
The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
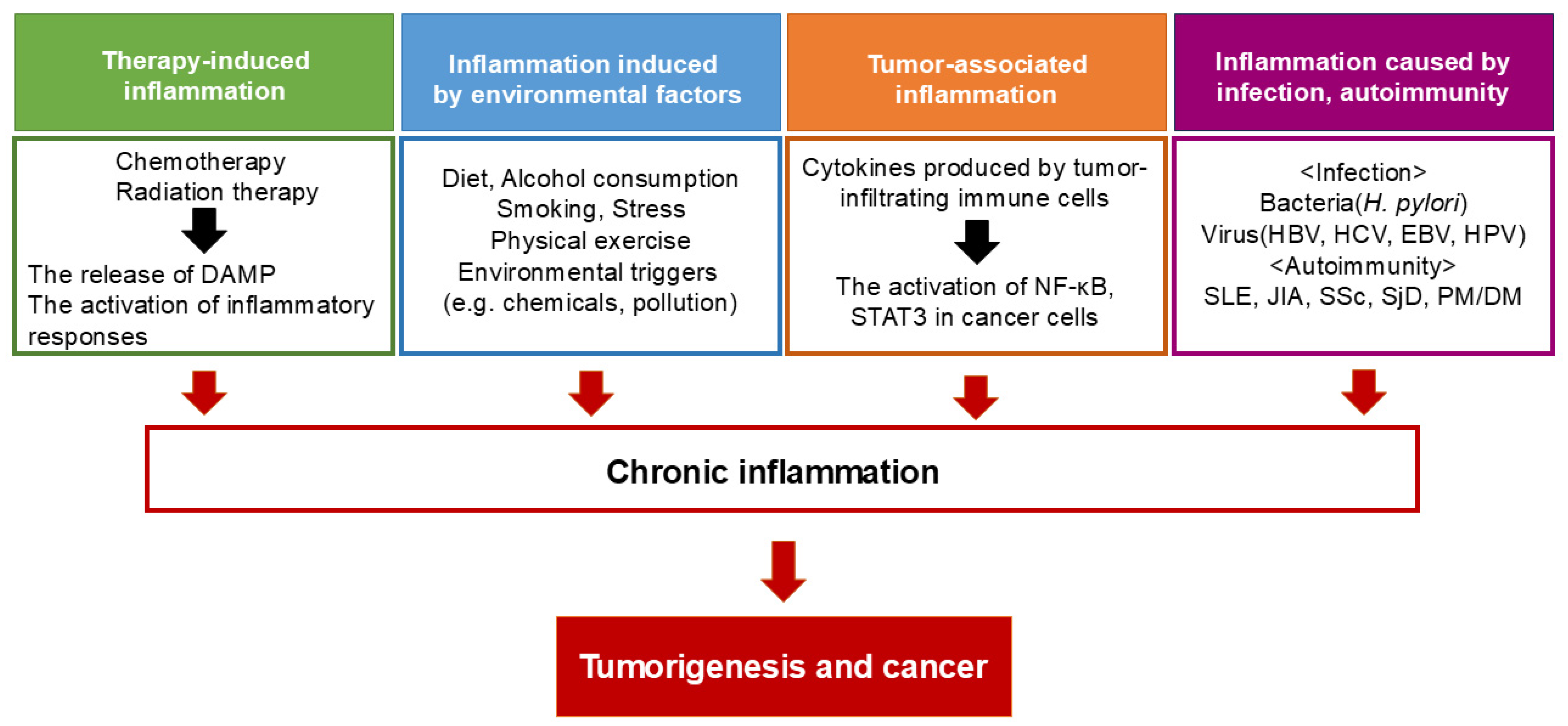
1. Introduction
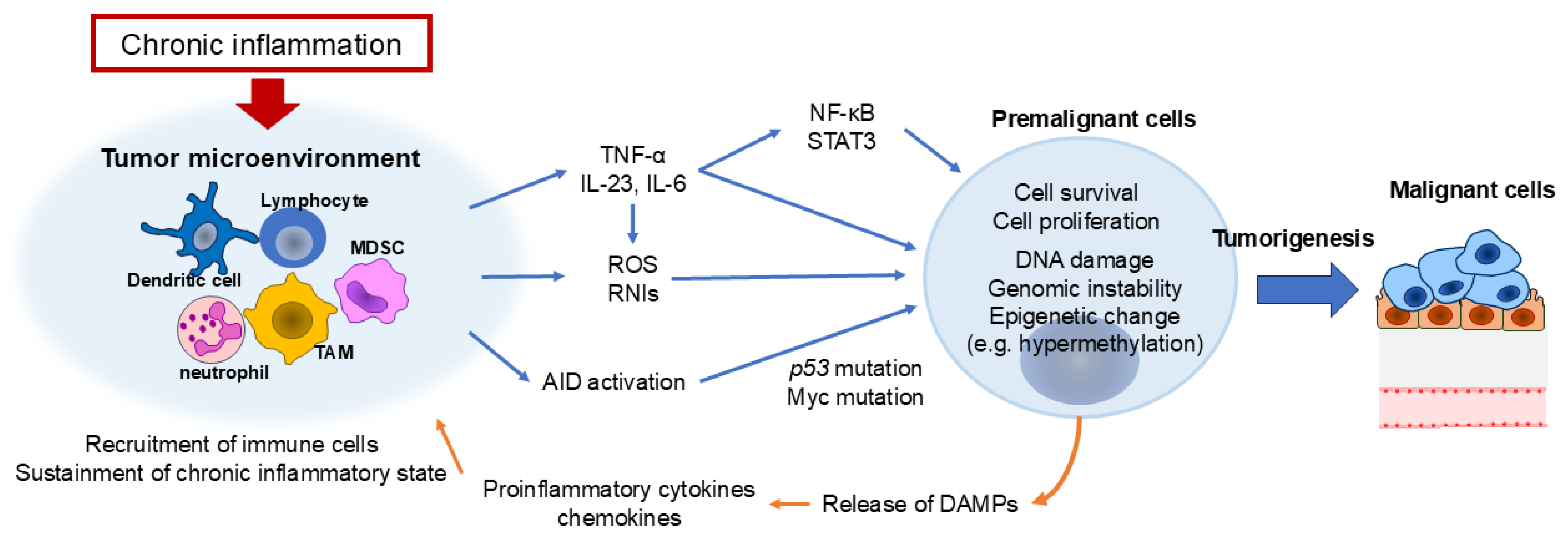
2. Inflammation and Tumor Initiation: From DNA Damage to the Tumor Microenvironment
2.1. Inflammation and Genetic Instability
2.2. Epigenetic Reprogramming and Tumor Promotion
2.3. Bidirectional Feedback Between Inflammation and DNA Damage
2.4. Role of Inflammation in Tumor Microenvironment and Metabolism
3. Inflammation as a Catalyst for Tumor Growth and Progression
3.1. Angiogenesis in Tumor Growth
3.2. Hypoxia and Immune Suppression in the Tumor Microenvironment
3.3. Immune Modulation in the Tumor Microenvironment (TME)
3.4. Metabolic Reprogramming and Therapy-Induced Inflammation
3.5. Tumor Communication and Invasion Mechanisms
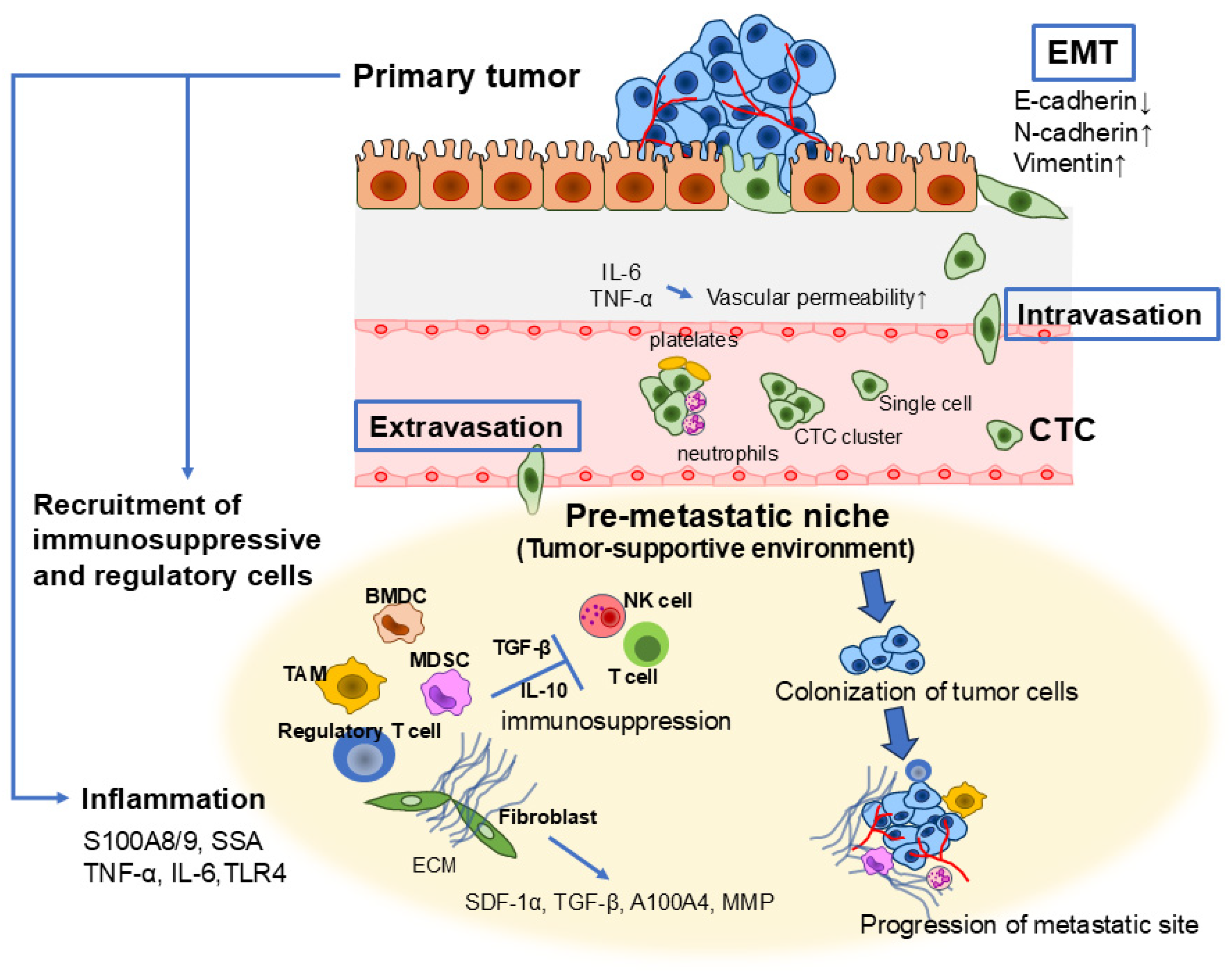
4. Inflammation as a Key Driver of Cancer Metastasis
4.1. Epithelial–Mesenchymal Transition (EMT) in Tumor Metastasis
4.2. Intravasation and Circulation of Cancer Cells
4.3. Extravasation of Cancer Cells and the Role of the Premetastatic Niche
4.4. Immune Modulation and Hypoxia in the Metastatic Niche
5. Cytokine Signaling in Tumor Promotion and Inflammation
5.1. Role of Cytokines in Tumor Promotion
5.2. Key Cytokines and Their Mechanisms
5.3. Immunosuppression and Therapeutic Implications
6. Other Factors Associated with Cancer Progression
6.1. Activin A
6.2. NRF2
7. Surgery Stress and Cancer Progression
7.1. Surgical Intervention and the Promotion of Micrometastatic and Residual Disease Growth
7.2. Metabolic Changes Following Surgery
8. Role of Anti-Inflammatory Drugs in Cancer Prevention and Treatment
8.1. Efficacy of Anti-Inflammatory Agents
8.2. Anti-Inflammatory Therapies Targeting Protumorigenic Inflammation
8.3. Emerging Therapies and the Role of STING Agonists
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic Inflammation in Cancer Development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef]
- Mantovani, A. Cancer: Inflammation by remote control. Nature 2005, 435, 752–753. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; DeLancey, J.O.; Center, M.M.; Jemal, A.; Ward, E.M. The global burden of cancer: Priorities for prevention. Carcinogenesis 2010, 31, 100–110. [Google Scholar] [CrossRef]
- Clinton, S.K.; Giovannucci, E.L.; Hursting, S.D. The World Cancer Research Fund/American Institute for Cancer Research Third Expert Report on Diet, Nutrition, Physical Activity, and Cancer: Impact and Future Directions. J. Nutr. 2020, 150, 663–671. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Wang, X.; He, J.; Pan, J. Editorial: Dual role of inflammatory mediators in cancer immunotherapy. Front. Immunol. 2023, 14, 1229355. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Derks, L.L.M.; van Boxtel, R. Stem cell mutations, associated cancer risk, and consequences for regenerative medicine. Cell Stem Cell 2023, 30, 1421–1433. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, M.; Jiang, D.; Su, Q.; Shi, J. The role of inflammation in autoimmune disease: A therapeutic target. Front. Immunol. 2023, 14, 1267091. [Google Scholar] [CrossRef]
