
Preclinical Models for Studying Fuchs Endothelial Corneal Dystrophy

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Cornea Anatomy and Functions
2.1. Stroma
2.2. Descemet’s Membrane
2.3. Endothelial Layer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Name | Prime Characteristic | Cellular/Acellular | Main Function | Developmental Origin | Ref. |
|---|---|---|---|---|---|
| Epithelium | Formed in three distinct cell layers: superficial cells, wing cells, and basal cells | cellular | Protect against pathogens and toxins, and frontal barrier | Derived from surface ectoderm at 5–6 weeks during development | [50] |
| Bowman’s layer | An acellular layer made up of collagen fibrils | acellular | Provides structural support to the corneal epithelium | Becomes noticeable at 13 and 19 weeks of embryo development | [50,53] |
| Stroma | The bulk of the cornea structure, highly organized | cellular | Provides corneal transparency and mechanical support | During the second wave of neural crest migration (at seven weeks), after primitive endothelium has formed | [50] |
| Descemet’s membrane (DM) | Structurally formed by the anterior banded layer (ABL) and the posterior non-banded layer (PNBL) | acellular | Supports the corneal endothelium structure | The anterior layer was formed before birth at the 8-week stage. Endothelial cells continuously secrete Descemet’s membrane before and after birth | [50] |
| Endothelium (CE) | A monolayer of cells hexagonal in shape that span along the DM | cellular | Keep the clarity of the entire cornea by maintaining the corneal hydration dynamically via a “pump-leak” process | Derived from Neural crest monolayer of cells | [50] |
2.4. Abnormal Physiology—FECD
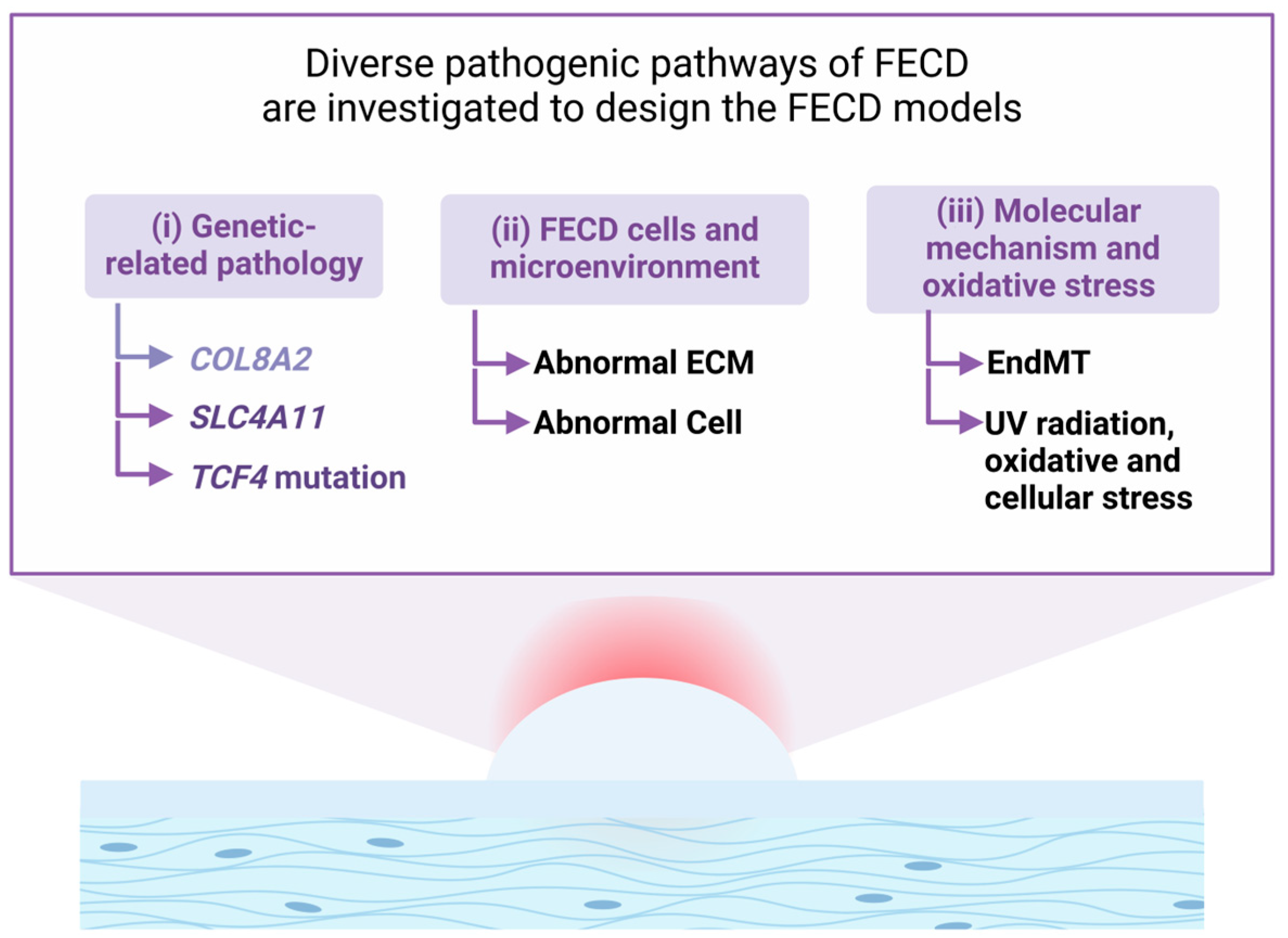
3. FECD Disease Pathology
3.1. Genetic-Related FECD Pathology
3.1.1. COL8A2
3.1.2. SLC4A11
3.1.3. TCF4 Mutation
3.2. FECD Cells and Microenvironment
3.2.1. Abnormal ECM (Guttata)
3.2.2. Abnormal Cell
Endothelial-to-Mesenchymal Transition (EndMT)
Cell Senescence
3.2.3. Oxidative Stress and EndMT
4. Models Used to Study FECD
4.1. In Vitro Models
4.1.1. Primary Human CEnC Models
Primary Human FECD CEnCs
4.1.2. Immortalized Human CEnCs
Immortalized CEnC Lines from Primary HCEnCs
Immortalized FECD Cell Line
4.1.3. Induced Pluripotent Stem Cells—Derived CEnCs
4.1.4. Primary HCEnCs on Biomimicking Synthetic Guttata
4.1.5. Animal Cell Culture Models
4.2. Ex Vivo Corneal Tissues
4.2.1. Ex Vivo Corneal Tissue
Human Ex Vivo Corneal Tissue
Ex Vivo Decellularized Tissue
4.2.2. Animal Ex Vivo Corneal Tissue
4.3. In Vivo Animal Models
4.3.1. Murine Models
4.3.2. Rabbit Models
4.3.3. Feline Models
4.3.4. Primate Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FECD | Fuchs Endothelial Corneal Dystrophy |
| CEnCs | corneal endothelial cells |
| CE | corneal endothelium |
| ECM | extracellular matrix |
| COL8A2 | collagen VIII α2-subunit chain |
| TCF4 | transcription factor 4 |
| DM | Descemet’s Membrane |
| ABL | anterior banded layer |
| PNBL | posterior non-banded layer |
| EC | endothelial cells |
| SLC4A11 | Solute carrier family 4-member 11 bicarbonate transport protein |
| EndMT | endothelial-mesenchymal transition |
| TGF-β | transforming growth factor beta |
| ZEB1 | E-box-binding homeobox 1 |
| LOXHD1 | lipoxygenase homology domains 1 |
| AGBL1 | ATP/GTP binding protein-like 1 |
| ER | endoplasmic reticulum |
| UPR | unfolded protein response |
| CTG | cytosine–thymine–guanine |
| TNR | trinucleotide repeat |
| E2-2 | E-protein family |
| MBNL1 | Muscleblind-like 1 protein |
| mRNA | messenger RNA |
| CHED2 | congenital hereditary endothelial corneal dystrophy type 2 |
| ROS | reactive oxygen species |
| EMT | epithelial-to-mesenchymal transition |
| UVA | ultraviolet A |
| CDKN1a | cyclin-dependent kinase inhibitor 1A protein |
| PRDX2 | Peroxiredoxin 2 |
| tBHP | tert-Butyl hydroperoxide |
| SOD2 | superoxide dismutase 2 |
| HCEnCs | human CEnCs |
| mtDNA | mitochondrial DNA |
| NCDP | neural crest-derived progenitor |
| iPSC | induced pluripotent stem cells |
| HPV | Human papillomavirus |
