
Impact of a High-Fat Diet on the Gut Microbiome: A Comprehensive Study of Microbial and Metabolite Shifts During Obesity



Abstract

1. Introduction
2. Materials and Methods
2.1. Animal and Diet Formulas
2.2. Fecal Sample Collection and DNA Isolation
2.3. Bacterial Amplicon Sequencing and Statistical Analysis
3. Results
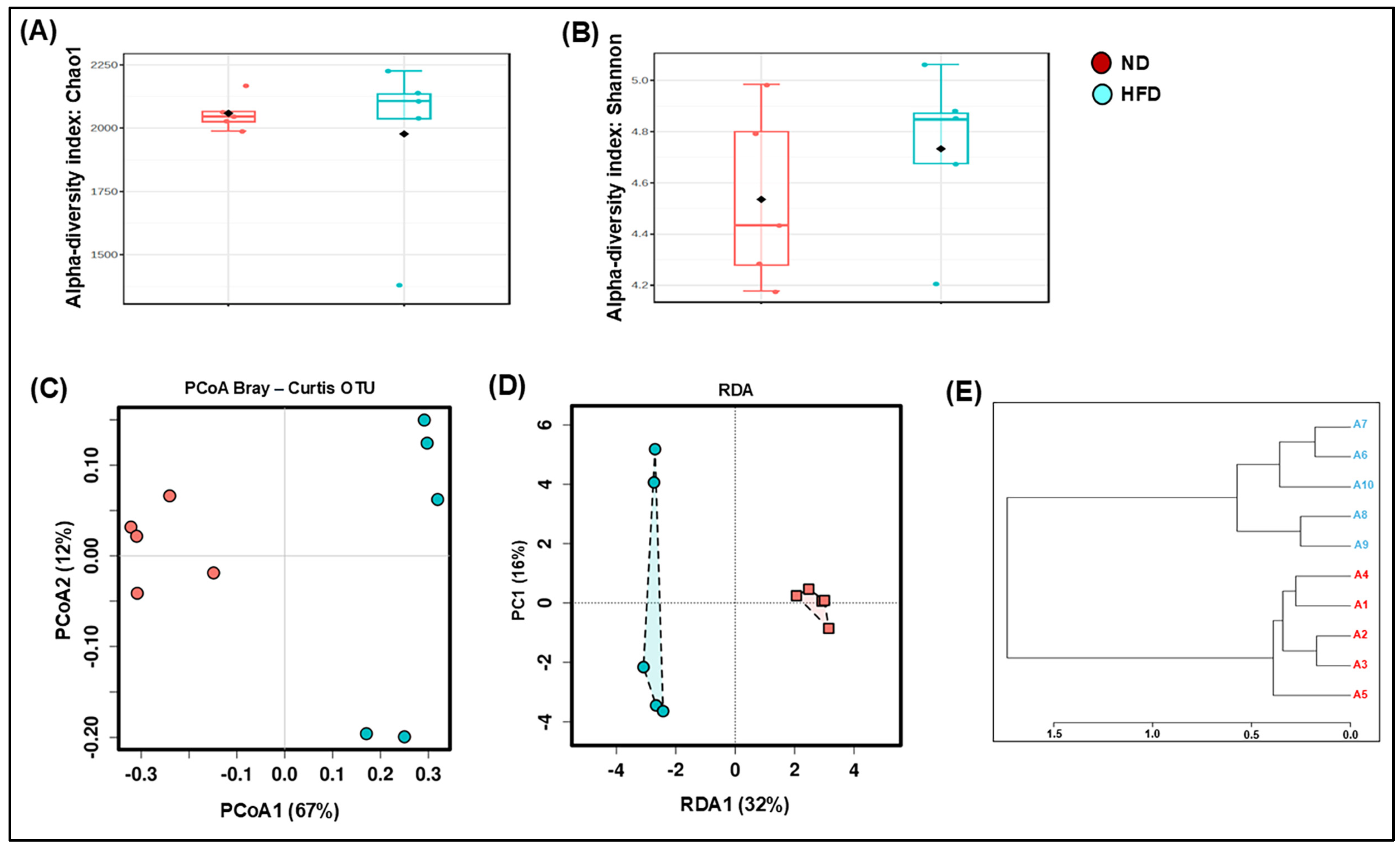
3.1. HFD Dysregulates the Composition and Decreases the Richness of the Gut Microbiota
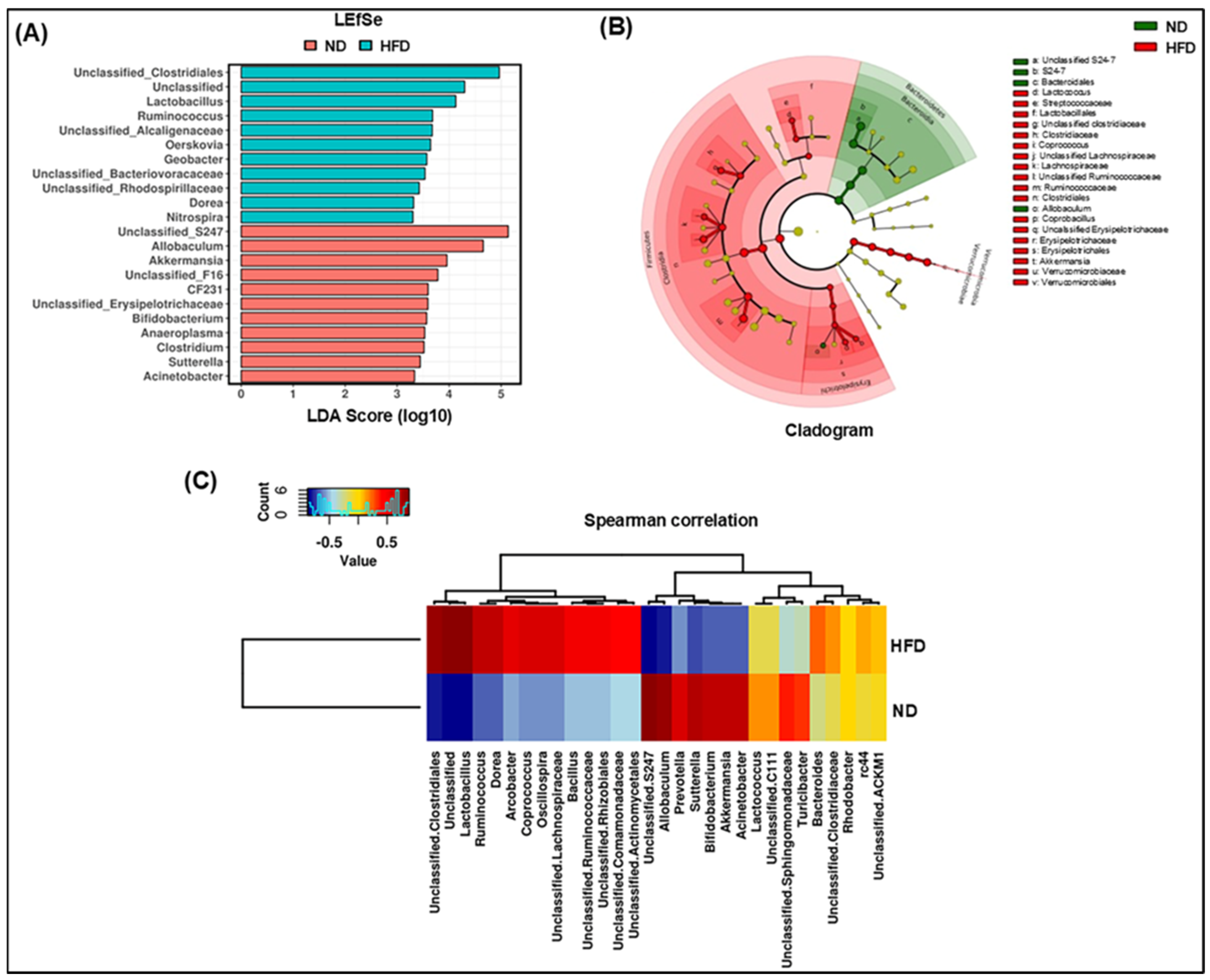
3.2. Modulation of Microbial Taxonomic Profiling in HFD-Fed Mice
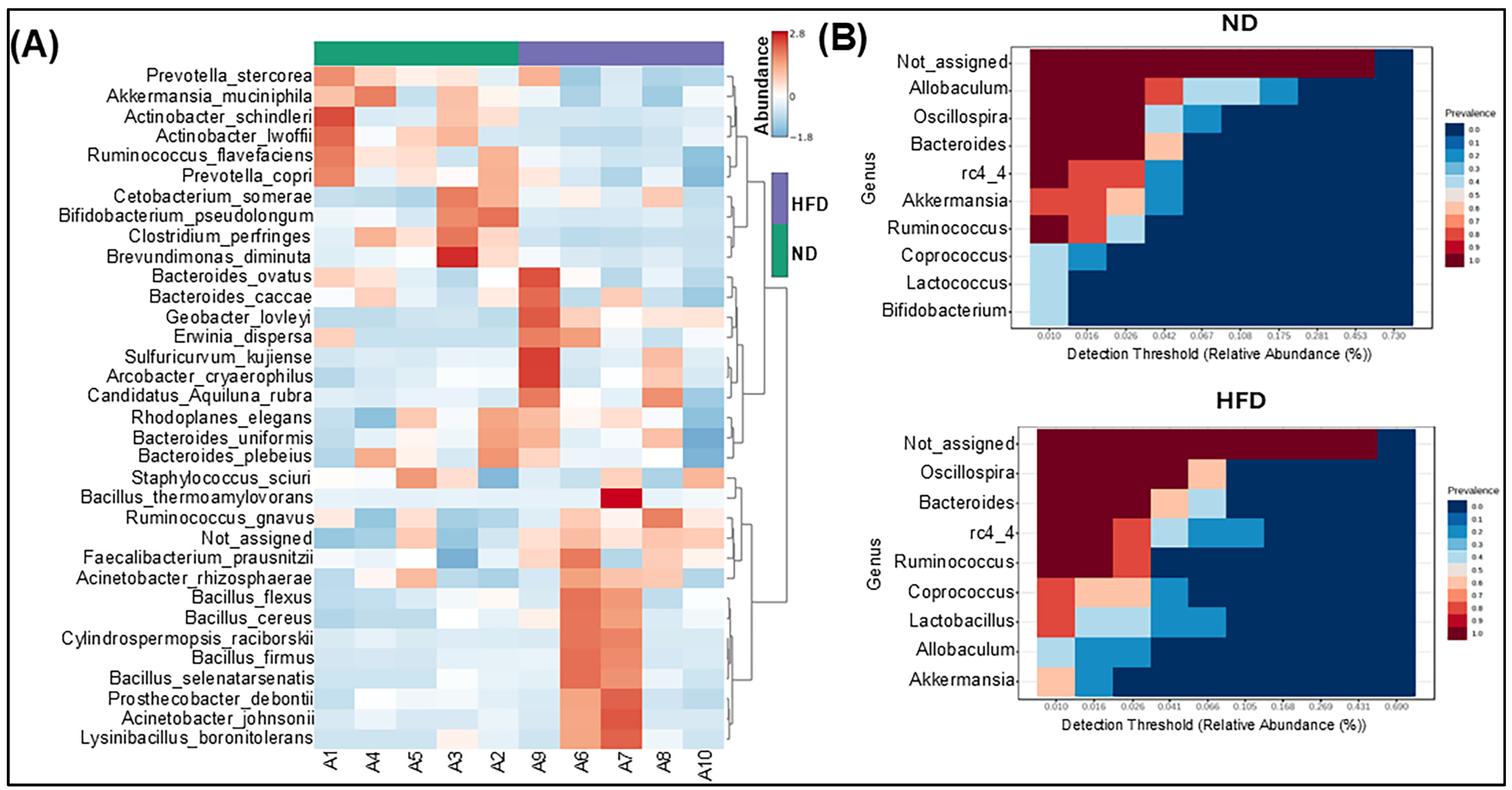
3.3. Specific Phylotype Alteration in the Fecal Microbiome in HFD-Fed Mice
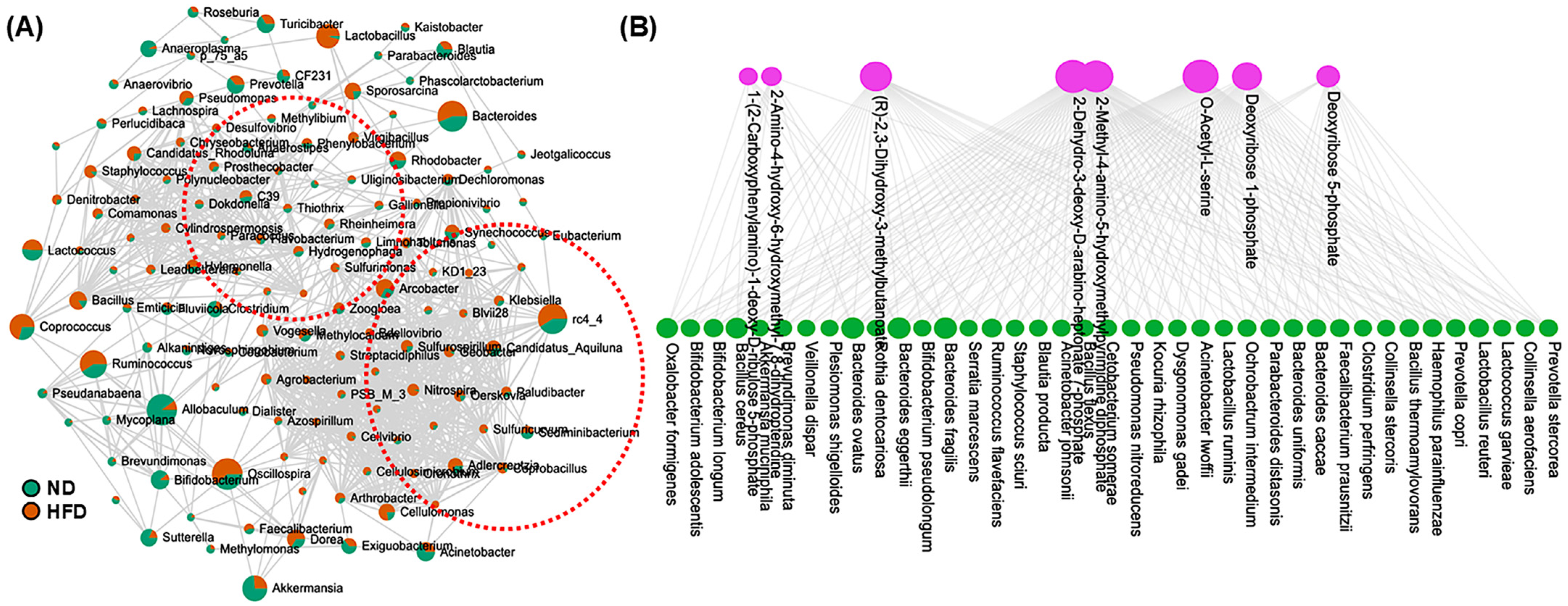
3.4. HFD Differentially Modulates Bacterial Populations
3.5. Correlation and Functional Enrichment Analysis of Microbiome–Metabolite Interaction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raoult, D.; Fournier, P.E.; Drancourt, M. What does the future hold for clinical microbiology? Nat. Rev. Microbiol. 2004, 2, 151–159. [Google Scholar] [PubMed]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottière, H.M.; Raes, J.; Ehrlich, D.; Doré, J. A metagenomic insight into our gut’s microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [PubMed]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar]
- Gu, Y.; Liu, C.; Zheng, N.; Jia, W.; Zhang, W.; Li, H. Metabolic and gut microbial characterization of obesity-prone mice under a high-fat diet. J. Proteome Res. 2019, 18, 1703–1714. [Google Scholar]
- Reddon, H.; Guéant, J.-L.; Meyre, D. The importance of gene–environment interactions in human obesity. Clin. Sci. 2016, 130, 1571–1597. [Google Scholar]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.; Koh, G.Y.; Nagy, A.; Semenkovich, C.; Gordon, J. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Thingholm, L.B.; Rühlemann, M.C.; Koch, M.; Fuqua, B.; Laucke, G.; Boehm, R.; Bang, C.; Franzosa, E.A.; Hübenthal, M.; Rahnavard, A. Obese individuals with and without type 2 diabetes show different gut microbial functional capacity and composition. Cell Host Microbe 2019, 26, 252–264.e210. [Google Scholar]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar]
- Zheng, R.; Xiang, X.; Shi, Y.; Xie, J.; Xing, L.; Zhang, T.; Zhou, Z.; Zhang, D. Gut microbiota and mycobiota change with feeding duration in mice on a high-fat and high-fructose diet. BMC Microbiol. 2024, 24, 504. [Google Scholar]
- Secchiero, P.; Rimondi, E.; Marcuzzi, A.; Longo, G.; Papi, C.; Manfredini, M.; Fields, M.; Caruso, L.; Di Caprio, R.; Balato, A. Metabolic Syndrome and Psoriasis: Pivotal Roles of Chronic Inflammation and Gut Microbiota. Int. J. Mol. Sci. 2024, 25, 8098. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L.; Bäckhed, F. Diet–microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 2016, 167, 1339–1353.e1321. [Google Scholar]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar]
- Khan, M.J.; Gerasimidis, K.; Edwards, C.A.; Shaikh, M.G. Role of gut microbiota in the aetiology of obesity: Proposed mechanisms and review of the literature. J. Obes. 2016, 2016, 7353642. [Google Scholar]
- Bliss, D.Z.; Weimer, P.J.; Jung, H.-J.G.; Savik, K. In vitro degradation and fermentation of three dietary fiber sources by human colonic bacteria. J. Agric. Food Chem. 2013, 61, 4614–4621. [Google Scholar]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar]
- Pindjakova, J.; Sartini, C.; Lo Re, O.; Rappa, F.; Coupe, B.; Lelouvier, B.; Pazienza, V.; Vinciguerra, M. Gut dysbiosis and adaptive immune response in diet-induced obesity vs. systemic inflammation. Front. Microbiol. 2017, 8, 1157. [Google Scholar]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar]