- Morgillo, F.; Dallio, M.; Della Corte, C.M.; Gravina, A.G.; Viscardi, G.; Loguercio, C.; Ciardiello, F.; Federico, A. Carcinogenesis as a Result of Multiple Inflammatory and Oxidative Hits: A Comprehensive Review from Tumor Microenvironment to Gut Microbiota. Neoplasia 2018, 20, 721–733. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Med. Cell. Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef]
- Zhou, R.W.; Harpaz, N.; Itzkowitz, S.H.; Parsons, R.E. Molecular mechanisms in colitis-associated colorectal cancer. Oncogenesis 2023, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Ranneh, Y.; Ali, F.; Akim, A.M.; Hamid, H.A.; Khazaai, H.; Fadel, A. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: A review. Appl. Biol. Chem. 2017, 60, 327–338. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Shimizu, T.; Marusawa, H.; Endo, Y.; Chiba, T. Inflammation-mediated genomic instability: Roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012, 103, 1201–1206. [Google Scholar] [CrossRef]
- Lee, D.S.W.; Rojas, O.L.; Gommerman, J.L. B cell depletion therapies in autoimmune disease: Advances and mechanistic insights. Nat. Rev. Drug Discov. 2021, 20, 179–199. [Google Scholar] [CrossRef]
- Das, D.; Karthik, N.; Taneja, R. Crosstalk Between Inflammatory Signaling and Methylation in Cancer. Front. Cell Dev. Biol. 2021, 9, 756458. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Li, W.; Liu, Q.; Shi, J.; Xu, X.; Xu, J. The role of TNF-α in the fate regulation and functional reprogramming of mesenchymal stem cells in an inflammatory microenvironment. Front. Immunol. 2023, 14, 1074863. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Kawazoe, T.; Kasahara, H.; Itoh, S.; Ishimoto, T.; Sakata-Yanagimoto, M.; Taniguchi, K. Inflammation-Induced Tumorigenesis and Metastasis. Int. J. Mol. Sci. 2021, 22, 5421. [Google Scholar] [CrossRef]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 23, 666–681. [Google Scholar] [CrossRef]
- Chen, M.; Ye, X.; Wang, R.; Poon, K. Research progress of cancer stem cells and IL-6/STAT3 signaling pathway in esophageal adenocarcinoma. Transl. Cancer Res. 2020, 9, 363–371. [Google Scholar] [CrossRef]
- Yang, K.; Wang, X.; Zhang, H.; Wang, Z.; Nan, G.; Li, Y.; Zhang, F.; Mohammed, M.K.; Haydon, R.C.; Luu, H.H.; et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: Implications in targeted cancer therapies. Lab. Investig. 2016, 96, 116–136. [Google Scholar] [CrossRef]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammation and oncogenesis: A vicious connection. Curr. Opin. Genet. Dev. 2010, 20, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, J.M.; Sheshadri, N.; Pan, J.A.; Sun, Y.; Shi, C.; Li, J.; Powers, R.S.; Crawford, H.C.; Zong, W.X. Oncogenic Ras induces inflammatory cytokine production by upregulating the squamous cell carcinoma antigens SerpinB3/B4. Nat. Commun. 2014, 5, 3729. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Varga, J.; Wang, T.C.; Greten, F.R. The Gastrointestinal Tumor Microenvironment. Gastroenterology 2013, 145, 63–78. [Google Scholar] [CrossRef]
- Turizo-Smith, A.D.; Córdoba-Hernandez, S.; Mejía-Guarnizo, L.V.; Monroy-Camacho, P.S.; Rodríguez-García, J.A. Inflammation and cancer: Friend or foe? Front. Pharmacol. 2024, 15, 1385479. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Sgarra, R.; Battista, S.; Cerchia, L.; Manfioletti, G. The Epithelial-Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression. Int. J. Mol. Sci. 2022, 23, 800. [Google Scholar] [CrossRef]