| hTERT | human Telomerase Reverse Transcriptase |
| MN | menadione |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| PDRN | polydeoxyribonucleotide |
| iFECDCs | Immortalized FECD cell lines |
| iHCEnCs | immortalization of CEnCs from healthy donors |
| Mfn2 | Mitofusin 2 |
| ATM | Ataxia-Telangiectasia Mutated |
| PI3K | phosphatidyl inositol 3-kinase |
| dCas9 | CRISPR-dead Cas9 |
| GelMA | gelatin methacrylate |
| BCE | bovine corneal endothelial |
| nDNA | nuclear DNA |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| SFN | sulforaphane |
| GFP | green fluorescent protein |
| LNP | lipid nanoparticles |
| DSO | Descemet Stripping Only |
| PPCD | posterior polymorphous corneal dystrophy |
| CYP1B1 | cytochrome P450 1B1 |
References
- Zhang, J.; McGhee, C.N.J.; Patel, D.V. The Molecular Basis of Fuchs’ Endothelial Corneal Dystrophy. Mol. Diagn. Ther. 2019, 23, 97–112. [Google Scholar] [CrossRef]
- Ong Tone, S.; Kocaba, V.; Bohm, M.; Wylegala, A.; White, T.L.; Jurkunas, U.V. Fuchs endothelial corneal dystrophy: The vicious cycle of Fuchs pathogenesis. Prog. Retin. Eye Res. 2021, 80, 100863. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetti, D.W.; Uotila, M.H.; Parikh, N.; Kaufman, H.E. Central cornea guttata. Incidence in the general population. Am. J. Ophthalmol. 1967, 64, 1155–1158. [Google Scholar]
- Zoega, G.M.; Fujisawa, A.; Sasaki, H.; Kubota, A.; Sasaki, K.; Kitagawa, K.; Jonasson, F. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology 2006, 113, 565–569. [Google Scholar] [CrossRef]
- Higa, A.; Sakai, H.; Sawaguchi, S.; Iwase, A.; Tomidokoro, A.; Amano, S.; Araie, M. Prevalence of and risk factors for cornea guttata in a population-based study in a southwestern island of Japan: The Kumejima study. Arch. Ophthalmol. 2011, 129, 332–336. [Google Scholar] [CrossRef]
- Kitagawa, K.; Kojima, M.; Sasaki, H.; Shui, Y.B.; Chew, S.J.; Cheng, H.M.; Ono, M.; Morikawa, Y.; Sasaki, K. Prevalence of primary cornea guttata and morphology of corneal endothelium in aging Japanese and Singaporean subjects. Ophthalmic Res. 2002, 34, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Iliff, B.W.; Riazuddin, S.A.; Gottsch, J.D. The genetics of Fuchs’ corneal dystrophy. Expert Rev. Ophthalmol. 2012, 7, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Klintworth, G.K. The molecular genetics of the corneal dystrophies—current status. Front. Biosci. 2003, 8, d687–d713. [Google Scholar] [CrossRef]
- Van Meter, W.; Mathews, P.; Philippy, B.; DeMatteo, J.; Eye Bank Association of America. 2023 Eye Banking Statistical Report—Executive Summary. Eye Bank. Corneal Transplant. 2024, 3, e0034. [Google Scholar] [CrossRef]
- Sarnicola, C.; Farooq, A.V.; Colby, K. Fuchs Endothelial Corneal Dystrophy: Update on Pathogenesis and Future Directions. Eye Contact Lens Sci. Clin. Pract. 2019, 45, 1–10. [Google Scholar] [CrossRef]
- Jun, A.S.; Meng, H.; Ramanan, N.; Matthaei, M.; Chakravarti, S.; Bonshek, R.; Black, G.C.; Grebe, R.; Kimos, M. An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis. Hum. Mol. Genet. 2012, 21, 384–393. [Google Scholar] [CrossRef] [PubMed]
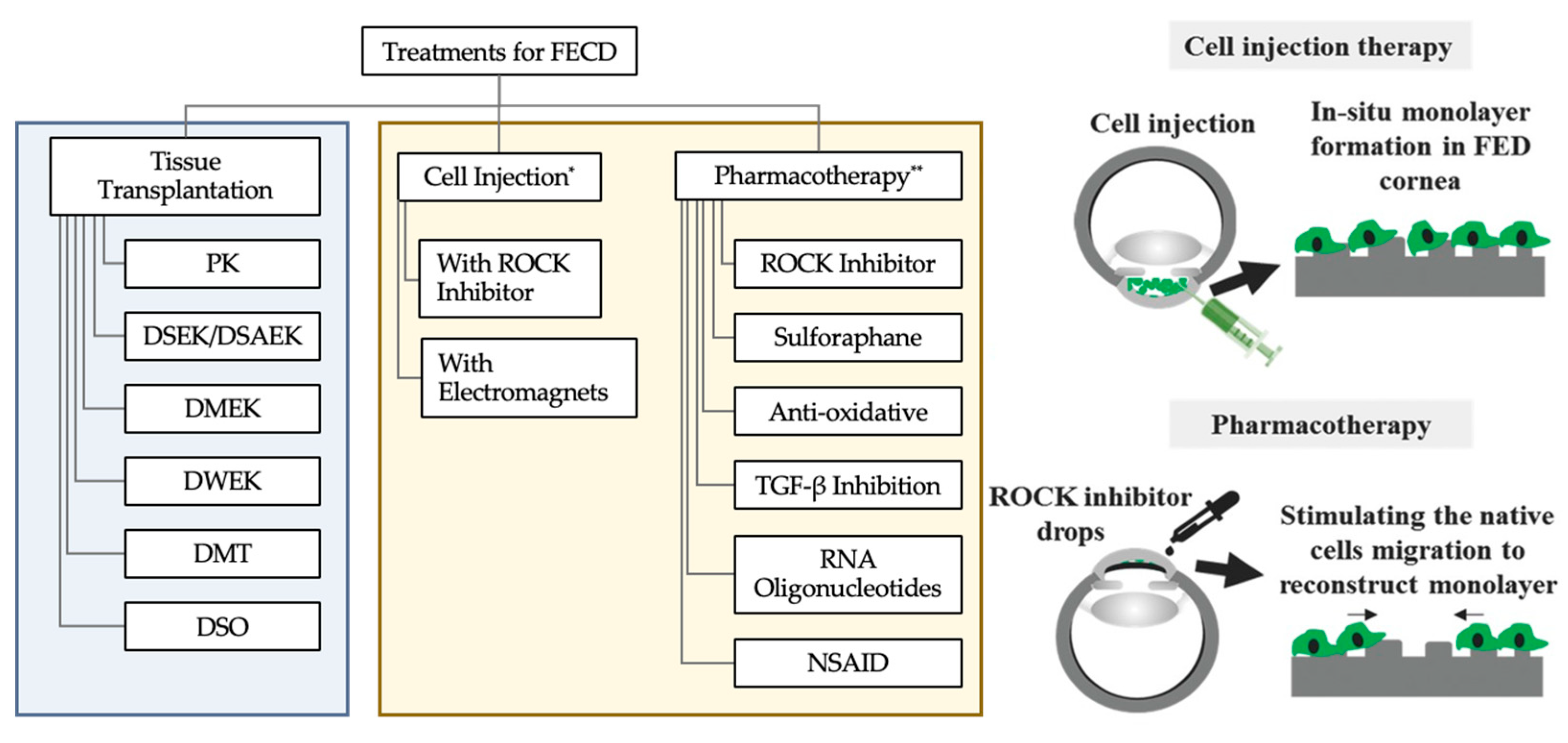
- Okumura, N.; Kinoshita, S.; Koizumi, N. Cell-based approach for treatment of corneal endothelial dysfunction. Cornea 2014, 33 (Suppl. S11), S37–S41. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, M.; Lwin, C.N.; Seah, X.Y.; Peh, G.; Mehta, J.S. Allogeneic Descemet’s Membrane Transplantation Enhances Corneal Endothelial Monolayer Formation and Restores Functional Integrity Following Descemet’s Stripping. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4249–4260. [Google Scholar] [CrossRef]
- Soh, Y.Q.; Mehta, J.S. Regenerative Therapy for Fuchs Endothelial Corneal Dystrophy. Cornea 2018, 37, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Kinoshita, S.; Koizumi, N. Application of Rho Kinase Inhibitors for the Treatment of Corneal Endothelial Diseases. J. Ophthalmol. 2017, 2017, 2646904. [Google Scholar] [CrossRef]