- Frost, R.A.; Nystrom, G.J.; Lang, C.H. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2002, 283, R698–R709. [Google Scholar] [PubMed]
- Li, J.; Lin, S.; Vanhoutte, P.M.; Woo, C.W.; Xu, A. Akkermansia muciniphila protects against atherosclerosis by preventing metabolic endotoxemia-induced inflammation in Apoe−/− mice. Circulation 2016, 133, 2434–2446. [Google Scholar] [PubMed]
- Brooks, S.P.; McAllister, M.; Sandoz, M.; Kalmokoff, M. Culture-independent phylogenetic analysis of the faecal flora of the rat. Can. J. Microbiol. 2003, 49, 589–601. [Google Scholar]
- Li, M.; Gu, D.; Xu, N.; Lei, F.; Du, L.; Zhang, Y.; Xie, W. Gut carbohydrate metabolism instead of fat metabolism regulated by gut microbes mediates high-fat diet-induced obesity. Benef. Microbes 2014, 5, 335–344. [Google Scholar] [PubMed]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; Gonzalez, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 36, 10.7.1–10.7.20. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.-J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar]
- Xc, M. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar]
- Duncan, S.H.; Lobley, G.; Holtrop, G.; Ince, J.; Johnstone, A.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Aguirre, M.; Venema, K. Does the gut microbiota contribute to obesity? Going beyond the gut feeling. Microorganisms 2015, 3, 213–235. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.; Gonzalez, A.; Ackermann, G.; Wendel, D.; Vázquez-Baeza, Y.; Jansson, J.K.; Gordon, J.I.; Knight, R. Meta-analyses of studies of the human microbiota. Genome Res. 2013, 23, 1704–1714. [Google Scholar]
- Liu, Z.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008, 36, e120. [Google Scholar]
- Choo, J.M.; Leong, L.E.; Rogers, G.B. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 2015, 5, 16350. [Google Scholar]
