biosensor Reveals Changes in Receptor–Microdomain Communication in Cardiac Disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Keravis, T.; Le Bec, A.; Pauvert, O.; Proteau, S.; Rousseau, E. Characterization of Cyclic Nucleotide Phosphodiesterase Isoforms Associated to Isolated Cardiac Nuclei. Biochim. Biophys. Acta (BBA) Gen. Subj. 1999, 1472, 431–446. [Google Scholar] [CrossRef]
- Haj Slimane, Z.; Bedioune, I.; Lechêne, P.; Varin, A.; Lefebvre, F.; Mateo, P.; Domergue-Dupont, V.; Dewenter, M.; Richter, W.; Conti, M.; et al. Control of Cytoplasmic and Nuclear Protein Kinase A by Phosphodiesterases and Phosphatases in Cardiac Myocytes. Cardiovasc. Res. 2014, 102, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Burmeister, B.T.; Johnson, K.R.; Baillie, G.S.; Karginov, A.V.; Skidgel, R.A.; O’Bryan, J.P.; Carnegie, G.K. UCR1C Is a Novel Activator of Phosphodiesterase 4 (PDE4) Long Isoforms and Attenuates Cardiomyocyte Hypertrophy. Cell. Signal. 2015, 27, 908–922. [Google Scholar] [CrossRef] [PubMed]
- McCartney, S.; Little, B.M.; Langeberg, L.K.; Scott, J.D. Cloning and Characterization of A-Kinase Anchor Protein 100 (AKAP100). J. Biol. Chem. 1995, 270, 9327–9333. [Google Scholar] [CrossRef]
- Huang, L.J.; Durick, K.; Weiner, J.A.; Chun, J.; Taylor, S.S. Identification of a Novel Protein Kinase A Anchoring Protein That Binds Both Type I and Type II Regulatory Subunits. J. Biol. Chem. 1997, 272, 8057–8064. [Google Scholar] [CrossRef]
- Huang, L.J.; Durick, K.; Weiner, J.A.; Chun, J.; Taylor, S.S. D-AKAP2, a Novel Protein Kinase A Anchoring Protein with a Putative RGS Domain. Proc. Natl. Acad. Sci. USA 1997, 94, 11184–11189. [Google Scholar] [CrossRef]
- Ginsberg, M.D.; Feliciello, A.; Jones, J.K.; Avvedimento, E.V.; Gottesman, M.E. PKA-Dependent Binding of mRNA to the Mitochondrial AKAP121 Protein. J. Mol. Biol. 2003, 327, 885–897. [Google Scholar] [CrossRef]
- Trendelenburg, G.; Hummel, M.; Riecken, E.-O.; Hanski, C. Molecular Characterization of AKAP149, a Novel A Kinase Anchor Protein with a KH Domain. Biochem. Biophys. Res. Commun. 1996, 225, 313–319. [Google Scholar] [CrossRef]
- Lin, R.-Y.; Moss, S.B.; Rubin, C.S. Characterization of S-AKAP84, a Novel Developmentally Regulated A Kinase Anchor Protein of Male Germ Cells. J. Biol. Chem. 1995, 270, 27804–27811. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Boccella, N.; Paolillo, R.; Cattaneo, F.; Trimarco, V.; Franzone, A.; D’Apice, S.; Giugliano, G.; Rinaldi, L.; Borzacchiello, D.; et al. Loss of Akap1 Exacerbates Pressure Overload-Induced Cardiac Hypertrophy and Heart Failure. Front. Physiol. 2018, 9, 558. [Google Scholar] [CrossRef]
- Xiang, H.; Xu, H.; Tan, B.; Yi, Q.; Zhang, X.; Wang, R.; Chen, T.; Xie, Q.; Tian, J.; Zhu, J. AKAP1 Regulates Mitochondrial Dynamics during the Fatty-Acid-Promoted Maturation of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes as Indicated by Proteomics Sequencing. Int. J. Mol. Sci. 2023, 24, 8112. [Google Scholar] [CrossRef] [PubMed]
- Maric, D.; Paterek, A.; Delaunay, M.; López, I.P.; Arambasic, M.; Diviani, D. A-Kinase Anchoring Protein 2 Promotes Protection against Myocardial Infarction. Cells 2021, 10, 2861. [Google Scholar] [CrossRef]
- Kashishian, A.; Howard, M.; Loh, C.; Gallatin, W.M.; Hoekstra, M.F.; Lai, Y. AKAP79 Inhibits Calcineurin through a Site Distinct from the Immunophilin-Binding Region. J. Biol. Chem. 1998, 273, 27412–27419. [Google Scholar] [CrossRef]