- Bostan, C.; Theriault, M.; Forget, K.J.; Doyon, C.; Cameron, J.D.; Proulx, S.; Brunette, I. In Vivo Functionality of a Corneal Endothelium Transplanted by Cell-Injection Therapy in a Feline Model. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1620–1634. [Google Scholar] [CrossRef]
- Kinoshita, S.; Koizumi, N.; Ueno, M.; Okumura, N.; Imai, K.; Tanaka, H.; Yamamoto, Y.; Nakamura, T.; Inatomi, T.; Bush, J.; et al. Injection of Cultured Cells with a ROCK Inhibitor for Bullous Keratopathy. N. Engl. J. Med. 2018, 378, 995–1003. [Google Scholar] [CrossRef]
- Haydari, M.N.; Perron, M.C.; Laprise, S.; Roy, O.; Cameron, J.D.; Proulx, S.; Brunette, I. A short-term in vivo experimental model for Fuchs endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6343–6354. [Google Scholar] [CrossRef]
- Okumura, N.; Ueno, M.; Koizumi, N.; Sakamoto, Y.; Hirata, K.; Hamuro, J.; Kinoshita, S. Enhancement on primate corneal endothelial cell survival in vitro by a ROCK inhibitor. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3680–3687. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef]
- Okumura, N.; Koizumi, N.; Ueno, M.; Sakamoto, Y.; Takahashi, H.; Hirata, K.; Torii, R.; Hamuro, J.; Kinoshita, S. Enhancement of corneal endothelium wound healing by Rho-associated kinase (ROCK) inhibitor eye drops. Br. J. Ophthalmol. 2011, 95, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Koizumi, N.; Ueno, M.; Sakamoto, Y.; Takahashi, H.; Tsuchiya, H.; Hamuro, J.; Kinoshita, S. ROCK inhibitor converts corneal endothelial cells into a phenotype capable of regenerating in vivo endothelial tissue. Am. J. Pathol. 2012, 181, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Koizumi, N.; Kay, E.P.; Ueno, M.; Sakamoto, Y.; Nakamura, S.; Hamuro, J.; Kinoshita, S. The ROCK inhibitor eye drop accelerates corneal endothelium wound healing. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2493–2502. [Google Scholar] [CrossRef]
- Okumura, N.; Inoue, R.; Okazaki, Y.; Nakano, S.; Nakagawa, H.; Kinoshita, S.; Koizumi, N. Effect of the Rho Kinase Inhibitor Y-27632 on Corneal Endothelial Wound Healing. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6067–6074. [Google Scholar] [CrossRef]
- Okumura, N.; Sakamoto, Y.; Fujii, K.; Kitano, J.; Nakano, S.; Tsujimoto, Y.; Nakamura, S.; Ueno, M.; Hagiya, M.; Hamuro, J.; et al. Rho kinase inhibitor enables cell-based therapy for corneal endothelial dysfunction. Sci. Rep. 2016, 6, 26113. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, N.; Okumura, N.; Ueno, M.; Kinoshita, S. New therapeutic modality for corneal endothelial disease using Rho-associated kinase inhibitor eye drops. Cornea 2014, 33 (Suppl. S11), S25–S31. [Google Scholar] [CrossRef]
- Guerra, F.P.; Anshu, A.; Price, M.O.; Giebel, A.W.; Price, F.W. Descemet’s membrane endothelial keratoplasty: Prospective study of 1-year visual outcomes, graft survival, and endothelial cell loss. Ophthalmology 2011, 118, 2368–2373. [Google Scholar] [CrossRef]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef]
- Alka, K.; Casey, J.R. Molecular phenotype of SLC4A11 missense mutants: Setting the stage for personalized medicine in corneal dystrophies. Hum. Mutat. 2018, 39, 676–690. [Google Scholar] [CrossRef]
- Lopez, I.A.; Rosenblatt, M.I.; Kim, C.; Galbraith, G.C.; Jones, S.M.; Kao, L.; Newman, D.; Liu, W.; Yeh, S.; Pushkin, A.; et al. Slc4a11 gene disruption in mice: Cellular targets of sensorineuronal abnormalities. J. Biol. Chem. 2009, 284, 26882–26896. [Google Scholar] [CrossRef]
- Mootha, V.V.; Hussain, I.; Cunnusamy, K.; Graham, E.; Gong, X.; Neelam, S.; Xing, C.; Kittler, R.; Petroll, W.M. TCF4 Triplet Repeat Expansion and Nuclear RNA Foci in Fuchs’ Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2003–2011. [Google Scholar] [CrossRef]
- Eye Bank Association of America. Eye Banking Statistical Report; Eye Bank Association of America: Washington, DC, USA, 2023. [Google Scholar]
- Foroutan, A.; Tabatabaei, S.A.; Behrouz, M.J.; Zarei, R.; Soleimani, M. Spontaneous wound dehiscence after penetrating keratoplasty. Int. J. Ophthalmol. 2014, 7, 905–908. [Google Scholar] [CrossRef]
