.png)
Huntingtin-Interacting Protein 1-Related (HIP1R) Regulates Rheumatoid Arthritis Synovial Fibroblast Invasiveness

Abstract
1. Introduction
2. Material and Methods
2.1. Isolation and Culture of RA Fibroblast-like Synoviocytes (FLS)
2.2. Immunofluorescence Microscopy
2.3. Western Blot
2.4. siRNA Knock Down
2.5. Invasion Assay
2.6. Adhesion to Matrigel
2.7. Migration Assay (Also Called Wound Healing or Scratch Assay)
2.8. Proliferation
2.9. RNA Extraction
2.10. RNA Sequencing and Analyses
3. Statistics
4. Results
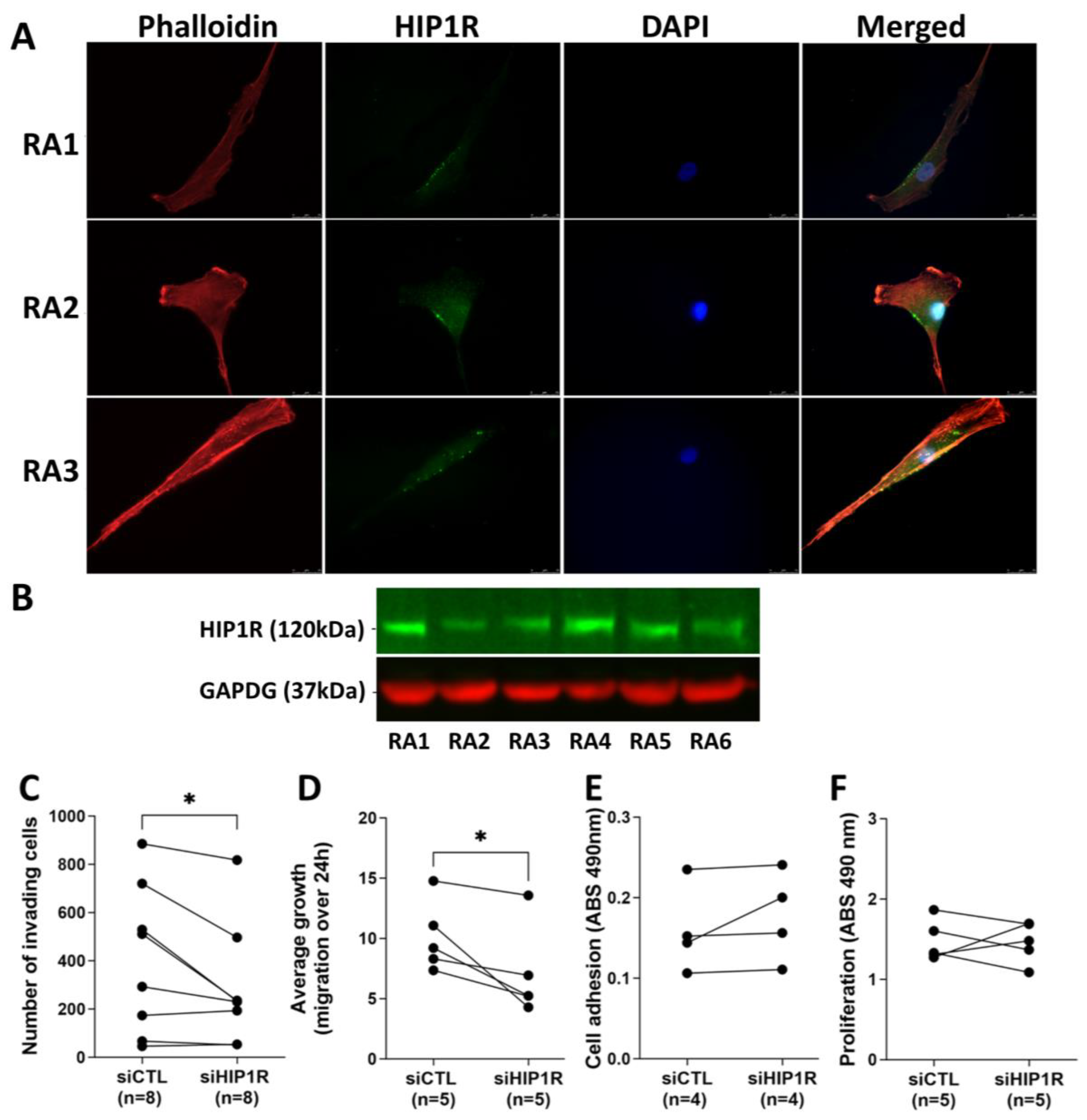
4.1. HIP1R Protein Is Present in RA FLS
4.2. siRNA Knockdown of HIP1R Significantly Decreased RA FLS Invasiveness and Migration
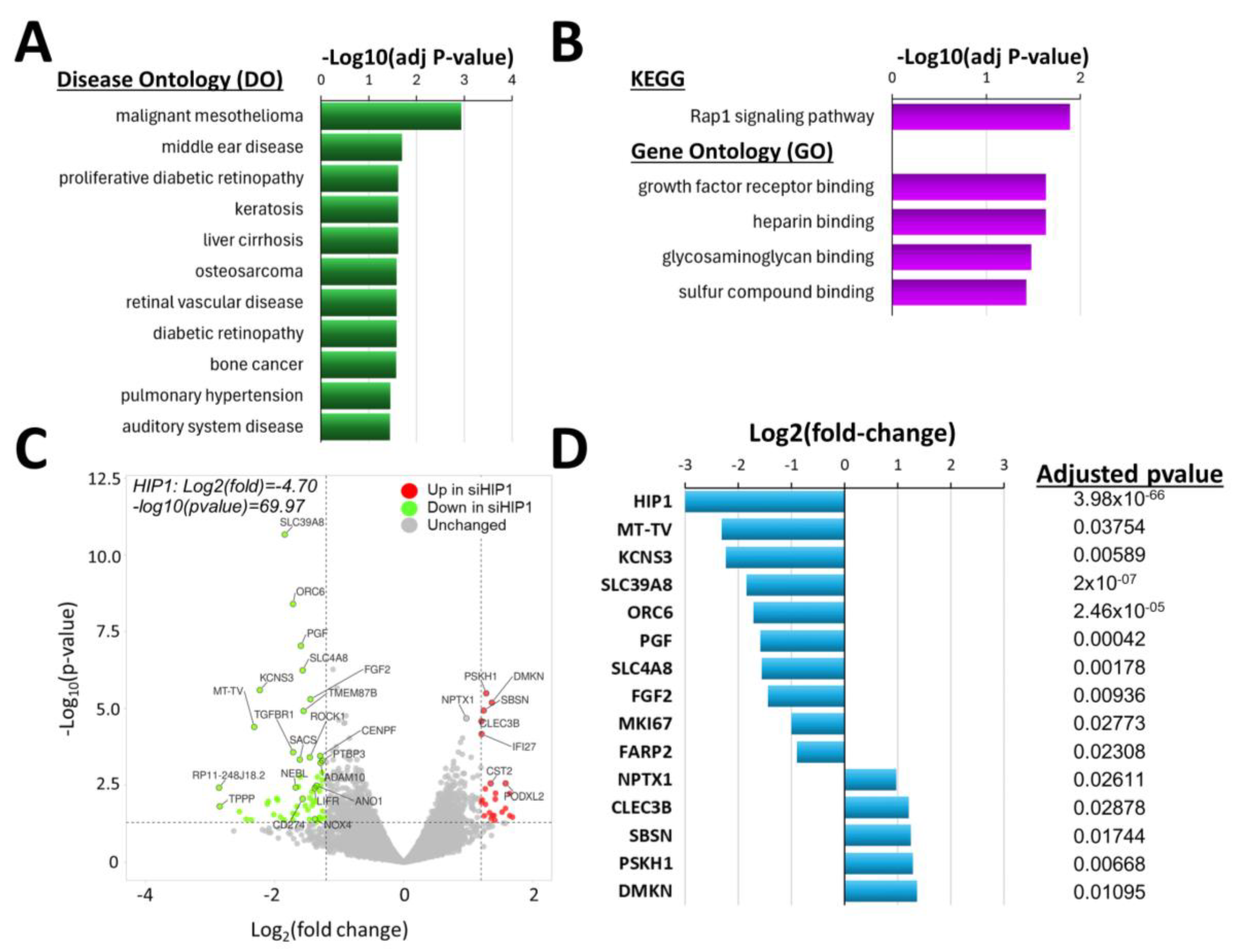
4.3. Knockdown of HIP1R Was Associated with Unique DEGs Also Involved in Cancer, and in FLS Growth and Invasion
4.4. Knockdown of HIP1 Caused Enrichment of DEGs Implicated in Different Diseases Such as Cancers and Unique Biologic Processes
4.5. Knockdown of HIP1 Significantly Changed Gene Expression, Including of Some FLS-Specific Genes
4.6. Comparison of the DEGs Lists of siRNA HIP1 Versus Control, and of siRNA HIP1R Versus Control
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Zeben, D.; Breedveld, F.C. Prognostic factors in rheumatoid arthritis. J. Rheumatol. Suppl. 1996, 44, 31–33. [Google Scholar] [PubMed]
- Gossec, L.; Dougados, M.; Goupille, P.; Cantagrel, A.; Sibilia, J.; Meyer, O.; Sany, J.; Daures, J.P.; Combe, B. Prognostic factors for remission in early rheumatoid arthritis: A multiparameter prospective study. Ann. Rheum. Dis. 2004, 63, 675–680. [Google Scholar] [PubMed]
- Wolfe, F.; Mitchell, D.M.; Sibley, J.T.; Fries, J.F.; Bloch, D.A.; Williams, C.A.; Spitz, P.W.; Haga, M.; Kleinheksel, S.M.; Cathey, M.A. The mortality of rheumatoid arthritis. Arthritis Rheum. 1994, 37, 481–494. [Google Scholar] [PubMed]
- Feldmann, M.; Maini, R.N. Lasker Clinical Medical Research Award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat. Med. 2003, 9, 1245–1250. [Google Scholar] [CrossRef]
- Nishimoto, N.; Hashimoto, J.; Miyasaka, N.; Yamamoto, K.; Kawai, S.; Takeuchi, T.; Murata, N.; van der Heijde, D.; Kishimoto, T. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): Evidence of clinical and radiographic benefit from an X ray reader-blinded randomised controlled trial of tocilizumab. Ann. Rheum. Dis. 2007, 66, 1162–1167. [Google Scholar]
- Edwards, J.C.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [Google Scholar]
- Genovese, M.C.; Becker, J.C.; Schiff, M.; Luggen, M.; Sherrer, Y.; Kremer, J.; Birbara, C.; Box, J.; Natarajan, K.; Nuamah, I.; et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N. Engl. J. Med. 2005, 353, 1114–1123. [Google Scholar] [CrossRef]
- Feldmann, M.; Maini, R.N. Perspectives from Masters in Rheumatology and Autoimmunity: Can We Get Closer to a Cure for Rheumatoid Arthritis? Arthritis Rheumatol. 2015, 67, 2283–2291. [Google Scholar]
- Laragione, T.; Brenner, M.; Mello, A.; Symons, M.; Gulko, P.S. The arthritis severity locus Cia5d is a novel genetic regulator of the invasive properties of synovial fibroblasts. Arthritis Rheum. 2008, 58, 2296–2306. [Google Scholar] [CrossRef]