- Hoshi, N.; Zhang, J.-S.; Omaki, M.; Takeuchi, T.; Yokoyama, S.; Wanaverbecq, N.; Langeberg, L.K.; Yoneda, Y.; Scott, J.D.; Brown, D.A.; et al. AKAP150 Signaling Complex Promotes Suppression of the M-Current by Muscarinic Agonists. Nat. Neurosci. 2003, 6, 564–571. [Google Scholar] [CrossRef]
- Li, Y.; Ndubuka, C.; Rubin, C.S. A Kinase Anchor Protein 75 Targets Regulatory (RII) Subunits of cAMP-Dependent Protein Kinase II to the Cortical Actin Cytoskeleton in Non-Neuronal Cells. J. Biol. Chem. 1996, 271, 16862–16869. [Google Scholar] [CrossRef]
- Nichols, C.B.; Rossow, C.F.; Navedo, M.F.; Westenbroek, R.E.; Catterall, W.A.; Santana, L.F.; McKnight, G.S. Sympathetic Stimulation of Adult Cardiomyocytes Requires Association of AKAP5 With a Subpopulation of L-Type Calcium Channels. Circ. Res. 2010, 107, 747–756. [Google Scholar] [CrossRef]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET Biosensor Uncovers cAMP Nano-Domains at β-Adrenergic Targets That Dictate Precise Tuning of Cardiac Contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef]
- Li, J.; Negro, A.; Lopez, J.; Bauman, A.L.; Henson, E.; Dodge-Kafka, K.; Kapiloff, M.S. The mAKAPβ Scaffold Regulates Cardiac Myocyte Hypertrophy via Recruitment of Activated Calcineurin. J. Mol. Cell. Cardiol. 2010, 48, 387–394. [Google Scholar] [CrossRef]
- Kapiloff, M.S.; Schillace, R.V.; Westphal, A.M.; Scott, J.D. mAKAP: An A-Kinase Anchoring Protein Targeted to the Nuclear Membrane of Differentiated Myocytes. J. Cell Sci. 1999, 112, 2725–2736. [Google Scholar] [CrossRef]
- Hakem Zadeh, F.; Teng, A.C.T.; Kuzmanov, U.; Chambers, P.J.; Tupling, A.R.; Gramolini, A.O. AKAP6 and Phospholamban Colocalize and Interact in HEK-293T Cells and Primary Murine Cardiomyocytes. Physiol. Rep. 2019, 7, e14144. [Google Scholar] [CrossRef] [PubMed]
- Vergarajauregui, S.; Becker, R.; Steffen, U.; Sharkova, M.; Esser, T.; Petzold, J.; Billing, F.; Kapiloff, M.S.; Schett, G.; Thievessen, I.; et al. AKAP6 Orchestrates the Nuclear Envelope Microtubule-Organizing Center by Linking Golgi and Nucleus via AKAP9. eLife 2020, 9, e61669. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.C.; Johnson, B.D.; Westenbroek, R.E.; Hays, L.G.; Yates, J.R.; Scheuer, T.; Catterall, W.A.; Murphy, B.J. Primary Structure and Function of an A Kinase Anchoring Protein Associated with Calcium Channels. Neuron 1998, 20, 1017–1026. [Google Scholar] [CrossRef]
- Gold, M.G.; Smith, F.D.; Scott, J.D.; Barford, D. AKAP18 Contains a Phosphoesterase Domain That Binds AMP. J. Mol. Biol. 2008, 375, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; et al. AKAP Complex Regulates Ca2+ Re-uptake into Heart Sarcoplasmic Reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef]
- Singh, A.; Redden, J.M.; Kapiloff, M.S.; Dodge-Kafka, K.L. The Large Isoforms of A-Kinase Anchoring Protein 18 Mediate the Phosphorylation of Inhibitor-1 by Protein Kinase A and the Inhibition of Protein Phosphatase 1 Activity. Mol. Pharmacol. 2011, 79, 533–540. [Google Scholar] [CrossRef]
- Seyler, C.; Scherer, D.; Köpple, C.; Kulzer, M.; Korkmaz, S.; Xynogalos, P.; Thomas, D.; Kaya, Z.; Scholz, E.; Backs, J.; et al. Role of Plasma Membrane-Associated AKAPs for the Regulation of Cardiac IK1 Current by Protein Kinase A. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 493–503. [Google Scholar] [CrossRef]