- Price, M.O.; Price, F.W., Jr. Endothelial keratoplasty—A review. Clin. Exp. Ophthalmol. 2010, 38, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.J.; Kane, S.; Dhaliwal, D.K. Descemetorhexis Without Endothelial Keratoplasty Versus DMEK for Treatment of Fuchs Endothelial Corneal Dystrophy. Cornea 2018, 37, 1479–1483. [Google Scholar] [CrossRef]
- Peh, G.S.L.; Ong, H.S.; Adnan, K.; Ang, H.P.; Lwin, C.N.; Seah, X.Y.; Lin, S.J.; Mehta, J.S. Functional Evaluation of Two Corneal Endothelial Cell-Based Therapies: Tissue-Engineered Construct and Cell Injection. Sci. Rep. 2019, 9, 6087. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, A.; Schmedt, T.; Chen, Y.; Jurkunas, U.V. Sulforaphane decreases endothelial cell apoptosis in fuchs endothelial corneal dystrophy: A novel treatment. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6724–6734. [Google Scholar] [CrossRef] [PubMed]
- Halilovic, A.; Schmedt, T.; Benischke, A.S.; Hamill, C.; Chen, Y.; Santos, J.H.; Jurkunas, U.V. Menadione-Induced DNA Damage Leads to Mitochondrial Dysfunction and Fragmentation During Rosette Formation in Fuchs Endothelial Corneal Dystrophy. Antioxid. Redox Signal. 2016, 24, 1072–1083. [Google Scholar] [CrossRef]
- Kim, E.C.; Meng, H.; Jun, A.S. N-Acetylcysteine increases corneal endothelial cell survival in a mouse model of Fuchs endothelial corneal dystrophy. Exp. Eye Res. 2014, 127, 20–25. [Google Scholar] [CrossRef]
- Kim, E.C.; Meng, H.; Jun, A.S. Lithium treatment increases endothelial cell survival and autophagy in a mouse model of Fuchs endothelial corneal dystrophy. Br. J. Ophthalmol. 2013, 97, 1068–1073. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Sanchez-Pintado, B.; Hafford Tear, N.J.; Klein, P.; Liskova, P.; Dulla, K.; Semo, M.; Vugler, A.A.; Muthusamy, K.; Dudakova, L.; et al. Antisense Therapy for a Common Corneal Dystrophy Ameliorates TCF4 Repeat Expansion-Mediated Toxicity. Am. J. Hum. Genet. 2018, 102, 528–539. [Google Scholar] [CrossRef]
- Soh, Y.Q.; Kocaba, V.; Pinto, M.; Mehta, J.S. Fuchs endothelial corneal dystrophy and corneal endothelial diseases: East meets West. Eye 2020, 34, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Parekh, M.; Miall, A.; Chou, A.; Buhl, L.; Deshpande, N.; Price, M.O.; Price, F.W.; Jurkunas, U.V. Enhanced Migration of Fuchs Corneal Endothelial Cells by Rho Kinase Inhibition: A Novel Ex Vivo Descemet’s Stripping Only Model. Cells 2024, 13, 1218. [Google Scholar] [CrossRef]
- Katikireddy, K.R.; White, T.L.; Miyajima, T.; Vasanth, S.; Raoof, D.; Chen, Y.; Price, M.O.; Price, F.W.; Jurkunas, V.U. NQO1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in Fuchs endothelial corneal dystrophy. Free. Radic. Biol. Med. 2018, 116, 19–30. [Google Scholar]
- Okumura, N.; Hashimoto, K.; Kitahara, M.; Okuda, H.; Ueda, E.; Watanabe, K.; Nakahara, M.; Sato, T.; Kinoshita, S.; Tourtas, T.; et al. Activation of TGF-beta signaling induces cell death via the unfolded protein response in Fuchs endothelial corneal dystrophy. Sci. Rep. 2017, 7, 6801. [Google Scholar] [CrossRef]
- Kim, E.C.; Toyono, T.; Berlinicke, C.A.; Zack, D.J.; Jurkunas, U.; Usui, T.; Jun, A.S. Screening and Characterization of Drugs That Protect Corneal Endothelial Cells Against Unfolded Protein Response and Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2017, 58, 892–900. [Google Scholar] [CrossRef]
- Xia, X.; Atkins, M.; Dalal, R.; Kuzmenko, O.; Chang, K.-C.; Sun, C.B.; Benatti, C.A.; Rak, D.J.; Nahmou, M.; Kunzevitzky, N.J.; et al. Magnetic Human Corneal Endothelial Cell Transplant: Delivery, Retention, and Short-Term Efficacy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2438–2448. [Google Scholar] [CrossRef]
- Rizwan, M.; Peh, G.S.; Adnan, K.; Naso, S.L.; Mendez, A.R.; Mehta, J.S.; Yim, E.K.F. In Vitro Topographical Model of Fuchs Dystrophy for Evaluation of Corneal Endothelial Cell Monolayer Formation. Adv. Healthc. Mater. 2016, 5, 2896–2910. [Google Scholar] [CrossRef]
- Sridhar, M.S. Anatomy of cornea and ocular surface. Indian J. Ophthalmol. 2018, 66, 190–194. [Google Scholar] [CrossRef]
- DelMonte, D.W.; Kim, T. Anatomy and physiology of the cornea. J. Cataract. Refract. Surg. 2011, 37, 588–598. [Google Scholar] [CrossRef]