- Lee, D.M.; Kiener, H.P.; Agarwal, S.K.; Noss, E.H.; Watts, G.F.; Chisaka, O.; Takeichi, M.; Brenner, M.B. Cadherin-11 in synovial lining formation and pathology in arthritis. Science 2007, 315, 1006–1010. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnaker, E.M.; Tarner, I.H.; Robbins, P.D.; et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Seemayer, C.A.; Kuchen, S.; Neidhart, M.; Kuenzler, P.; Rihoskova, V.; Neumann, E.; Pruschy, M.; Aicher, W.K.; Muller-Ladner, U.; Gay, R.E.; et al. p53 in rheumatoid arthritis synovial fibroblasts at sites of invasion. Ann. Rheum. Dis. 2003, 62, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Seemayer, C.A.; Kuchen, S.; Kuenzler, P.; Rihoskova, V.; Rethage, J.; Aicher, W.K.; Michel, B.A.; Gay, R.E.; Kyburz, D.; Neidhart, M.; et al. Cartilage destruction mediated by synovial fibroblasts does not depend on proliferation in rheumatoid arthritis. Am. J. Pathol. 2003, 162, 1549–1557. [Google Scholar] [CrossRef]
- Laragione, T.; Brenner, M.; Li, W.; Gulko, P.S. Cia5d regulates a new fibroblast-like synoviocyte invasion-associated gene expression signature. Arthritis Res. Ther. 2008, 10, R92. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef]
- Friscic, J.; Bottcher, M.; Reinwald, C.; Bruns, H.; Wirth, B.; Popp, S.J.; Walker, K.I.; Ackermann, J.A.; Chen, X.; Turner, J.; et al. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity 2021, 54, 1002–1021.e1010. [Google Scholar] [CrossRef]
- Tolboom, T.C.; van der Helm-Van Mil, A.H.; Nelissen, R.G.; Breedveld, F.C.; Toes, R.E.; Huizinga, T.W. Invasiveness of fibroblast-like synoviocytes is an individual patient characteristic associated with the rate of joint destruction in patients with rheumatoid arthritis. Arthritis Rheum. 2005, 52, 1999–2002. [Google Scholar] [CrossRef]
- Vijaykrishnaraj, M.; Patil, P.; Ghate, S.D.; Bhandary, A.K.; Haridas, V.M.; Shetty, P. Efficacy of HDAC inhibitors and epigenetic modulation in the amelioration of synovial inflammation, cellular invasion, and bone erosion in rheumatoid arthritis pathogenesis. Int. Immunopharmacol. 2023, 122, 110644. [Google Scholar] [CrossRef]
- Laragione, T.; Cheng, K.F.; Tanner, M.R.; He, M.; Beeton, C.; Al-Abed, Y.; Gulko, P.S. The cation channel Trpv2 is a new suppressor of arthritis severity, joint damage, and synovial fibroblast invasion. Clin. Immunol. 2015, 158, 183–192. [Google Scholar] [CrossRef]
- Wang, A.Z.; Wang, J.C.; Fisher, G.W.; Diamond, H.S. Interleukin-1β-stimulated invasion of articular cartilage by rheumatoid synovial fibroblasts is inhibited by antibodies to specific integrin receptors and by collagenase inhibitors. Arthritis Rheum. 1997, 40, 1298–1307. [Google Scholar] [PubMed]
- Chang, S.K.; Noss, E.H.; Chen, M.; Gu, Z.; Townsend, K.; Grenha, R.; Leon, L.; Lee, S.Y.; Lee, D.M.; Brenner, M.B. Cadherin-11 regulates fibroblast inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 8402–8407. [Google Scholar] [CrossRef] [PubMed]
- Laragione, T.; Brenner, M.; Lahiri, A.; Gao, E.; Harris, C.; Gulko, P.S. Huntingtin-interacting protein 1 (HIP1) regulates arthritis severity and synovial fibroblast invasiveness by altering PDGFR and Rac1 signalling. Ann. Rheum. Dis. 2018, 77, 1627–1635. [Google Scholar]
- Ai, R.; Laragione, T.; Hammaker, D.; Boyle, D.L.; Wildberg, A.; Maeshima, K.; Palescandolo, E.; Krishna, V.; Pocalyko, D.; Whitaker, J.W.; et al. Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat. Commun. 2018, 9, 1921. [Google Scholar] [CrossRef]
- Hyun, T.S.; Ross, T.S. HIP1: Trafficking roles and regulation of tumorigenesis. Trends Mol. Med. 2004, 10, 194–199. [Google Scholar]
- Bradley, S.V.; Smith, M.R.; Hyun, T.S.; Lucas, P.C.; Li, L.; Antonuk, D.; Joshi, I.; Jin, F.; Ross, T.S. Aberrant Huntingtin interacting protein 1 in lymphoid malignancies. Cancer Res. 2007, 67, 8923–8931. [Google Scholar]