- Lin, J.W.; Wyszynski, M.; Madhavan, R.; Sealock, R.; Kim, J.U.; Sheng, M. Yotiao, a Novel Protein of Neuromuscular Junction and Brain That Interacts with Specific Splice Variants of NMDA Receptor Subunit NR1. J. Neurosci. 1998, 18, 2017–2027. [Google Scholar] [CrossRef]
- Shanks, R.A.; Steadman, B.T.; Schmidt, P.H.; Goldenring, J.R. AKAP350 at the Golgi Apparatus. J. Biol. Chem. 2002, 277, 40967–40972. [Google Scholar] [CrossRef]
- Gillingham, A.K.; Munro, S. The PACT Domain, a Conserved Centrosomal Targeting Motif in the Coiled-coil Proteins AKAP450 and Pericentrin. EMBO Rep. 2000, 1, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Kurokawa, J.; Reiken, S.; Motoike, H.; D’Armiento, J.; Marks, A.R.; Kass, R.S. Requirement of a Macromolecular Signaling Complex for β Adrenergic Receptor Modulation of the KCNQ1-KCNE1 Potassium Channel. Science 2002, 295, 496–499. [Google Scholar] [CrossRef]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-Kinase Anchoring Protein Yotiao Facilitates Complex Formation between Adenylyl Cyclase Type 9 and the IKs Potassium Channel in Heart. J. Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.S.; Park, K.-H.; El Harchi, A.; Camonis, J.; Kass, R.S.; Escande, D.; Mérot, J.; Loussouarn, G.; Le Bouffant, F.; Baró, I. I Ks Response to Protein Kinase A-Dependent KCNQ1 Phosphorylation Requires Direct Interaction with Microtubules. Cardiovasc. Res. 2008, 79, 427–435. [Google Scholar] [CrossRef]
- Nauert, J.B.; Klauck, T.M.; Langeberg, L.K.; Scott, J.D. Gravin, an Autoantigen Recognized by Serum from Myasthenia Gravis Patients, Is a Kinase Scaffold Protein. Curr. Biol. 1997, 7, 52–62. [Google Scholar] [CrossRef]
- Xia, W.; Unger, P.; Miller, L.; Nelson, J.; Gelman, I.H. The Src-Suppressed C Kinase Substrate, SSeCKS, Is a Potential Metastasis Inhibitor in Prostate Cancer. Cancer Res. 2001, 61, 5644–5651. [Google Scholar]
- Tao, J. Protein Kinase A Regulates AKAP250 (Gravin) Scaffold Binding to the 2-Adrenergic Receptor. EMBO J. 2003, 22, 6419–6429. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, Q.-H.; Chu, Y.; Wu, W.-M.; Song, J.-X.; Zhu, X.-B.; Wang, Q. Blockage of AKAP12 Accelerates Angiotensin II (Ang II)-Induced Cardiac Injury in Mice by Regulating the Transforming Growth Factor Β1 (TGF-Β1) Pathway. Biochem. Biophys. Res. Commun. 2018, 499, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Soderling, J.; Scott, J.D. AKAP-Lbc Anchors Protein Kinase A and Nucleates Gα12-Selective Rho-Mediated Stress Fiber Formation. J. Biol. Chem. 2001, 276, 44247–44257. [Google Scholar] [CrossRef]
- Carnegie, G.K.; Soughayer, J.; Smith, F.D.; Pedroja, B.S.; Zhang, F.; Diviani, D.; Bristow, M.R.; Kunkel, M.T.; Newton, A.C.; Langeberg, L.K.; et al. AKAP-Lbc Mobilizes a Cardiac Hypertrophy Signaling Pathway. Mol. Cell 2008, 32, 169–179. [Google Scholar] [CrossRef]
- Appert-Collin, A.; Cotecchia, S.; Nenniger-Tosato, M.; Pedrazzini, T.; Diviani, D. The A-Kinase Anchoring Protein (AKAP)-Lbc-Signaling Complex Mediates A1 Adrenergic Receptor-Induced Cardiomyocyte Hypertrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 10140–10145. [Google Scholar] [CrossRef] [PubMed]