- El Tekle, G.; Garrett, W.S. Bacteria in cancer initiation, promotion and progression. Nat. Rev. Cancer 2023, 23, 600–618. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- van Hinsbergh, V.W.; Koolwijk, P. Endothelial sprouting and angiogenesis: Matrix metalloproteinases in the lead. Cardiovasc. Res. 2008, 78, 203–212. [Google Scholar] [CrossRef]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef]
- Azzi, S.; Hebda, J.K.; Gavard, J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Schito, L. Hypoxia-Dependent Angiogenesis and Lymphangiogenesis in Cancer. Adv. Exp. Med. Biol. 2019, 1136, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef]
- Werno, C.; Menrad, H.; Weigert, A.; Dehne, N.; Goerdt, S.; Schledzewski, K.; Kzhyshkowska, J.; Brüne, B. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 2010, 31, 1863–1872. [Google Scholar] [CrossRef]
- White, J.R.; Harris, R.A.; Lee, S.R.; Craigon, M.H.; Binley, K.; Price, T.; Beard, G.L.; Mundy, C.R.; Naylor, S. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics 2004, 83, 1–8. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Mancino, A.; Schioppa, T.; Larghi, P.; Pasqualini, F.; Nebuloni, M.; Chen, I.H.; Sozzani, S.; Austyn, J.M.; Mantovani, A.; Sica, A. Divergent effects of hypoxia on dendritic cell functions. Blood 2008, 112, 3723–3734. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular Matrices and Cancer-Associated Fibroblasts: Targets for Cancer Diagnosis and Therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Siska, P.J.; Singer, K.; Evert, K.; Renner, K.; Kreutz, M. The immunological Warburg effect: Can a metabolic-tumor-stroma score (MeTS) guide cancer immunotherapy? Immunol. Rev. 2020, 295, 187–202. [Google Scholar] [CrossRef]
- Burns, J.S.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 2755. [Google Scholar] [CrossRef]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yuan, H.; Li, L.; Li, Q.; Lin, P.; Li, K. Oxidative Stress and Reprogramming of Lipid Metabolism in Cancers. Antioxidants 2025, 14, 201. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell. Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef]
- Wu, Y.; Pu, X.; Wang, X.; Xu, M. Reprogramming of lipid metabolism in the tumor microenvironment: A strategy for tumor immunotherapy. Lipids Health Dis. 2024, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, H.; Jounaidi, Y. Comprehensive snapshots of natural killer cells functions, signaling, molecular mechanisms and clinical utilization. Signal Transduct. Target. Ther. 2024, 9, 302. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Farhood, B.; Eleojo Musa, A.; Taeb, S.; Najafi, M. Damage-associated molecular patterns in tumor radiotherapy. Int. Immunopharmacol. 2020, 86, 106761. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef]
- Sato, A.; Rahman, N.I.A.; Shimizu, A.; Ogita, H. Cell-to-cell contact-mediated regulation of tumor behavior in the tumor microenvironment. Cancer Sci. 2021, 112, 4005–4012. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Van Vactor, D. The trip of the tip: Understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 2009, 10, 332–343. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Jung, E.; Osswald, M.; Blaes, J.; Wiestler, B.; Sahm, F.; Schmenger, T.; Solecki, G.; Deumelandt, K.; Kurz, F.T.; Xie, R.; et al. Tweety-Homolog 1 Drives Brain Colonization of Gliomas. J. Neurosci. 2017, 37, 6837–6850. [Google Scholar] [CrossRef] [PubMed]