- Rao, D.S.; Hyun, T.S.; Kumar, P.D.; Mizukami, I.F.; Rubin, M.A.; Lucas, P.C.; Sanda, M.G.; Ross, T.S. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J. Clin. Investig. 2002, 110, 351–360. [Google Scholar] [CrossRef]
- Rao, D.S.; Bradley, S.V.; Kumar, P.D.; Hyun, T.S.; Saint-Dic, D.; Oravecz-Wilson, K.; Kleer, C.G.; Ross, T.S. Altered receptor trafficking in Huntingtin Interacting Protein 1-transformed cells. Cancer Cell 2003, 3, 471–482. [Google Scholar] [CrossRef]
- Laragione, T.; Harris, C.; Gulko, P.S. KIF1C and new Huntingtin-interacting protein 1 binding proteins regulate rheumatoid arthritis fibroblast-like synoviocytes’ phenotypes. Front. Immunol. 2024, 15, 1323410. [Google Scholar]
- Laragione TH, C.; Rice, N.; Gulko, P.S. DUSP6 deletion protects mice and reduces disease severity in autoimmune arthritis. iScience 2024, 27, 110158. [Google Scholar]
- Bradley, S.V.; Hyun, T.S.; Oravecz-Wilson, K.I.; Li, L.; Waldorff, E.I.; Ermilov, A.N.; Goldstein, S.A.; Zhang, C.X.; Drubin, D.G.; Varela, K.; et al. Degenerative phenotypes caused by the combined deficiency of murine HIP1 and HIP1r are rescued by human HIP1. Hum. Mol. Genet. 2007, 16, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Hyun, T.S.; Rao, D.S.; Saint-Dic, D.; Michael, L.E.; Kumar, P.D.; Bradley, S.V.; Mizukami, I.F.; Oravecz-Wilson, K.I.; Ross, T.S. HIP1 and HIP1r stabilize receptor tyrosine kinases and bind 3-phosphoinositides via epsin N-terminal homology domains. J. Biol. Chem. 2004, 279, 14294–14306. [Google Scholar] [CrossRef] [PubMed]
- Hyun, T.S.; Li, L.; Oravecz-Wilson, K.I.; Bradley, S.V.; Provot, M.M.; Munaco, A.J.; Mizukami, I.F.; Sun, H.; Ross, T.S. Hip1-related mutant mice grow and develop normally but have accelerated spinal abnormalities and dwarfism in the absence of HIP1. Mol. Cell Biol. 2004, 24, 4329–4340. [Google Scholar] [CrossRef]
- Yang, N.; Liang, Y.; Yang, P.; Jiang, L. Flurbiprofen inhibits cell proliferation in thyroid cancer through interrupting HIP1R-induced endocytosis of PTEN. Eur. J. Med. Res. 2022, 27, 29. [Google Scholar] [CrossRef]
- Felix, M.; Friedel, D.; Jayavelu, A.K.; Filipski, K.; Reinhardt, A.; Warnken, U.; Stichel, D.; Schrimpf, D.; Korshunov, A.; Wang, Y.; et al. HIP1R and vimentin immunohistochemistry predict 1p/19q status in IDH-mutant glioma. Neuro Oncol. 2022, 24, 2121–2132. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kumkumian, G.K.; Lafyatis, R.; Remmers, E.F.; Case, J.P.; Kim, S.J.; Wilder, R.L. Platelet-derived growth factor and IL-1 interactions in rheumatoid arthritis. Regulation of synoviocyte proliferation, prostaglandin production, and collagenase transcription. J. Immunol. 1989, 143, 833–837. [Google Scholar] [CrossRef]
- Mills, I.G.; Gaughan, L.; Robson, C.; Ross, T.; McCracken, S.; Kelly, J.; Neal, D.E. Huntingtin interacting protein 1 modulates the transcriptional activity of nuclear hormone receptors. J. Cell Biol. 2005, 170, 191–200. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Neumann, E.; Lefevre, S.; Zimmermann, B.; Gay, S.; Muller-Ladner, U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol. Med. 2010, 16, 458–468. [Google Scholar] [CrossRef]
- Han, Z.; Boyle, D.L.; Chang, L.; Bennett, B.; Karin, M.; Yang, L.; Manning, A.M.; Firestein, G.S. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Investig. 2001, 108, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhang, S.; Fang, X.; Jiang, Y.; Fang, T.; Liu, J.; Lu, K. Therapeutic Advances of Rare ALK Fusions in Non-Small Cell Lung Cancer. Curr. Oncol. 2022, 29, 7816–7831. [Google Scholar] [CrossRef] [PubMed]