- Caso, S.; Maric, D.; Arambasic, M.; Cotecchia, S.; Diviani, D. AKAP-Lbc Mediates Protection against Doxorubicin-Induced Cardiomyocyte Toxicity. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 2336–2346. [Google Scholar] [CrossRef]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating Cardiac PIP3 and cAMP Signaling through a PKA Anchoring Function of P110γ. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Murabito, A.; Sala, V.; Pisano, A.R.; Bertolini, S.; Gianotti, A.; Caci, E.; Montresor, A.; Premchandar, A.; Pirozzi, F.; et al. A PI3Kγ Mimetic Peptide Triggers CFTR Gating, Bronchodilation, and Reduced Inflammation in Obstructive Airway Diseases. Sci. Transl. Med. 2022, 14, eabl6328. [Google Scholar] [CrossRef]
- Ghigo, A.; Perino, A.; Mehel, H.; Zahradníková, A.; Morello, F.; Leroy, J.; Nikolaev, V.O.; Damilano, F.; Cimino, J.; De Luca, E.; et al. Phosphoinositide 3-Kinase γ Protects Against Catecholamine-Induced Ventricular Arrhythmia Through Protein Kinase A–Mediated Regulation of Distinct Phosphodiesterases. Circulation 2012, 126, 2073–2083. [Google Scholar] [CrossRef]
- Ndongson-Dongmo, B.; Heller, R.; Hoyer, D.; Brodhun, M.; Bauer, M.; Winning, J.; Hirsch, E.; Wetzker, R.; Schlattmann, P.; Bauer, R. Phosphoinositide 3-Kinase Gamma Controls Inflammation-Induced Myocardial Depression via Sequential cAMP and iNOS Signalling. Cardiovasc. Res. 2015, 108, 243–253. [Google Scholar] [CrossRef]
- Kang, M.; Otani, Y.; Guo, Y.; Yan, J.; Goult, B.T.; Howe, A.K. The Focal Adhesion Protein Talin Is a Mechanically Gated A-Kinase Anchoring Protein. Proc. Natl. Acad. Sci. USA 2024, 121, e2314947121. [Google Scholar] [CrossRef]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central Role for G Protein-Coupled Phosphoinositide 3-Kinase γ in Inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects from Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical Regulation of a Molecular Clutch Defines Force Transmission and Transduction in Response to Matrix Rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef]
- Haining, A.W.M.; Rahikainen, R.; Cortes, E.; Lachowski, D.; Rice, A.; Von Essen, M.; Hytönen, V.P.; Del Río Hernández, A. Mechanotransduction in Talin through the Interaction of the R8 Domain with DLC1. PLoS Biol. 2018, 16, e2005599. [Google Scholar] [CrossRef]
- Terrenoire, C.; Houslay, M.D.; Baillie, G.S.; Kass, R.S. The Cardiac IKs Potassium Channel Macromolecular Complex Includes the Phosphodiesterase PDE4D3. J. Biol. Chem. 2009, 284, 9140–9146. [Google Scholar] [CrossRef]
- Terrin, A.; Monterisi, S.; Stangherlin, A.; Zoccarato, A.; Koschinski, A.; Surdo, N.C.; Mongillo, M.; Sawa, A.; Jordanides, N.E.; Mountford, J.C.; et al. PKA and PDE4D3 Anchoring to AKAP9 Provides Distinct Regulation of cAMP Signals at the Centrosome. J. Cell Biol. 2012, 198, 607–621. [Google Scholar] [CrossRef]
- Kapiloff, M.S.; Jackson, N.; Airhart, N. mAKAP and the Ryanodine Receptor Are Part of a Multi-Component Signaling Complex on the Cardiomyocyte Nuclear Envelope. J. Cell Sci. 2001, 114, 3167–3176. [Google Scholar] [CrossRef] [PubMed]
- Carlisle Michel, J.J.; Dodge, K.L.; Wong, W.; Mayer, N.C.; Langeberg, L.K.; Scott, J.D. PKA-Phosphorylation of PDE4D3 Facilitates Recruitment of the mAKAP Signalling Complex. Biochem. J. 2004, 381, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Dodge, K.L. mAKAP Assembles a Protein Kinase A/PDE4 Phosphodiesterase cAMP Signaling Module. EMBO J. 2001, 20, 1921–1930. [Google Scholar] [CrossRef]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The Protein Kinase A Anchoring Protein mAKAP Coordinates Two Integrated cAMP Effector Pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef]