- Melwani, P.K.; Pandey, B.N. Tunneling nanotubes: The intercellular conduits contributing to cancer pathogenesis and its therapy. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 189028. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Mercurio, A.M. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar] [CrossRef]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef]
- Zhu, Y.; Cheng, Y.; Guo, Y.; Chen, J.; Chen, F.; Luo, R.; Li, A. Protein kinase D2 contributes to TNF-α-induced epithelial mesenchymal transition and invasion via the PI3K/GSK-3β/β-catenin pathway in hepatocellular carcinoma. Oncotarget 2016, 7, 5327–5341. [Google Scholar] [CrossRef]
- Cohen, E.N.; Gao, H.; Anfossi, S.; Mego, M.; Reddy, N.G.; Debeb, B.; Giordano, A.; Tin, S.; Wu, Q.; Garza, R.J.; et al. Inflammation Mediated Metastasis: Immune Induced Epithelial-To-Mesenchymal Transition in Inflammatory Breast Cancer Cells. PLoS ONE 2015, 10, e0132710. [Google Scholar] [CrossRef]
- Song, J.; Feng, L.; Zhong, R.; Xia, Z.; Zhang, L.; Cui, L.; Yan, H.; Jia, X.; Zhang, Z. Icariside II inhibits the EMT of NSCLC cells in inflammatory microenvironment via down-regulation of Akt/NF-κB signaling pathway. Mol. Carcinog. 2017, 56, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Lv, N.; Gao, Y.; Guan, H.; Wu, D.; Ding, S.; Teng, W.; Shan, Z. Inflammatory mediators, tumor necrosis factor-α and interferon-γ, induce EMT in human PTC cell lines. Oncol. Lett. 2015, 10, 2591–2597. [Google Scholar] [CrossRef]
- Sun, K.H.; Sun, G.H.; Wu, Y.C.; Ko, B.J.; Hsu, H.T.; Wu, S.T. TNF-α augments CXCR2 and CXCR3 to promote progression of renal cell carcinoma. J. Cell. Mol. Med. 2016, 20, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Tebaldi, T.; Viero, G.; Spartà, A.M.; Quattrone, A.; Provenzani, A. Downregulation of HuR as a new mechanism of doxorubicin resistance in breast cancer cells. Mol. Cancer 2012, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2009, 100, 134–144. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Bourcy, M.; Lesage, J.; Leroi, N.; Syne, L.; Blacher, S.; Hubert, P.; Erpicum, C.; Foidart, J.M.; Delvenne, P.; et al. Soluble factors regulated by epithelial-mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J. Pathol. 2015, 236, 491–504. [Google Scholar] [CrossRef]
- Lim, S.; Becker, A.; Zimmer, A.; Lu, J.; Buettner, R.; Kirfel, J. SNAI1-mediated epithelial-mesenchymal transition confers chemoresistance and cellular plasticity by regulating genes involved in cell death and stem cell maintenance. PLoS ONE 2013, 8, e66558. [Google Scholar] [CrossRef]
- Chen, D.; Li, W.; Liu, S.; Su, Y.; Han, G.; Xu, C.; Liu, H.; Zheng, T.; Zhou, Y.; Mao, C. Interleukin-23 promotes the epithelial-mesenchymal transition of oesophageal carcinoma cells via the Wnt/β-catenin pathway. Sci. Rep. 2015, 5, 8604. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2017, 36, 687–699. [Google Scholar] [CrossRef]
- Jiang, Y.X.; Yang, S.W.; Li, P.A.; Luo, X.; Li, Z.Y.; Hao, Y.X.; Yu, P.W. The promotion of the transformation of quiescent gastric cancer stem cells by IL-17 and the underlying mechanisms. Oncogene 2017, 36, 1256–1264. [Google Scholar] [CrossRef]