- Mohler, H.; Benke, D.; Fritschy, J.M. GABA(B)-receptor isoforms molecular architecture and distribution. Life Sci. 2001, 68, 2297–2300. [Google Scholar] [PubMed]
- Tamura, S.; Watanabe, M.; Kanbara, K.; Yanagawa, T.; Watanabe, K.; Otsuki, Y.; Kinoshita, M. Expression and distribution of GABAergic system in rat knee joint synovial membrane. Histol. Histopathol. 2009, 24, 1009–1019. [Google Scholar]
- Kelley, J.M.; Hughes, L.B.; Bridges, S.L., Jr. Does γ-aminobutyric acid (GABA) influence the development of chronic inflammation in rheumatoid arthritis? J. Neuroinflamm. 2008, 5, 1. [Google Scholar] [CrossRef]
- Tian, J.; Yong, J.; Dang, H.; Kaufman, D.L. Oral GABA treatment downregulates inflammatory responses in a mouse model of rheumatoid arthritis. Autoimmunity 2011, 44, 465–470. [Google Scholar] [CrossRef]
- Bhat, R.; Axtell, R.; Mitra, A.; Miranda, M.; Lock, C.; Tsien, R.W.; Steinman, L. Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2580–2585. [Google Scholar] [CrossRef]
- Bassi, G.S.; Malvar, D.D.C.; Cunha, T.M.; Cunha, F.Q.; Kanashiro, A. Spinal GABA-B receptor modulates neutrophil recruitment to the knee joint in zymosan-induced arthritis. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 851–861. [Google Scholar] [CrossRef]
- Chiou, B.; Gao, C.; Giera, S.; Folts, C.J.; Kishore, P.; Yu, D.; Oak, H.C.; Jiang, R.; Piao, X. Cell type-specific evaluation of ADGRG1/GPR56 function in developmental central nervous system myelination. Glia 2021, 69, 413–423. [Google Scholar] [CrossRef]
- Schacke, S.; Kirkpatrick, J.; Stocksdale, A.; Bauer, R.; Hagel, C.; Riecken, L.B.; Morrison, H. Ezrin deficiency triggers glial fibrillary acidic protein upregulation and a distinct reactive astrocyte phenotype. Glia 2022, 70, 2309–2329. [Google Scholar] [CrossRef]
- Laragione, T.; Gulko, P.S. mTOR regulates the invasive properties of synovial fibroblasts in rheumatoid arthritis. Mol. Med. 2010, 16, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hei, Y.; Wang, B.; Tian, S.; Chen, X.; Zhang, J.; Wang, F. TXNDC12 inhibits pancreatic tumor cells ferroptosis by regulating GSH/GGT7 and promotes its growth and metastasis. J. Cancer 2024, 15, 3913–3929. [Google Scholar] [PubMed]
- Fan, H.N.; Chen, Z.Y.; Chen, X.Y.; Chen, M.; Yi, Y.C.; Zhu, J.S.; Zhang, J. METTL14-mediated m(6)A modification of circORC5 suppresses gastric cancer progression by regulating miR-30c-2-3p/AKT1S1 axis. Mol. Cancer 2022, 21, 51. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, Q.; Tang, Y.; Liu, Z.; Sun, G.; Lu, Z.; Chen, Y. Identification and validation of a cigarette smoke-related five-gene signature as a prognostic biomarker in kidney renal clear cell carcinoma. Sci. Rep. 2022, 12, 2189. [Google Scholar]
- Krishnan, K.; Bruce, B.; Hewitt, S.; Thomas, D.; Khanna, C.; Helman, L.J. Ezrin mediates growth and survival in Ewing’s sarcoma through the AKT/mTOR, but not the MAPK, signaling pathway. Clin. Exp. Metastasis 2006, 23, 227–236. [Google Scholar]
- Kim, M.S.; Cho, W.H.; Song, W.S.; Lee, S.Y.; Jeon, D.G. Prognostic significance of ezrin expression in pleomorphic malignant fibrous histiocytoma. Anticancer. Res. 2007, 27, 1171–1178. [Google Scholar]
- Tynninen, O.; Carpen, O.; Jaaskelainen, J.; Paavonen, T.; Paetau, A. Ezrin expression in tissue microarray of primary and recurrent gliomas. Neuropathol. Appl. Neurobiol. 2004, 30, 472–477. [Google Scholar] [CrossRef]
- Wick, W.; Grimmel, C.; Wild-Bode, C.; Platten, M.; Arpin, M.; Weller, M. Ezrin-dependent promotion of glioma cell clonogenicity, motility, and invasion mediated by BCL-2 and transforming growth factor-β2. J. Neurosci. 2001, 21, 3360–3368. [Google Scholar]