- Fei, N.; Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013, 7, 880–884. [Google Scholar] [PubMed]
- Rastogi, S.; Singh, A. Gut microbiome and human health: Exploring how the probiotic genus Lactobacillus modulate immune responses. Front. Pharmacol. 2022, 13, 1042189. [Google Scholar]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef]
- Velazquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef]
- Wolters, M.; Ahrens, J.; Romaní-Pérez, M.; Watkins, C.; Sanz, Y.; Benítez-Páez, A.; Stanton, C.; Günther, K. Dietary fat, the gut microbiota, and metabolic health–A systematic review conducted within the MyNewGut project. Clin. Nutr. 2019, 38, 2504–2520. [Google Scholar]
- Matey-Hernandez, M.L.; Williams, F.M.; Potter, T.; Valdes, A.M.; Spector, T.D.; Menni, C. Genetic and microbiome influence on lipid metabolism and dyslipidemia. Physiol. Genom. 2018, 50, 117–126. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar]
- Li, Y.; Liu, Q.; Peng, C.; Ruan, B. Both gut microbiota and differentially expressed proteins are relevant to the development of obesity. BioMed Res. Int. 2020, 2020, 5376108. [Google Scholar]
- de Noronha, S.I.R.; de Moraes, L.A.G.; Hassell, J.E., Jr.; Stamper, C.E.; Arnold, M.R.; Heinze, J.D.; Foxx, C.L.; Lieb, M.M.; Cler, K.E.; Karns, B.L. High-fat diet, microbiome-gut-brain axis signaling, and anxiety-like behavior in male rats. Biol. Res. 2024, 57, 23. [Google Scholar]
- Scheithauer, T.P.; Dallinga-Thie, G.M.; de Vos, W.M.; Nieuwdorp, M.; van Raalte, D.H. Causality of small and large intestinal microbiota in weight regulation and insulin resistance. Mol. Metab. 2016, 5, 759–770. [Google Scholar]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lan, C.; Li, H.; Ouyang, Q.; Kong, F.; Wu, A.; Ren, Z.; Tian, G.; Cai, J.; Yu, B. Rational consideration of Akkermansia muciniphila targeting intestinal health: Advantages and challenges. npj Biofilms Microbiomes 2022, 8, 81. [Google Scholar]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar]