- Elgundi, Z.; Papanicolaou, M.; Major, G.; Cox, T.R.; Melrose, J.; Whitelock, J.M.; Farrugia, B.L. Cancer Metastasis: The Role of the Extracellular Matrix and the Heparan Sulfate Proteoglycan Perlecan. Front. Oncol. 2019, 9, 1482. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Yi, M.; Li, T.; Niu, M.; Zhang, H.; Wu, Y.; Wu, K.; Dai, Z. Targeting cytokine and chemokine signaling pathways for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Bergers, G. Regulation of Blood and Lymphatic Vessels by Immune Cells in Tumors and Metastasis. Annu. Rev. Physiol. 2019, 81, 535–560. [Google Scholar] [CrossRef]
- Gu, X.; Wei, S.; Lv, X. Circulating tumor cells: From new biological insights to clinical practice. Signal Transduct. Target. Ther. 2024, 9, 226. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Leach, J.; Morton, J.P.; Sansom, O.J. Neutrophils: Homing in on the myeloid mechanisms of metastasis. Mol. Immunol. 2019, 110, 69–76. [Google Scholar] [CrossRef]
- Gaertner, F.; Massberg, S. Patrolling the vascular borders: Platelets in immunity to infection and cancer. Nat. Rev. Immunol. 2019, 19, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Stark, K.; Schubert, I.; Joshi, U.; Kilani, B.; Hoseinpour, P.; Thakur, M.; Grünauer, P.; Pfeiler, S.; Schmidergall, T.; Stockhausen, S.; et al. Distinct Pathogenesis of Pancreatic Cancer Microvesicle–Associated Venous Thrombosis Identifies New Antithrombotic Targets In Vivo. Arter. Thromb. Vasc. Biol. 2018, 38, 772–786. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y.; et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 2016, 94, 509–522. [Google Scholar] [CrossRef]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre–metastatic Niche. Cancer Res. 2016, 76, 1335–1347. [Google Scholar] [CrossRef]
- Yamamura, Y.; Asai, N.; Enomoto, A.; Kato, T.; Mii, S.; Kondo, Y.; Ushida, K.; Niimi, K.; Tsunoda, N.; Nagino, M.; et al. Akt–Girdin Signaling in Cancer-Associated Fibroblasts Contributes to Tumor Progression. Cancer Res. 2015, 75, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8–serum amyloid A3–TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Sakurai, Y.; Ishibashi, S.; Maru, Y. Imbalance of Clara cell-mediated homeostatic inflammation is involved in lung metastasis. Oncogene 2011, 30, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.T.; Forst, B.; Cremers, N.; Quagliata, L.; Ambartsumian, N.; Grum-Schwensen, B.; Klingelhöfer, J.; Abdul-Al, A.; Herrmann, P.; Osterland, M.; et al. A link between inflammation and metastasis: Serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene 2015, 34, 424–435. [Google Scholar] [CrossRef]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef]
- Yan, H.H.; Pickup, M.; Pang, Y.; Gorska, A.E.; Li, Z.; Chytil, A.; Geng, Y.; Gray, J.W.; Moses, H.L.; Yang, L. Gr-1+CD11b+ Myeloid Cells Tip the Balance of Immune Protection to Tumor Promotion in the Premetastatic Lung. Cancer Res. 2010, 70, 6139–6149. [Google Scholar] [CrossRef]
- Rutkowski, M.R.; Stephen, T.L.; Svoronos, N.; Allegrezza, M.J.; Tesone, A.J.; Perales-Puchalt, A.; Brencicova, E.; Escovar-Fadul, X.; Nguyen, J.M.; Cadungog, M.G.; et al. Microbially Driven TLR5-Dependent Signaling Governs Distal Malignant Progression through Tumor-Promoting Inflammation. Cancer Cell 2015, 27, 27–40. [Google Scholar] [CrossRef]