- Fields, L.A.; Koschinski, A.; Zaccolo, M. Sustained Exposure to Catecholamines Affects cAMP/PKA Compartmentalised Signalling in Adult Rat Ventricular Myocytes. Cell. Signal. 2016, 28, 725–732. [Google Scholar] [CrossRef]
- Abi-Gerges, A.; Richter, W.; Lefebvre, F.; Mateo, P.; Varin, A.; Heymes, C.; Samuel, J.-L.; Lugnier, C.; Conti, M.; Fischmeister, R.; et al. Decreased Expression and Activity of cAMP Phosphodiesterases in Cardiac Hypertrophy and Its Impact on β-Adrenergic cAMP Signals. Circ. Res. 2009, 105, 784–792. [Google Scholar] [CrossRef]
- Qvigstad, E.; Moltzau, L.R.; Aronsen, J.M.; Nguyen, C.H.T.; Hougen, K.; Sjaastad, I.; Levy, F.O.; Skomedal, T.; Osnes, J.-B. Natriuretic Peptides Increase Β1-Adrenoceptor Signalling in Failing Hearts through Phosphodiesterase 3 Inhibition. Cardiovasc. Res. 2010, 85, 763–772. [Google Scholar] [CrossRef]
- Colombe, A.-S.; Pidoux, G. Cardiac cAMP-PKA Signaling Compartmentalization in Myocardial Infarction. Cells 2021, 10, 922. [Google Scholar] [CrossRef]
- Pavlaki, N.; Froese, A.; Li, W.; De Jong, K.A.; Geertz, B.; Subramanian, H.; Mohagaonkar, S.; Luo, X.; Schubert, M.; Wiegmann, R.; et al. Gene Therapy with Phosphodiesterases 2A and 4B Ameliorates Heart Failure and Arrhythmias by Improving Subcellular cAMP Compartmentation. Cardiovasc. Res. 2024, 120, 1011–1023. [Google Scholar] [CrossRef]
- Abrenica, B.; AlShaaban, M.; Czubryt, M.P. The A-Kinase Anchor Protein AKAP121 Is a Negative Regulator of Cardiomyocyte Hypertrophy. J. Mol. Cell. Cardiol. 2009, 46, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Kelley, G.G.; Kapiloff, M.S.; Smrcka, A.V. Phospholipase C∈ Scaffolds to Muscle-Specific A Kinase Anchoring Protein (mAKAPβ) and Integrates Multiple Hypertrophic Stimuli in Cardiac Myocytes. J. Biol. Chem. 2011, 286, 23012–23021. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.; Gildart, M.; Tokarski, K.; Kapiloff, M.S. mAKAPβ Signalosomes—A Nodal Regulator of Gene Transcription Associated with Pathological Cardiac Remodeling. Cell. Signal. 2019, 63, 109357. [Google Scholar] [CrossRef]
- Li, J.; Aponte Paris, S.; Thakur, H.; Kapiloff, M.S.; Dodge-Kafka, K.L. Muscle A-Kinase–Anchoring Protein-β–Bound Calcineurin Toggles Active and Repressive Transcriptional Complexes of Myocyte Enhancer Factor 2D. J. Biol. Chem. 2019, 294, 2543–2554. [Google Scholar] [CrossRef]
- Kritzer, M.D.; Li, J.; Passariello, C.L.; Gayanilo, M.; Thakur, H.; Dayan, J.; Dodge-Kafka, K.; Kapiloff, M.S. The Scaffold Protein Muscle A-Kinase Anchoring Protein β Orchestrates Cardiac Myocyte Hypertrophic Signaling Required for the Development of Heart Failure. Circ. Heart Fail. 2014, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.; Hille, S.S.; Subramanian, H.; Wiegmann, R.; Roser, P.; Müller, O.J.; Nikolaev, V.O.; De Jong, K.A. Remodelling of cAMP Dynamics within the SERCA2a Microdomain in Heart Failure with Preserved Ejection Fraction Caused by Obesity and Type 2 Diabetes. Cardiovasc. Res. 2024, 120, 273–285. [Google Scholar] [CrossRef]
- Soilness, J.E.; Maslen, C.; Webber, S.; Foster, M.; Raeburn, D.; Palfreyman, M.N.; Ashton, M.J.; Karlsson, J. Suppression of Eosinophil Function by RP 73401, a Potent and Selective Inhibitor of Cyclic AMP-specific Phosphodiesterase: Comparison with Rolipram. Br. J Pharmacol. 1995, 115, 39–46. [Google Scholar] [CrossRef]