- Lagkouvardos, I.; Lesker, T.R.; Hitch, T.C.; Gálvez, E.J.; Smit, N.; Neuhaus, K.; Wang, J.; Baines, J.F.; Abt, B.; Stecher, B. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 2019, 7, 28. [Google Scholar] [CrossRef]
- Zhang, X.-S.; Li, J.; Krautkramer, K.A.; Badri, M.; Battaglia, T.; Borbet, T.C.; Koh, H.; Ng, S.; Sibley, R.A.; Li, Y. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. eLife 2018, 7, e37816. [Google Scholar]
- Mousavinasab, F.; Karimi, R.; Taheri, S.; Ahmadvand, F.; Sanaaee, S.; Najafi, S.; Halvaii, M.S.; Haghgoo, A.; Zamany, M.; Majidpoor, J. Microbiome modulation in inflammatory diseases: Progress to microbiome genetic engineering. Cancer Cell Int. 2023, 23, 271. [Google Scholar] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [PubMed]
- Brunner, K.T.; Henneberg, C.J.; Wilechansky, R.M.; Long, M.T. Nonalcoholic fatty liver disease and obesity treatment. Curr. Obes. Rep. 2019, 8, 220–228. [Google Scholar] [PubMed]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar]
- Chawla, A.; Nguyen, K.D.; Goh, Y.S. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar]
- Zacarías, M.F.; Collado, M.C.; Gomez-Gallego, C.; Flinck, H.; Aittoniemi, J.; Isolauri, E.; Salminen, S. Pregestational overweight and obesity are associated with differences in gut microbiota composition and systemic inflammation in the third trimester. PLoS ONE 2018, 13, e0200305. [Google Scholar] [CrossRef]
- Maffei, M.; Funicello, M.; Vottari, T.; Gamucci, O.; Costa, M.; Lisi, S.; Viegi, A.; Ciampi, O.; Bardi, G.; Vitti, P. The obesity and inflammatory marker haptoglobin attracts monocytes via interaction with chemokine (CC motif) receptor 2 (CCR2). BMC Biol. 2009, 7, 87. [Google Scholar]