- Zhang, X.; Zhang, R.; Zheng, Y.; Shen, J.; Xiao, D.; Li, J.; Shi, X.; Huang, L.; Tang, H.; Liu, J.; et al. Expression of γ-aminobutyric acid receptors on neoplastic growth and prediction of prognosis in non-small cell lung cancer. J. Transl. Med. 2013, 11, 102. [Google Scholar] [CrossRef]
- Choi, S.; Lee, S.; Han, Y.H.; Choi, J.; Kim, I.; Lee, J.; An, H.J. miR-31-3p functions as a tumor suppressor by directly targeting GABBR2 in prostate cancer. Front. Oncol. 2022, 12, 945057. [Google Scholar] [CrossRef]
- Ji, B.; Feng, Y.; Sun, Y.; Ji, D.; Qian, W.; Zhang, Z.; Wang, Q.; Zhang, Y.; Zhang, C.; Sun, Y. GPR56 promotes proliferation of colorectal cancer cells and enhances metastasis via epithelial-mesenchymal transition through PI3K/AKT signaling activation. Oncol. Rep. 2018, 40, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, Z.; Yang, W.; Li, Z.; Xing, S.; Li, H.; Hu, B.; Li, P. Expression of orphan GPR56 correlates with tumor progression in human epithelial ovarian cancer. Neoplasma 2017, 64, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.Y.; Peng, Y.M.; Juang, H.H.; Chen, T.C.; Pan, H.L.; Chang, G.W.; Lin, H.H. GPR56/ADGRG1 Activation Promotes Melanoma Cell Migration via NTF Dissociation and CTF-Mediated Gα(12/13)/RhoA Signaling. J. Investig. Dermatol. 2017, 137, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Rodriguez-Tomas, E.; Ostman, A. Functional and clinical roles of stromal PDGF receptors in tumor biology. Cancer Metastasis Rev. 2024, 43, 1593–1609. [Google Scholar]
- Zhao, Y.; Yun, D.; Zou, X.; Jiang, T.; Li, G.; Hu, L.; Chen, J.; Xu, J.; Mao, Y.; Chen, H.; et al. Whole exome-wide association study identifies a missense variant in SLC2A4RG associated with glioblastoma risk. Am. J. Cancer Res. 2017, 7, 1937–1947. [Google Scholar]
- Charbonneau, M.; Lavoie, R.R.; Lauzier, A.; Harper, K.; McDonald, P.P.; Dubois, C.M. Platelet-Derived Growth Factor Receptor Activation Promotes the Prodestructive Invadosome-Forming Phenotype of Synoviocytes from Patients with Rheumatoid Arthritis. J. Immunol. 2016, 196, 3264–3275. [Google Scholar] [CrossRef]
- Koyama, K.; Hatsushika, K.; Ando, T.; Sakuma, M.; Wako, M.; Kato, R.; Haro, H.; Sugiyama, H.; Hamada, Y.; Ogawa, H.; et al. Imatinib mesylate both prevents and treats the arthritis induced by type II collagen antibody in mice. Mod. Rheumatol. 2007, 17, 306–310. [Google Scholar] [CrossRef]
- Rubin, K.; Terracio, L.; Ronnstrand, L.; Heldin, C.H.; Klareskog, L. Expression of platelet-derived growth factor receptors is induced on connective tissue cells during chronic synovial inflammation. Scand. J. Immunol. 1988, 27, 285–294. [Google Scholar]
- Sugiura, T.; Kamino, H.; Nariai, Y.; Murakawa, Y.; Kondo, M.; Kawakami, M.; Ikeda, N.; Uchio, Y.; Urano, T. Screening of a Panel of Low Molecular Weight Compounds That Inhibit Synovial Fibroblast Invasion in Rheumatoid Arthritis. J. Immunol. 2020, 205, 3277–3290. [Google Scholar]
- Osanai, M.; Lee, G.H. The retinoic acid-metabolizing enzyme CYP26A1 upregulates fascin and promotes the malignant behavior of breast carcinoma cells. Oncol. Rep. 2015, 34, 850–858. [Google Scholar]
- Tang, Y.; Wang, Y.; Xu, X.; Sun, H.; Tang, W. STEAP4 promoter methylation correlates with tumorigenesis of hepatocellular carcinoma. Pathol. Res. Pract. 2022, 233, 153870. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, Z.; Song, Z.; Wu, Y.; Jin, Q.; Zhao, Z. Predictive potential of STEAP family for survival, immune microenvironment and therapy response in glioma. Int. Immunopharmacol. 2021, 101, 108183. [Google Scholar] [PubMed]