- Onal, S.; Turker-Burhan, M.; Bati-Ayaz, G.; Yanik, H.; Pesen-Okvur, D. Breast cancer cells and macrophages in a paracrine-juxtacrine loop. Biomaterials 2021, 267, 120412. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Huang, B.; Lang, X.; Li, X. The role of IL-6/JAK2/STAT3 signaling pathway in cancers. Front. Oncol. 2022, 12, 1023177. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Li, X.; Zhong, J.; Deng, X.; Guo, X.; Lu, Y.; Lin, J.; Huang, X.; Wang, C. Targeting Myeloid-Derived Suppressor Cells to Enhance the Antitumor Efficacy of Immune Checkpoint Blockade Therapy. Front. Immunol. 2021, 12, 754196. [Google Scholar] [CrossRef]
- Kartikasari, A.E.R.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front. Oncol. 2021, 11, 692142. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Xu, W.; Teupser, D.; auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: Role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999, 274, 33627–33636. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef]
- Elsby, R.; Kitteringham, N.R.; Goldring, C.E.; Lovatt, C.A.; Chamberlain, M.; Henderson, C.J.; Wolf, C.R.; Park, B.K. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J. Biol. Chem. 2003, 278, 22243–22249. [Google Scholar] [CrossRef]
- Rushworth, S.A.; MacEwan, D.J. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood 2008, 111, 3793–3801. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef]
- Kaur, T.; Khanduja, K.L.; Gupta, R.; Gupta, N.M.; Vaiphei, K. Changes in antioxidant defense status in response to cisplatin and 5-FU in esophageal carcinoma. Dis. Esophagus 2008, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Takahashi, Y.; Takayama, T.; Kura, T.; Katahira, T.; Sakamaki, S.; Niitsu, Y. Transfection of glutathione S-transferase (GST)-pi antisense complementary DNA increases the sensitivity of a colon cancer cell line to adriamycin, cisplatin, melphalan, and etoposide. Cancer Res. 1996, 56, 3577–3582. [Google Scholar] [PubMed]
- Cho, J.M.; Manandhar, S.; Lee, H.R.; Park, H.M.; Kwak, M.K. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: Implication to cancer cell resistance. Cancer Lett. 2008, 260, 96–108. [Google Scholar] [CrossRef]
- Aleksunes, L.M.; Slitt, A.L.; Maher, J.M.; Augustine, L.M.; Goedken, M.J.; Chan, J.Y.; Cherrington, N.J.; Klaassen, C.D.; Manautou, J.E. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol. Appl. Pharmacol. 2008, 226, 74–83. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lim, M.J.; Kim, M.H.; Yu, C.H.; Yun, Y.S.; Ahn, J.; Song, J.Y. An effective strategy for increasing the radiosensitivity of Human lung Cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radic. Biol. Med. 2012, 53, 807–816. [Google Scholar] [CrossRef]
- Singh, A.; Bodas, M.; Wakabayashi, N.; Bunz, F.; Biswal, S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox Signal. 2010, 13, 1627–1637. [Google Scholar] [CrossRef]
- Michelson, S.; Leith, J.T. Dormancy, regression, and recurrence: Towards a unifying theory of tumor growth control. J. Theor. Biol. 1994, 169, 327–338. [Google Scholar] [CrossRef]
- Horowitz, M.; Neeman, E.; Sharon, E.; Ben-Eliyahu, S. Exploiting the critical perioperative period to improve long-term cancer outcomes. Nat. Rev. Clin. Oncol. 2015, 12, 213–226. [Google Scholar] [CrossRef]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef]