- Chiellini, C.; Santini, F.; Marsili, A.; Berti, P.; Bertacca, A.; Pelosini, C.; Scartabelli, G.; Pardini, E.; Lopez-Soriano, J.; Centoni, R. Serum haptoglobin: A novel marker of adiposity in humans. J. Clin. Endocrinol. Metab. 2004, 89, 2678–2683. [Google Scholar]
- Perumal, N.L.; Mufida, A.; Yadav, A.K.; Son, D.-G.; Ryoo, Y.-W.; Kim, S.-A.; Jang, B.-C. Suppression of Lipid Accumulation in the Differentiation of 3T3-L1 Preadipocytes and Human Adipose Stem Cells into Adipocytes by TAK-715, a Specific Inhibitor of p38 MAPK. Life 2023, 13, 412. [Google Scholar] [CrossRef]
- Luck, H.; Tsai, S.; Chung, J.; Clemente-Casares, X.; Ghazarian, M.; Revelo, X.S.; Lei, H.; Luk, C.T.; Shi, S.Y.; Surendra, A. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. 2015, 21, 527–542. [Google Scholar]
- Tanaka, M. Molecular mechanism of obesity-induced adipose tissue inflammation; the role of Mincle in adipose tissue fibrosis and ectopic lipid accumulation. Endocr. J. 2020, 67, 107–111. [Google Scholar] [PubMed]
- Wang, S.; Miura, M.; Jung, Y.-K.; Zhu, H.; Li, E.; Yuan, J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 1998, 92, 501–509. [Google Scholar] [PubMed]
- Kang, S.; Wang, S.; Kuida, K.; Yuan, J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 2002, 9, 1115–1125. [Google Scholar]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar]
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The gut microbiota and inflammation: An overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar]
- Yu, Z.; Chen, W.; Zhang, L.; Chen, Y.; Chen, W.; Meng, S.; Lu, L.; Han, Y.; Shi, J. Gut-derived bacterial LPS attenuates incubation of methamphetamine craving via modulating microglia. Brain Behav. Immun. 2023, 111, 101–115. [Google Scholar]