- Rolan, P.; Hutchinson, M.; Johnson, K. Ibudilast: A Review of Its Pharmacology, Efficacy and Safety in Respiratory and Neurological Disease. Expert. Opin. Pharmacother. 2009, 10, 2897–2904. [Google Scholar] [CrossRef]
- Titus, D.J.; Wilson, N.M.; Alcazar, O.; Calixte, D.A.; Dietrich, W.D.; Gurney, M.E.; Atkins, C.M. A Negative Allosteric Modulator of PDE4D Enhances Learning after Traumatic Brain Injury. Neurobiol. Learn. Mem. 2018, 148, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Nugent, R.A.; Mo, X.; Sindac, J.A.; Hagen, T.J.; Fox, D.; O’Donnell, J.M.; Zhang, C.; Xu, Y.; Zhang, H.-T.; et al. Design and Synthesis of Selective Phosphodiesterase 4D (PDE4D) Allosteric Inhibitors for the Treatment of Fragile X Syndrome and Other Brain Disorders. J. Med. Chem. 2019, 62, 4884–4901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, Y.; Chowdhary, A.; Fox, D.; Gurney, M.E.; Zhang, H.-T.; Auerbach, B.D.; Salvi, R.J.; Yang, M.; Li, G.; et al. Memory Enhancing Effects of BPN14770, an Allosteric Inhibitor of Phosphodiesterase-4D, in Wild-Type and Humanized Mice. Neuropsychopharmacol. 2018, 43, 2299–2309. [Google Scholar] [CrossRef]
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S.; et al. Design of Phosphodiesterase 4D (PDE4D) Allosteric Modulators for Enhancing Cognition with Improved Safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef]
- McDowell, L.; Olin, B. Crisaborole: A Novel Nonsteroidal Topical Treatment for Atopic Dermatitis. J. Pharm. Technol. 2019, 35, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F. Update on Roflumilast, a Phosphodiesterase 4 Inhibitor for the Treatment of Chronic Obstructive Pulmonary Disease. Br. J. Pharmacol. 2011, 163, 53–67. [Google Scholar] [CrossRef]
- Schafer, P.H.; Parton, A.; Capone, L.; Cedzik, D.; Brady, H.; Evans, J.F.; Man, H.-W.; Muller, G.W.; Stirling, D.I.; Chopra, R. Apremilast Is a Selective PDE4 Inhibitor with Regulatory Effects on Innate Immunity. Cell. Signal. 2014, 26, 2016–2029. [Google Scholar] [CrossRef]
- Tralau-Stewart, C.J.; Williamson, R.A.; Nials, A.T.; Gascoigne, M.; Dawson, J.; Hart, G.J.; Angell, A.D.R.; Solanke, Y.E.; Lucas, F.S.; Wiseman, J.; et al. GSK256066, an Exceptionally High-Affinity and Selective Inhibitor of Phosphodiesterase 4 Suitable for Administration by Inhalation: In Vitro, Kinetic, and In Vivo Characterization. J. Pharmacol. Exp. Ther. 2011, 337, 145–154. [Google Scholar] [CrossRef]
- Warren, R.B.; Strober, B.; Silverberg, J.I.; Guttman, E.; Andres, P.; Felding, J.; Tutkunkardas, D.; Kjøller, K.; Sommer, M.O.A.; French, L.E. Oral Orismilast: Efficacy and Safety in Moderate-to-severe Psoriasis and Development of Modified Release Tablets. Acad. Dermatol. Venereol. 2023, 37, 711–720. [Google Scholar] [CrossRef]
- White, W.B.; Cooke, G.E.; Kowey, P.R.; Calverley, P.M.A.; Bredenbröker, D.; Goehring, U.-M.; Zhu, H.; Lakkis, H.; Mosberg, H.; Rowe, P.; et al. Cardiovascular Safety in Patients Receiving Roflumilast for the Treatment of COPD. Chest 2013, 144, 758–765. [Google Scholar] [CrossRef]
- Dong, C.; Virtucio, C.; Zemska, O.; Baltazar, G.; Zhou, Y.; Baia, D.; Jones-Iatauro, S.; Sexton, H.; Martin, S.; Dee, J.; et al. Treatment of Skin Inflammation with Benzoxaborole Phosphodiesterase Inhibitors: Selectivity, Cellular Activity, and Effect on Cytokines Associated with Skin Inflammation and Skin Architecture Changes. J. Pharmacol. Exp. Ther. 2016, 358, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.Y.; Giblin, A.; Judina, A.; Rujirachaivej, P.; Corral, L.G.; Glennon, E.; Tai, Z.X.; Feng, T.; Torres, E.; Zorn, A.; et al. Targeted Protein Degradation of PDE4 Shortforms by a Novel Proteolysis Targeting Chimera. FEBS J. 2024, febs.17359. [Google Scholar] [CrossRef]