- Eghrari, A.O.; Riazuddin, S.A.; Gottsch, J.D. Overview of the Cornea: Structure, Function, and Development. Prog. Mol. Biol. Transl. Sci. 2015, 134, 7–23. [Google Scholar] [CrossRef]
- Yee, R.W.; Matsuda, M.; Schultz, R.O.; Edelhauser, H.F. Changes in the normal corneal endothelial cellular pattern as a function of age. Curr. Eye Res. 1985, 4, 671–678. [Google Scholar] [CrossRef]
- Tisdale, A.; Spurr-Michaud, S.; Rodrigues, M.; Hackett, J.; Krachmer, J.; Gipson, I.K. Development of the anchoring structures of the epithelium in rabbit and human fetal corneas. Investig. Ophthalmol. Vis. Sci. 1988, 29, 727–736. [Google Scholar]
- Eghrari, A.O.; Riazuddin, S.A.; Gottsch, J.D. Fuchs Corneal Dystrophy. Prog. Mol. Biol. Transl. Sci. 2015, 134, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Borderie, V.M.; Baudrimont, M.; Vallee, A.; Ereau, T.L.; Gray, F.; Laroche, L. Corneal endothelial cell apoptosis in patients with Fuchs’ dystrophy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2501–2505. [Google Scholar]
- Louttit, M.D.; Kopplin, L.J.; Igo, R.P., Jr.; Fondran, J.R.; Tagliaferri, A.; Bardenstein, D.; Aldave, A.J.; Croasdale, C.R.; Price, M.O.; Rosenwasser, G.O.; et al. A multicenter study to map genes for Fuchs endothelial corneal dystrophy: Baseline characteristics and heritability. Cornea 2012, 31, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, M.; Hribek, A.; Clahsen, T.; Bachmann, B.; Cursiefen, C.; Jun, A.S. Fuchs Endothelial Corneal Dystrophy: Clinical, Genetic, Pathophysiologic, and Therapeutic Aspects. Annu. Rev. Vis. Sci. 2019, 5, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Elhalis, H.; Azizi, B.; Jurkunas, U.V. Fuchs endothelial corneal dystrophy. Ocul. Surf. 2010, 8, 173–184. [Google Scholar] [CrossRef]
- Okumura, N.; Minamiyama, R.; Ho, L.T.; Kay, E.P.; Kawasaki, S.; Tourtas, T.; Schlotzer-Schrehardt, U.; Kruse, F.E.; Young, R.D.; Quantock, A.J.; et al. Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Lab. Investig. 2015, 95, 1291–1304. [Google Scholar] [CrossRef]
- Kocaba, V.; Katikireddy, K.R.; Gipson, I.; Price, M.O.; Price, F.W.; Jurkunas, U.V. Association of the Gutta-Induced Microenvironment with Corneal Endothelial Cell Behavior and Demise in Fuchs Endothelial Corneal Dystrophy. JAMA Ophthalmol. 2018, 136, 886–892. [Google Scholar] [CrossRef]
- Liu, C.; Gao, Z.-Q.; Li, J.; Zhou, Q. Identification of novel therapeutic targets for Fuchs’ endothelial corneal dystrophy based on gene bioinformatics analysis. PLoS ONE 2022, 17, e0264018. [Google Scholar] [CrossRef]
- Afshari, N.A.; Li, Y.J.; Pericak-Vance, M.A.; Gregory, S.; Klintworth, G.K. Genome-wide linkage scan in fuchs endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1093–1097. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, W.; Yan, X.M.; Wang, L.P.; Dong, H.; Shou, T.; Lei, H.; Guo, Q. Analysis of SLC4A11, ZEB1, LOXHD1, COL8A2 and TCF4 gene sequences in a multi-generational family with late-onset Fuchs corneal dystrophy. Int. J. Mol. Med. 2016, 37, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.A.; Vasanth, S.; Katsanis, N.; Gottsch, J.D. Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am. J. Hum. Genet. 2013, 93, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Wieben, E.D.; Aleff, R.A.; Tosakulwong, N.; Butz, M.L.; Highsmith, W.E.; Edwards, A.O.; Baratz, K.H. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS ONE 2012, 7, e49083. [Google Scholar] [CrossRef]
- Biswas, S.; Munier, F.L.; Yardley, J.; Hart-Holden, N.; Perveen, R.; Cousin, P.; Sutphin, J.E.; Noble, B.; Batterbury, M.; Kielty, C.; et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 2001, 10, 2415–2423. [Google Scholar] [CrossRef]
- Gottsch, J.D.; Zhang, C.; Sundin, O.H.; Bell, W.R.; Stark, W.J.; Green, W.R. Fuchs corneal dystrophy: Aberrant collagen distribution in an L450W mutant of the COL8A2 gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4504–4511. [Google Scholar] [CrossRef]