- Mohr, A.E.; Crawford, M.S.; Jasbi, P.; Fessler, S.; Sweazea, K.L. Lipopolysaccharide and the gut microbiota: Considering structural variation. FEBS Lett. 2022, 596, 849–875. [Google Scholar] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [PubMed]
- Hills, R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut microbiome: Profound implications for diet and disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef]
- Sochocka, M.; Donskow-Łysoniewska, K.; Diniz, B.S.; Kurpas, D.; Brzozowska, E.; Leszek, J. The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease—A critical review. Mol. Neurobiol. 2019, 56, 1841–1851. [Google Scholar]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar]
- Sánchez-Alcoholado, L.; Ordóñez, R.; Otero, A.; Plaza-Andrade, I.; Laborda-Illanes, A.; Medina, J.A.; Ramos-Molina, B.; Gómez-Millán, J.; Queipo-Ortuño, M.I. Gut microbiota-mediated inflammation and gut permeability in patients with obesity and colorectal cancer. Int. J. Mol. Sci. 2020, 21, 6782. [Google Scholar] [CrossRef] [PubMed]
- Luu, T.H.; Michel, C.; Bard, J.-M.; Dravet, F.; Nazih, H.; Bobin-Dubigeon, C. Intestinal proportion of Blautia sp. is associated with clinical stage and histoprognostic grade in patients with early-stage breast cancer. Nutr. Cancer 2017, 69, 267–275. [Google Scholar]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar]
- Blaut, M. Gut microbiota and energy balance: Role in obesity. Proc. Nutr. Soc. 2015, 74, 227–234. [Google Scholar] [PubMed]
- Yu, D.; Richardson, N.E.; Green, C.L.; Spicer, A.B.; Murphy, M.E.; Flores, V.; Jang, C.; Kasza, I.; Nikodemova, M.; Wakai, M.H. The adverse metabolic effects of branched-chain amino acids are mediated by isoleucine and valine. Cell Metab. 2021, 33, 905–922.e906. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway (KEGG) | Total | Expected | Hits | p-Value | FDR | Features |