- Murabito, A.; Cnudde, S.; Hirsch, E.; Ghigo, A. Potential Therapeutic Applications of AKAP Disrupting Peptides. Clin. Sci. 2020, 134, 3259–3282. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, A.; Tasca, L.; Butnarasu, C.; Sala, V.; Prono, G.; Murabito, A.; Garbero, O.V.; Millo, E.; Terranova, L.; Blasi, F.; et al. A Nonnatural Peptide Targeting the A-Kinase Anchoring Function of PI3Kγ for Therapeutic cAMP Modulation in Pulmonary Cells. J. Biol. Chem. 2024, 300, 107873. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Category | PDE Family | Isoforms | References |
|---|---|---|---|
| cAMP-specific PDEs | PDE4 | PDE4A, PDE4B, PDE4C, PDE4D | [14,15] |
| PDE7 | PDE7A, PDE7B | [16,17] | |
| PDE8 | PDE8A, PDE8B | [18,19,20] | |
| cGMP-specific PDEs | PDE5 | PDE5A | [21,22,23] |
| PDE6 | PDE6A, PDE6B, PDE6C | [24,25] | |
| PDE9 | PDE9A | [26,27,28] | |
| Dual-substrate PDEs | PDE1 | PDE1A, PDE1B, PDE1C | [29,30,31] |
| PDE2 | PDE2A | [32] | |
| PDE3 | PDE3A, PDE3B | [33,34,35,36] | |
| PDE10 | PDE10A | [37,38] | |
| PDE11 | PDE11A | [39,40] |
| Category | PDE Family | Main Functions and Localisation | Km (μmol/l) | References | |
|---|---|---|---|---|---|
| cAMP | cGMP | ||||
| cAMP-specific PDEs | PDE4 | Plays a key role when cAMP levels are elevated. Detected in cardiomyocytes and fibroblasts. | 1–6 | NA | [15,47,48,49,50,51,52] |
| PDE8 | Controls ICa,L current. Detected in cardiomyocytes. | 0.1–0.6 | NA | [19,20,51,53,54] | |
| cGMP-specific PDEs | PDE5 | Preferentially regulates a pool of cGMP produced by soluble GC. Detected in cardiomyocytes and fibroblasts. | 201 | 1–6 | [2,21,22,23,51,55,56] |
| PDE9 | Preferentially regulates the NP-induced cGMP. Detected in cardiomyocytes and not detected in fibroblasts. | 230 | 0.1–0.4 | [2,26,27,28,51,57,58] | |
| Dual-substrate PDEs | PDE1 | Regulation of calcium/calmodulin. Detected in cardiomyocytes and fibroblasts. | 1–125 | 1–8 | [29,51,59,60] |
| PDE2 | Regulates local mitochondria-related cAMP pools. More abundantly expressed in cardiac fibroblasts compared to cardiomyocytes. | 30–112 | 10–31 | [6,51,61,62] | |
| PDE3 | Responsible for the tonic effects in the myocardium. PDE3 is the most important in cardiomyocytes. | 0.1–0.8 | 0.1–0.8 | [33,34,36,60,63,64,65] | |
| PDE10 | cAMP regulates PDE10 biphasically, modulating cGMP hydrolysis. Detected in cardiomyocytes and fibroblasts. | 0.2–0.3 | 1.1–7.2 | [38,45,51,66] | |
| AKAP | Aliases | Functions in Cardiomyocytes |
|---|---|---|
| AKAP1 | D-AKAP1 [139], AKAP121 [141], AKAP149 [142], S-AKAP84 [143], mitoAKAP [144] | Regulates mitochondrial dynamics, oxidative phosphorylation, and cardiomyocyte survival, playing a protective role against cardiac hypertrophy and heart failure [144,145]. |
| AKAP2 | D-AKAP2 [140] | Organises a signalling complex with PKA and Src3, promoting anti-apoptotic and pro-angiogenic responses essential for myocardial infarction recovery [146]. |
| AKAP5 | AKAP79 [147], AKAP150 [148], AKAP75 [149] | Coordinates PKA signalling in T-tubules and plasma membrane, regulating calcium channels and cardiac contractility under sympathetic stimulation [150,151]. |