- Leonard, B.C.; Jalilian, I.; Raghunathan, V.K.; Wang, W.; Jun, A.S.; Murphy, C.J.; Thomasy, S.M. Biomechanical changes to Descemet’s membrane precede endothelial cell loss in an early-onset murine model of Fuchs endothelial corneal dystrophy. Exp. Eye Res. 2019, 180, 18–22. [Google Scholar] [CrossRef]
- Vilas, G.L.; Loganathan, S.K.; Quon, A.; Sundaresan, P.; Vithana, E.N.; Casey, J. Oligomerization of SLC4A11 protein and the severity of FECD and CHED2 corneal dystrophies caused by SLC4A11 mutations. Hum. Mutat. 2012, 33, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Jung, M.; Fecher-Trost, C.; Lovatt, M.; Peh, G.S.L.; Noskov, S.; Mehta, J.S.; Zimmermann, R.; Casey, J.R. Defective cell adhesion function of solute transporter, SLC4A11, in endothelial corneal dystrophies. Hum. Mol. Genet. 2020, 29, 97–116. [Google Scholar] [CrossRef]
- Roy, S.; Praneetha, D.C.; Vendra, V.P. Mutations in the Corneal Endothelial Dystrophy-Associated Gene SLC4A11 Render the Cells More Vulnerable to Oxidative Insults. Cornea 2015, 34, 668–674. [Google Scholar] [CrossRef]
- Du, J.; Aleff, R.A.; Soragni, E.; Kalari, K.; Nie, J.; Tang, X.; Davila, J.; Kocher, J.P.; Patel, S.V.; Gottesfeld, J.M.; et al. RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J. Biol. Chem. 2015, 290, 5979–5990. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Hayashi, R.; Koizumi, N. Perspective of Future Potent Therapies for Fuchs Endothelial Corneal Dystrophy. Open Ophthalmol. J. 2018, 12, 154–163. [Google Scholar] [CrossRef]
- Wieben, E.D.; Aleff, R.A.; Tang, X.; Butz, M.L.; Kalari, K.R.; Highsmith, E.W.; Jen, J.; Vasmatzis, G.; Patel, S.V.; Maguire, L.J.; et al. Trinucleotide Repeat Expansion in the Transcription Factor 4 (TCF4) Gene Leads to Widespread mRNA Splicing Changes in Fuchs’ Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 343–352. [Google Scholar] [CrossRef]
- Wieben, E.D.; Aleff, R.A.; Tang, X.; Kalari, K.R.; Maguire, L.J.; Patel, S.V.; Baratz, K.H.; Fautsch, M.P. Gene expression in the corneal endothelium of Fuchs endothelial corneal dystrophy patients with and without expansion of a trinucleotide repeat in TCF4. PLoS ONE 2018, 13, e0200005. [Google Scholar] [CrossRef]
- Nakano, M.; Okumura, N.; Nakagawa, H.; Koizumi, N.; Ikeda, Y.; Ueno, M.; Yoshii, K.; Adachi, H.; Aleff, R.A.; Butz, M.L.; et al. Trinucleotide Repeat Expansion in the TCF4 Gene in Fuchs’ Endothelial Corneal Dystrophy in Japanese. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4865–4869. [Google Scholar] [CrossRef]
- Xing, C.; Gong, X.; Hussain, I.; Khor, C.C.; Tan, D.T.; Aung, T.; Mehta, J.S.; Vithana, E.N.; Mootha, V.V. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs’ corneal dystrophy in Chinese implies common causal variant. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7073–7078. [Google Scholar] [CrossRef]
- Baratz, K.H.; Tosakulwong, N.; Ryu, E.; Brown, W.L.; Branham, K.; Chen, W.; Tran, K.D.; Schmid-Kubista, K.E.; Heckenlively, J.R.; Swaroop, A.; et al. E2-2 protein and Fuchs’s corneal dystrophy. N. Engl. J. Med. 2010, 363, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gong, X.; Johnson, S.T.; Corey, D.R.; Mootha, V.V. The TCF4 Trinucleotide Repeat Expansion of Fuchs’ Endothelial Corneal Dystrophy: Implications for the Anterior Segment of the Eye. Investig. Ophthalmol. Vis. Sci. 2023, 64, 16. [Google Scholar] [CrossRef]
- Bhattacharyya, N.; Chai, N.; Hafford-Tear, N.J.; Sadan, A.N.; Szabo, A.; Zarouchlioti, C.; Jedlickova, J.; Leung, S.K.; Liao, T.; Dudakova, L.; et al. Deciphering novel TCF4-driven mechanisms underlying a common triplet repeat expansion-mediated disease. PLoS Genet. 2024, 20, e1011230. [Google Scholar] [CrossRef]
- Rong, Z.; Gong, X.; Hulleman, J.D.; Corey, D.R.; Mootha, V.V. Trinucleotide Repeat-Targeting dCas9 as a Therapeutic Strategy for Fuchs’ Endothelial Corneal Dystrophy. Transl. Vis. Sci. Technol. 2020, 9, 47. [Google Scholar] [CrossRef]
- Hu, J.; Rong, Z.; Gong, X.; Zhou, Z.; Sharma, V.K.; Xing, C.; Watts, J.K.; Corey, D.R.; Mootha, V.V. Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs’ dystrophy. Hum. Mol. Genet. 2018, 27, 1015–1026. [Google Scholar] [CrossRef]