- Coffey, J.C.; Smith, M.J.; Wang, J.H.; Bouchier-Hayes, D.; Cotter, T.G.; Redmond, H.P. Cancer surgery: Risks and opportunities. Bioessays 2006, 28, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Berguer, R.; Bravo, N.; Bowyer, M.; Egan, C.; Knolmayer, T.; Ferrick, D. Major surgery suppresses maximal production of helper T-cell type 1 cytokines without potentiating the release of helper T-cell type 2 cytokines. Arch. Surg. 1999, 134, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Antonio, N.; Bønnelykke-Behrndtz, M.L.; Ward, L.C.; Collin, J.; Christensen, I.J.; Steiniche, T.; Schmidt, H.; Feng, Y.; Martin, P. The wound inflammatory response exacerbates growth of pre-neoplastic cells and progression to cancer. EMBO J. 2015, 34, 2219–2236. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Chang, S.H.; Liu, C.H.; Conway, R.; Han, D.K.; Nithipatikom, K.; Trifan, O.C.; Lane, T.F.; Hla, T. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc. Natl. Acad. Sci. USA 2004, 101, 591–596. [Google Scholar] [CrossRef]
- Zhang, S.; Da, L.; Yang, X.; Feng, D.; Yin, R.; Li, M.; Zhang, Z.; Jiang, F.; Xu, L. Celecoxib potentially inhibits metastasis of lung cancer promoted by surgery in mice, via suppression of the PGE2-modulated β-catenin pathway. Toxicol. Lett. 2014, 225, 201–207. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef]
- Mao, J.T.; Smoake, J.; Park, H.K.; Lu, Q.-Y.; Xue, B. Grape Seed Procyanidin Extract Mediates Antineoplastic Effects against Lung Cancer via Modulations of Prostacyclin and 15-HETE Eicosanoid Pathways. Cancer Prev. Res. 2016, 9, 925–932. [Google Scholar] [CrossRef]
- Hager, P.; Permert, J.; Wikström, A.C.; Herrington, M.K.; Ostenson, C.G.; Strömmer, L. Preoperative glucocorticoid administration attenuates the systemic stress response and hyperglycemia after surgical trauma in the rat. Metabolism 2009, 58, 449–455. [Google Scholar] [CrossRef]
- Desborough, J.P. The stress response to trauma and surgery. Br. J. Anaesth. 2000, 85, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Dungan, K.M.; Braithwaite, S.S.; Preiser, J.-C. Stress hyperglycaemia. Lancet 2009, 373, 1798–1807. [Google Scholar] [CrossRef]
- Hou, Y.; Zhou, M.; Xie, J.; Chao, P.; Feng, Q.; Wu, J. High glucose levels promote the proliferation of breast cancer cells through GTPases. Breast Cancer 2017, 9, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, W.; Li, J.; Liu, M.; Wei, M. Resveratrol suppresses the STAT3 signaling pathway and inhibits proliferation of high glucose-exposed HepG2 cells partly through SIRT1. Oncol. Rep. 2013, 30, 2820–2828. [Google Scholar] [CrossRef]
- Cao, L.; Chen, X.; Xiao, X.; Ma, Q.; Li, W. Resveratrol inhibits hyperglycemia-driven ROS-induced invasion and migration of pancreatic cancer cells via suppression of the ERK and p38 MAPK signaling pathways. Int. J. Oncol. 2016, 49, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; D’Orazi, G. High glucose dephosphorylates serine 46 and inhibits p53 apoptotic activity. J. Exp. Clin. Cancer Res. 2014, 33, 79. [Google Scholar] [CrossRef]
- Biernacka, K.M.; Uzoh, C.C.; Zeng, L.; Persad, R.A.; Bahl, A.; Gillatt, D.; Perks, C.M.; Holly, J.M. Hyperglycaemia-induced chemoresistance of prostate cancer cells due to IGFBP2. Endocr. Relat. Cancer 2013, 20, 741–751. [Google Scholar] [CrossRef]