| AKAP6 | mAKAPβ [152], AKAP100 [138], mAKAP [153] | Regulates calcium handling by interacting with PLN and organises the nuclear envelope microtubule-organisng centre through centrosomal and Golgi-associated proteins [154,155]. |
| AKAP7 | AKAP15 [156], AKAP18 [157] | Localises PKA to the plasma membrane, regulating membrane events like cardiac IK1 currents [158,159,160]. |
| AKAP9 | Yotiao [161], AKAP350 [162], AKAP450 [163] | Coordinates β-adrenergic regulation of the IKs potassium channel by assembling PKA, PP1, AC9, and PDE4D3 into a macromolecular complex. Disruptions are linked to long-QT syndrome and impaired cardiac repolarisation [164,165,166]. |
| AKAP12 | Gravin [167], SSeCKS [168], AKAP250 [169] | Mitigates maladaptive remodelling, oxidative stress, and fibrosis by inhibiting Ang-II-induced TGFβ1 signalling. Also regulates cardiac contractility and calcium handling during isoproterenol stimulation [53,170]. |
| AKAP13 | AKAP-Lbc [171] | Coordinates cardiomyocyte signalling pathways involved in protection against doxorubicin toxicity, pathological hypertrophy, and α1-adrenergic receptor-mediated RhoA activation [172,173,174]. |
| Drug Name | PDE4 Specificity | Disease | Phase | NCT Number * |
|---|---|---|---|---|
| Roflumilast [208] | Pan-PDE4 | Polycystic Ovary Syndrome | IV | NCT02037672; NCT02187250 |
| Chronic Hand Eczema | IV | NCT05682859 | ||
| Ulcerative Colitis | IV | NCT05684484 | ||
| Chronic Obstructive Pulmonary Disease | IV | NCT01595750 | ||
| Apremilast [209] | Pan-PDE4 | Recurrent Aphthous Stomatitis (RAS) | IV | NCT03690544 |
| Alopecia Areata | IV | NCT05926882 | ||
| Chronic and Recurrent Erythema Nodosum Leprosum | IV | NCT04822909 | ||
| Oral Lichen Planus | IV | NCT06260904 | ||
| Crisaborole [207] | Pan-PDE4 | Moderate Atopic Dermatitis | IV | NCT04214197 |
| Seborrheic Dermatitis | IV | NCT03567980 | ||
| Hemay005 [211] | Pan-PDE4 | Behçet’s Disease | III | NCT06145893 |
| Severe Plaque Psoriasis | III | NCT04839328 | ||
| Cilomilast [208] | Pan-PDE4, more selective for PDE4D | Chronic Obstructive Pulmonary Disease | III | NCT00103922 |
| Tanimilast [211] | Pan-PDE4 | Chronic Obstructive Pulmonary Disease and Chronic Bronchitis | III | NCT04636801 |
| GSK256066 [210] | PDE4B | Chronic Obstructive Pulmonary Disease | II | NCT00549679 |
| BPN14770 [205] | PDE4D | Fragile X Syndrome | II | NCT03569631 |
| KIT2014 [176] | PDE4B and PDE4D | Healthy Subjects | I | NCT06659757 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherstnev, I.; Judina, A.; Luciani, G.B.; Ghigo, A.; Hirsch, E.; Gorelik, J. Role of PDE4 Family in Cardiomyocyte Physiology and Heart Failure. Cells 2025, 14, 460. https://doi.org/10.3390/cells14060460
Sherstnev I, Judina A, Luciani GB, Ghigo A, Hirsch E, Gorelik J. Role of PDE4 Family in Cardiomyocyte Physiology and Heart Failure. Cells. 2025; 14(6):460. https://doi.org/10.3390/cells14060460
Chicago/Turabian StyleSherstnev, Ivan, Aleksandra Judina, Giovanni Battista Luciani, Alessandra Ghigo, Emilio Hirsch, and Julia Gorelik. 2025. "Role of PDE4 Family in Cardiomyocyte Physiology and Heart Failure" Cells 14, no. 6: 460. https://doi.org/10.3390/cells14060460
APA StyleSherstnev, I., Judina, A., Luciani, G. B., Ghigo, A., Hirsch, E., & Gorelik, J. (2025). Role of PDE4 Family in Cardiomyocyte Physiology and Heart Failure. Cells, 14(6), 460. https://doi.org/10.3390/cells14060460