- Chen, F.-M.; Chen, R.; Wang, X.-J.; Sun, H.-H.; Wu, Z.-F. In vitro cellular responses to scaffolds containing two microencapulated growth factors. Biomaterials 2009, 30, 5215–5224. [Google Scholar] [CrossRef]
- Lee, J.G.; Jung, E.; Heur, M. Fibroblast growth factor 2 induces proliferation and fibrosis via SNAI1-mediated activation of CDK2 and ZEB1 in corneal endothelium. J. Biol. Chem. 2018, 293, 3758–3769. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, M.; Meng, H.; Meeker, A.K.; Eberhart, C.G.; Jun, A.S. Endothelial Cdkn1a (p21) overexpression and accelerated senescence in a mouse model of Fuchs endothelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6718–6727. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Deshpande, N.; Jurkunas, U.V. UV-A light induces G2/M phase arrest and subsequent endothelial-mesenchymal transition in Fuchs Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2179. [Google Scholar]
- Cadet, J.; Gentner, N.E.; Rozga, B.; Paterson, M.C. Rapid quantitation of ultraviolet-induced thymine-containing dimers in human cell DNA by reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1983, 280, 99–108. [Google Scholar] [CrossRef]
- Jurkunas, U.V.; Bitar, M.S.; Funaki, T.; Azizi, B. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am. J. Pathol. 2010, 177, 2278–2289. [Google Scholar] [CrossRef]
- Gottsch, J.D.; Bowers, A.L.; Margulies, E.H.; Seitzman, G.D.; Kim, S.W.; Saha, S.; Jun, A.S.; Stark, W.J.; Liu, S.H. Serial analysis of gene expression in the corneal endothelium of Fuchs’ dystrophy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 594–599. [Google Scholar]
- Hartness, E.M.; Shevalye, H.; Skeie, J.M.; Schmidt, G.; Greiner, M.A. Fuchs Endothelial Corneal Dystrophy Involves Decreased Expression of Genes for Fe-S Cluster Synthesis. Investig. Ophthalmol. Vis. Sci. 2023, 64, 656. [Google Scholar]
- Saha, S.; Skeie, J.M.; Schmidt, G.A.; Eggleston, T.; Shevalye, H.; Sales, C.S.; Phruttiwanichakun, P.; Dusane, A.; Field, M.G.; Rinkoski, T.A.; et al. TCF4 trinucleotide repeat expansions and UV irradiation increase susceptibility to ferroptosis in Fuchs endothelial corneal dystrophy. Redox Biol. 2024, 77, 103348. [Google Scholar] [CrossRef]
- Chi, M.; Zhao, Y.; Yuan, B.; Qiu, Z.; Peng, R.; Hong, J. MiR-23a-3p targets PTEN as a novel anti-ferroptosis regulator in Fuchs endothelial corneal dystrophy. Exp. Eye Res. 2025, 250, 110180. [Google Scholar] [CrossRef] [PubMed]
- Azizi, B.; Ziaei, A.; Fuchsluger, T.; Schmedt, T.; Chen, Y.; Jurkunas, U.V. p53-regulated increase in oxidative-stress—induced apoptosis in Fuchs endothelial corneal dystrophy: A native tissue model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9291–9297. [Google Scholar] [CrossRef]
- Lovatt, M.; Adnan, K.; Kocaba, V.; Dirisamer, M.; Peh, G.S.; Mehta, J.S. Peroxiredoxin-1 regulates lipid peroxidation in corneal endothelial cells. Redox Biol. 2019, 30, 101417. [Google Scholar] [CrossRef]
- Vacanti, C.A. The history of tissue engineering. J. Cell Mol. Med. 2006, 10, 569–576. [Google Scholar] [CrossRef]
- Griffith, M.; Osborne, R.; Munger, R.; Xiong, X.; Doillon, C.J.; Laycock, N.L.; Hakim, M.; Song, Y.; Watsky, M.A. Functional human corneal equivalents constructed from cell lines. Science 1999, 286, 2169–2172. [Google Scholar] [CrossRef] [PubMed]
- Katikireddy, K.R.; Schmedt, T.; Price, M.O.; Price, F.W.; Jurkunas, U.V. Existence of Neural Crest-Derived Progenitor Cells in Normal and Fuchs Endothelial Dystrophy Corneal Endothelium. Am. J. Pathol. 2016, 186, 2736–2750. [Google Scholar] [CrossRef]
- Zaniolo, K.; Bostan, C.; Rochette Drouin, O.; Deschambeault, A.; Perron, M.C.; Brunette, I.; Proulx, S. Culture of human corneal endothelial cells isolated from corneas with Fuchs endothelial corneal dystrophy. Exp. Eye Res. 2012, 94, 22–31. [Google Scholar] [CrossRef]
- Gendron, S.P.; Theriault, M.; Proulx, S.; Brunette, I.; Rochette, P.J. Restoration of Mitochondrial Integrity, Telomere Length, and Sensitivity to Oxidation by In Vitro Culture of Fuchs’ Endothelial Corneal Dystrophy Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5926–5934. [Google Scholar] [CrossRef]