- Kelwick, R.; Wagstaff, L.; Decock, J.; Roghi, C.; Cooley, L.S.; Robinson, S.D.; Arnold, H.; Gavrilovic, J.; Jaworski, D.M.; Yamamoto, K.; et al. Metalloproteinase-dependent and -independent processes contribute to inhibition of breast cancer cell migration, angiogenesis and liver metastasis by a disintegrin and metalloproteinase with thrombospondin motifs-15. Int. J. Cancer 2015, 136, E14–E26. [Google Scholar] [PubMed]
- Pap, T.; Franz, J.K.; Hummel, K.M.; Jeisy, E.; Gay, R.; Gay, S. Activation of synovial fibroblasts in rheumatoid arthritis: Lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res. 2000, 2, 59–64. [Google Scholar]
- Muller-Ladner, U.; Kriegsmann, J.; Gay, R.E.; Gay, S. Oncogenes in rheumatoid arthritis. Rheum. Dis. Clin. N. Am. 1995, 21, 675–690. [Google Scholar]
- Hsu, C.Y.; Lin, C.H.; Jan, Y.H.; Su, C.Y.; Yao, Y.C.; Cheng, H.C.; Hsu, T.I.; Wang, P.S.; Su, W.P.; Yang, C.J.; et al. Huntingtin-Interacting Protein-1 Is an Early-Stage Prognostic Biomarker of Lung Adenocarcinoma and Suppresses Metastasis via Akt-mediated Epithelial-Mesenchymal Transition. Am. J. Respir. Crit. Care Med. 2016, 193, 869–880. [Google Scholar] [CrossRef]
- Bradley, S.V.; Holland, E.C.; Liu, G.Y.; Thomas, D.; Hyun, T.S.; Ross, T.S. Huntingtin interacting protein 1 is a novel brain tumor marker that associates with epidermal growth factor receptor. Cancer Res. 2007, 67, 3609–3615. [Google Scholar]
- Abreu, J.R.; Krausz, S.; Dontje, W.; Grabiec, A.M.; de Launay, D.; Nolte, M.A.; Tak, P.P.; Reedquist, K.A. Sustained T cell Rap1 signaling is protective in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 3289–3299. [Google Scholar]
- Salinas, G.F.; Krausz, S.; Dontje, W.; Evavold, B.D.; Tak, P.P.; Baeten, D.L.; Reedquist, K.A. Sustained Rap1 activation in autoantigen-specific T lymphocytes attenuates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2012, 250, 35–43. [Google Scholar]
- Tang, S.; Chen, T.; Yu, Z.; Zhu, X.; Yang, M.; Xie, B.; Li, N.; Cao, X.; Wang, J. RasGRP3 limits Toll-like receptor-triggered inflammatory response in macrophages by activating Rap1 small GTPase. Nat. Commun. 2014, 5, 4657. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Wang, R.C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90–99. [Google Scholar] [PubMed]
- Oura, H.; Bertoncini, J.; Velasco, P.; Brown, L.F.; Carmeliet, P.; Detmar, M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood 2003, 101, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.J.; Lin, T.H.; Chiu, Y.C.; Tang, C.H.; Yang, R.S.; Fu, W.M. Enhancement of placenta growth factor expression by oncostatin M in human rheumatoid arthritis synovial fibroblasts. J. Cell Physiol. 2013, 228, 983–990. [Google Scholar] [CrossRef]
- Yoo, S.A.; Park, J.H.; Hwang, S.H.; Oh, S.M.; Lee, S.; Cicatiello, V.; Rho, S.; De Falco, S.; Hwang, D.; Cho, C.S.; et al. Placental growth factor-1 and -2 induce hyperplasia and invasiveness of primary rheumatoid synoviocytes. J. Immunol. 2015, 194, 2513–2521. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, T.; Lou, Y.; Yan, B.; Cui, S.; Jiang, L.; Han, B. Placental growth factor promotes metastases of non-small cell lung cancer through MMP9. Cell Physiol. Biochem. 2015, 37, 1210–1218. [Google Scholar]
- Taylor, A.P.; Leon, E.; Goldenberg, D.M. Placental growth factor (PlGF) enhances breast cancer cell motility by mobilising ERK1/2 phosphorylation and cytoskeletal rearrangement. Br. J. Cancer 2010, 103, 82–89. [Google Scholar]