|---|---|---|---|---|---|---|
| Pentose phosphate pathway | 35 | 0.0453 | 2 | 0.000853 | 0.156 | C00672 |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 35 | 0.0453 | 2 | 0.000853 | 0.156 | C00673 |
| Sulfur relay system | 11 | 0.0142 | 1 | 0.0142 | 1 | C01302 |
| Valine, leucine, and isoleucine biosynthesis | 23 | 0.0298 | 1 | 0.0294 | 1 | C04272 |
| Pantothenate and CoA biosynthesis | 28 | 0.0362 | 1 | 0.0357 | 1 | C04272 |
| Thiamine metabolism | 31 | 0.0401 | 1 | 0.0394 | 1 | C04752 |
| Sulfur metabolism | 33 | 0.0427 | 1 | 0.0419 | 1 | C00979 |
| Zeatin biosynthesis | 39 | 0.0505 | 1 | 0.0494 | 1 | C00979 |
| Folate biosynthesis | 57 | 0.0738 | 1 | 0.0715 | 1 | C01300 |
| Cysteine and methionine metabolism | 63 | 0.0815 | 1 | 0.0787 | 1 | C00979 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamun, M.A.A.; Rakib, A.; Mandal, M.; Singh, U.P. Impact of a High-Fat Diet on the Gut Microbiome: A Comprehensive Study of Microbial and Metabolite Shifts During Obesity. Cells 2025, 14, 463. https://doi.org/10.3390/cells14060463
Mamun MAA, Rakib A, Mandal M, Singh UP. Impact of a High-Fat Diet on the Gut Microbiome: A Comprehensive Study of Microbial and Metabolite Shifts During Obesity. Cells. 2025; 14(6):463. https://doi.org/10.3390/cells14060463
Chicago/Turabian StyleMamun, Md Abdullah Al, Ahmed Rakib, Mousumi Mandal, and Udai P. Singh. 2025. "Impact of a High-Fat Diet on the Gut Microbiome: A Comprehensive Study of Microbial and Metabolite Shifts During Obesity" Cells 14, no. 6: 463. https://doi.org/10.3390/cells14060463
APA StyleMamun, M. A. A., Rakib, A., Mandal, M., & Singh, U. P. (2025). Impact of a High-Fat Diet on the Gut Microbiome: A Comprehensive Study of Microbial and Metabolite Shifts During Obesity. Cells, 14(6), 463. https://doi.org/10.3390/cells14060463