- Sato, K.; Hikita, H.; Myojin, Y.; Fukumoto, K.; Murai, K.; Sakane, S.; Tamura, T.; Yamai, T.; Nozaki, Y.; Yoshioka, T.; et al. Hyperglycemia enhances pancreatic cancer progression accompanied by elevations in phosphorylated STAT3 and MYC levels. PLoS ONE 2020, 15, e0235573. [Google Scholar] [CrossRef]
- Webster, K.A. Stress hyperglycemia and enhanced sensitivity to myocardial infarction. Curr. Hypertens. Rep. 2008, 10, 78–84. [Google Scholar] [CrossRef]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Miret Durazo, C.I.; Zachariah Saji, S.; Rawat, A.; Motiño Villanueva, A.L.; Bhandari, A.; Nurjanah, T.; Ryali, N.; Zepeda Martínez, I.G.; Cruz Santiago, J.A. Exploring Aspirin’s Potential in Cancer Prevention: A Comprehensive Review of the Current Evidence. Cureus 2024, 16, e70005. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.-G.; Ma, W.; Drew, D.A.; Cao, Y.; Nguyen, L.H.; Joshi, A.D.; Ng, K.; Ogino, S.; Meyerhardt, J.A.; Song, M.; et al. Aspirin Use and Risk of Colorectal Cancer Among Older Adults. JAMA Oncol. 2021, 7, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Elwood, P.; Morgan, G.; Watkins, J.; Protty, M.; Mason, M.; Adams, R.; Dolwani, S.; Pickering, J.; Delon, C.; Longley, M. Aspirin and cancer treatment: Systematic reviews and meta-analyses of evidence: For and against. Br. J. Cancer 2024, 130, 3–8. [Google Scholar] [CrossRef]
- Baker, A.; Kartsonaki, C. Aspirin Use and Survival Among Patients With Breast Cancer: A Systematic Review and Meta-Analysis. Oncologist 2023, 29, e1–e14. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Ballman, K.V.; Partridge, A.H.; Hahn, O.M.; Briccetti, F.M.; Irvin, W.J.; Symington, B.; Visvanathan, K.; Pohlmann, P.R.; Openshaw, T.H.; et al. Aspirin vs Placebo as Adjuvant Therapy for Breast Cancer: The Alliance A011502 Randomized Trial. JAMA 2024, 331, 1714–1721. [Google Scholar] [CrossRef]
- Drew, D.A.; Chan, A.T. Aspirin in the Prevention of Colorectal Neoplasia. Annu. Rev. Med. 2021, 72, 415–430. [Google Scholar] [CrossRef]
- Hou, J.; Karin, M.; Sun, B. Targeting cancer-promoting inflammation—Have anti-inflammatory therapies come of age? Nat. Rev. Clin. Oncol. 2021, 18, 261–279. [Google Scholar] [CrossRef]
- Brown, M.; Cohen, J.; Arun, P.; Chen, Z.; Van Waes, C. NF-kappaB in carcinoma therapy and prevention. Expert. Opin. Ther. Targets 2008, 12, 1109–1122. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez-Rodriguez, A.P.; Martínez-Pérez, A.; Rodrigo, J.P.; García-Pedrero, J.M.; Gonzalez, S. Chemo-Immunotherapy: A New Trend in Cancer Treatment. Cancers 2023, 15, 2912. [Google Scholar] [CrossRef]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra252. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, A.; Andoh, A. The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis. Cells 2025, 14, 488. https://doi.org/10.3390/cells14070488
Nishida A, Andoh A. The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis. Cells. 2025; 14(7):488. https://doi.org/10.3390/cells14070488
Chicago/Turabian StyleNishida, Atsushi, and Akira Andoh. 2025. "The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis" Cells 14, no. 7: 488. https://doi.org/10.3390/cells14070488
APA StyleNishida, A., & Andoh, A. (2025). The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis. Cells, 14(7), 488. https://doi.org/10.3390/cells14070488