- Snuderl, M.; Batista, A.; Kirkpatrick, N.D.; Ruiz de Almodovar, C.; Riedemann, L.; Walsh, E.C.; Anolik, R.; Huang, Y.; Martin, J.D.; Kamoun, W.; et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 2013, 152, 1065–1076. [Google Scholar]
- Wang, Y.; Sun, Q.; Ye, Y.; Sun, X.; Xie, S.; Zhan, Y.; Song, J.; Fan, X.; Zhang, B.; Yang, M.; et al. FGF-2 signaling in nasopharyngeal carcinoma modulates pericyte-macrophage crosstalk and metastasis. JCI Insight 2022, 7, e157874. [Google Scholar]
- Huang, Y.-C.; Chen, W.-C.; Yu, C.-L.; Chang, T.-K.; Wei, A.I.-C.; Chang, T.-M.; Liu, J.-F.; Wang, S.-W. FGF2 drives osteosarcoma metastasis through activating FGFR1-4 receptor pathway-mediated ICAM-1 expression. Biochem. Pharmacol. 2023, 218, 115853. [Google Scholar]
- Andres, G.; Leali, D.; Mitola, S.; Coltrini, D.; Camozzi, M.; Corsini, M.; Belleri, M.; Hirsch, E.; Schwendener, R.A.; Christofori, G.; et al. A pro-inflammatory signature mediates FGF2-induced angiogenesis. J. Cell Mol. Med. 2009, 13, 2083–2108. [Google Scholar]
- Bocelli-Tyndall, C.; Trella, E.; Frachet, A.; Zajac, P.; Pfaff, D.; Geurts, J.; Heiler, S.; Barbero, A.; Mumme, M.; Resink, T.J.; et al. FGF2 induces RANKL gene expression as well as IL1β regulated MHC class II in human bone marrow-derived mesenchymal progenitor stromal cells. Ann. Rheum. Dis. 2015, 74, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Chen, S.; Yang, D.; Cao, M.; Yao, Y.; Wu, Z.; Li, N.; Shen, N.; Li, X.; Song, X.; et al. FGF2 cooperates with IL-17 to promote autoimmune inflammation. Sci. Rep. 2017, 7, 7024. [Google Scholar] [CrossRef]
- Dong, P.; Tang, X.; Wang, J.; Zhu, B.; Li, Z. miR-653-5p suppresses the viability and migration of fibroblast-like synoviocytes by targeting FGF2 and inactivation of the Wnt/β-catenin pathway. J. Orthop. Surg. Res. 2022, 17, 5. [Google Scholar] [CrossRef]
- Kang, J.A.; Kwak, J.S.; Park, S.H.; Sim, K.Y.; Kim, S.K.; Shin, Y.; Jung, I.J.; Yang, J.I.; Chun, J.S.; Park, S.G. ZIP8 exacerbates collagen-induced arthritis by increasing pathogenic T cell responses. Exp. Mol. Med. 2021, 53, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Yan, P.; Wang, Y.; Liu, S.; He, F. Knockdown of zinc transporter ZIP8 expression inhibits neuroblastoma progression and metastasis in vitro. Mol. Med. Rep. 2018, 18, 477–485. [Google Scholar] [CrossRef]
- Xie, Z.; Lu, H.; Zheng, J.; Song, J.; Sun, K. Origin recognition complex subunit 6 (ORC6) is a key mediator of LPS-induced NFκB activation and the pro-inflammatory response. Cell Commun. Signal 2024, 22, 399. [Google Scholar] [CrossRef]
- Miagkov, A.V.; Kovalenko, D.V.; Brown, C.E.; Didsbury, J.R.; Cogswell, J.P.; Stimpson, S.A.; Baldwin, A.S.; Makarov, S.S. NF-κB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proc. Natl. Acad. Sci. USA 1998, 95, 13859–13864. [Google Scholar] [CrossRef]
- Muller-Ladner, U.; Gay, R.E.; Gay, S. Role of nuclear factor κB in synovial inflammation. Curr. Rheumatol. Rep. 2002, 4, 201–207. [Google Scholar] [CrossRef]
- Chen, H.; Bao, L.; Hu, J.; Wu, D.; Tong, X. ORC6, Negatively Regulated by miR-1-3p, Promotes Proliferation, Migration, and Invasion of Hepatocellular Carcinoma Cells. Front. Cell Dev. Biol. 2021, 9, 652292. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, D.; Chen, Y.; Zhu, H.; Liu, Z.; Yu, Z.; Xie, J. ORC6 acts as an effective prognostic predictor for non-small cell lung cancer and is closely associated with tumor progression. Oncol. Lett. 2024, 27, 96. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.W.; Zhu, X.D.; Wang, Y.Q.; Wang, X.W.; Zheng, B.S.; Chen, B.C.; Chen, Z.J. Overexpression of Dermokine-α enhances the proliferation and epithelial-mesenchymal transition of pancreatic tumor cells. Cell Signal 2022, 99, 110439. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, H.; Bhadel, S.; Ivey, M.; Conaway, M.; Spencer, A.; Hernan, R.; Holemon, H.; Gioeli, D. Identification of kinases regulating prostate cancer cell growth using an RNAi phenotypic screen. PLoS ONE 2012, 7, e38950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.; Yin, J.; Xu, R. MiR-566 mediates cell migration and invasion in colon cancer cells by direct targeting of PSKH1. Cancer Cell Int. 2019, 19, 333. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Diao, C.Y.; Huang, X.; Tan, W.X.; Chen, Y.B.; Qian, X.Y.; Gao, J.; Zhao, D.B. RhoA Promotes Synovial Proliferation and Bone Erosion in Rheumatoid Arthritis through Wnt/PCP Pathway. Mediators Inflamm. 2023, 2023, 5057009. [Google Scholar] [CrossRef]
- Sen, M.; Chamorro, M.; Reifert, J.; Corr, M.; Carson, D.A. Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 2001, 44, 772–781. [Google Scholar] [CrossRef]
- de Rooy, D.P.; Yeremenko, N.G.; Wilson, A.G.; Knevel, R.; Lindqvist, E.; Saxne, T.; Krabben, A.; Leijsma, M.K.; Daha, N.A.; Tsonaka, S.; et al. Genetic studies on components of the Wnt signalling pathway and the severity of joint destruction in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 769–775. [Google Scholar] [CrossRef]
- Sen, M.; Lauterbach, K.; El-Gabalawy, H.; Firestein, G.S.; Corr, M.; Carson, D.A. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 2791–2796. [Google Scholar] [CrossRef]
- Sen, M. Wnt signalling in rheumatoid arthritis. Rheumatology 2005, 44, 708–713. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Donor | Diagnosis | Gender | Ethnicity | Age | Disease Duration (years) | RF | Medication (s) |
|---|---|---|---|---|---|---|---|
| RA47 | RA | Female | Caucasian | 66 | 12 | + | Plaquenil (previous MTX use) |
| RA48 | RA | Male | Caucasian | 82 | 20 | + | Leflunomide (past Methotrexate) |
| RA69 | RA | M | Caucasian | 80 | 12 | − | NSAID, MTX, tofacitinib, simvastatin |
| RA70 | RA | F | Caucasian | 69 | 11 | + | Prednisone, MTX |
| RA72 | RA | F | Caucasian | 61 | 22 | + | Oxycodone, prednisone |
| RA73 | RA | M | Caucasian | 74 | 32 | + | MTX, etanercept |
| RA2354 | RA | F | NA | 67 | NA | NA | - |
| RA2383 | RA | F | NA | 51 | NA | NA | Abatacept |
| RA 2384 | RA | NA | NA | NA | NA | NA | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laragione, T.; Harris, C.; Gulko, P.S. Huntingtin-Interacting Protein 1-Related (HIP1R) Regulates Rheumatoid Arthritis Synovial Fibroblast Invasiveness. Cells 2025, 14, 483. https://doi.org/10.3390/cells14070483
Laragione T, Harris C, Gulko PS. Huntingtin-Interacting Protein 1-Related (HIP1R) Regulates Rheumatoid Arthritis Synovial Fibroblast Invasiveness. Cells. 2025; 14(7):483. https://doi.org/10.3390/cells14070483
Chicago/Turabian StyleLaragione, Teresina, Carolyn Harris, and Percio S. Gulko. 2025. "Huntingtin-Interacting Protein 1-Related (HIP1R) Regulates Rheumatoid Arthritis Synovial Fibroblast Invasiveness" Cells 14, no. 7: 483. https://doi.org/10.3390/cells14070483
APA StyleLaragione, T., Harris, C., & Gulko, P. S. (2025). Huntingtin-Interacting Protein 1-Related (HIP1R) Regulates Rheumatoid Arthritis Synovial Fibroblast Invasiveness. Cells, 14(7), 483. https://doi.org/10.3390/cells14070483