- Peh, G.S.; Chng, Z.; Ang, H.P.; Cheng, T.Y.; Adnan, K.; Seah, X.Y.; George, B.L.; Toh, K.P.; Tan, D.T.; Yam, G.H.; et al. Propagation of human corneal endothelial cells: A novel dual media approach. Cell Transpl. 2015, 24, 287–304. [Google Scholar] [CrossRef]
- Muhammad, R.; Peh, G.S.; Adnan, K.; Law, J.B.; Mehta, J.S.; Yim, E.K. Micro- and nano-topography to enhance proliferation and sustain functional markers of donor-derived primary human corneal endothelial cells. Acta Biomater. 2015, 19, 138–148. [Google Scholar] [CrossRef]
- Palchesko, R.N.; Carrasquilla, S.D.; Feinberg, A.W. Natural Biomaterials for Corneal Tissue Engineering, Repair, and Regeneration. Adv. Healthc. Mater. 2018, 7, e1701434. [Google Scholar] [CrossRef] [PubMed]
- Frausto, R.F.; Le, D.J.; Aldave, A.J. Transcriptomic Analysis of Cultured Corneal Endothelial Cells as a Validation for Their Use in Cell Replacement Therapy. Cell Transpl. 2016, 25, 1159–1176. [Google Scholar] [CrossRef] [PubMed]
- Bednarz, J.; Teifel, M.; Friedl, P.; Engelmann, K. Immortalization of human corneal endothelial cells using electroporation protocol optimized for human corneal endothelial and human retinal pigment epithelial cells. Acta Ophthalmol. Scand. 2000, 78, 130–136. [Google Scholar] [CrossRef]
- Valtink, M.; Gruschwitz, R.; Funk, R.H.; Engelmann, K. Two clonal cell lines of immortalized human corneal endothelial cells show either differentiated or precursor cell characteristics. Cells Tissues Organs 2008, 187, 286–294. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Weng, J.; Li, Q.; Knauf, H.P.; Wilson, S.E. Fuchs’ corneal endothelial cells transduced with the human papilloma virus E6/E7 oncogenes. Exp. Eye Res. 1997, 65, 135–142. [Google Scholar] [CrossRef]
- Yokoi, T.; Seko, Y.; Yokoi, T.; Makino, H.; Hatou, S.; Yamada, M.; Kiyono, T.; Umezawa, A.; Nishina, H.; Azuma, N. Establishment of functioning human corneal endothelial cell line with high growth potential. PLoS ONE 2012, 7, e29677. [Google Scholar] [CrossRef]
- Liu, C.; Vojnovic, D.; Kochevar, I.E.; Jurkunas, U.V. UV-A Irradiation Activates Nrf2-Regulated Antioxidant Defense and Induces p53/Caspase3-Dependent Apoptosis in Corneal Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2319–2327. [Google Scholar] [CrossRef]
- Belair, C.D.; Yeager, T.R.; Lopez, P.M.; Reznikoff, C.A. Telomerase activity: A biomarker of cell proliferation, not malignant transformation. Proc. Natl. Acad. Sci. USA 1997, 94, 13677–13682. [Google Scholar] [CrossRef]
- Schmedt, T.; Chen, Y.; Nguyen, T.T.; Li, S.; Bonanno, J.A.; Jurkunas, U.V. Telomerase immortalization of human corneal endothelial cells yields functional hexagonal monolayers. PLoS ONE 2012, 7, e51427. [Google Scholar] [CrossRef]
- Di Donna, S.; Mamchaoui, K.; Cooper, R.N.; Seigneurin-Venin, S.; Tremblay, J.; Butler-Browne, G.S.; Mouly, V. Telomerase can extend the proliferative capacity of human myoblasts, but does not lead to their immortalization. Mol. Cancer Res. 2003, 1, 643–653. [Google Scholar]
- Sheerin, A.N.; Smith, S.K.; Jennert-Burston, K.; Brook, A.J.; Allen, M.C.; Ibrahim, B.; Jones, D.; Wallis, C.; Engelmann, K.; Rhys-Williams, W.; et al. Characterization of cellular senescence mechanisms in human corneal endothelial cells. Aging Cell 2012, 11, 234–240. [Google Scholar] [CrossRef]
- Tone, S.O.; Wylegala, A.; Böhm, M.; Melangath, G.; Deshpande, N.; Jurkunas, U.V. Increased corneal endothelial cell migration in Fuchs endothelial corneal dystrophy: A live cell imaging study. Ophthalmol. Sci. 2021, 1, 100006. [Google Scholar] [CrossRef] [PubMed]
- Benischke, A.S.; Vasanth, S.; Miyai, T.; Katikireddy, K.R.; White, T.; Chen, Y.; Halilovic, A.; Price, M.; Price, F., Jr.; Liton, P.B.; et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci. Rep. 2017, 7, 6656. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, S.; Inagaki, E.; Sayano, T.; Yamazaki, R.; Fusaki, N.; Hatou, S.; Hirayama, M.; Tsubota, K.; Negishi, K.; Okano, H.; et al. Non-apoptotic regulated cell death in Fuchs endothelial corneal dystrophy. Regen. Ther. 2023, 24, 592–601. [Google Scholar] [CrossRef]
- Ng, X.Y.; Peh, G.S.L.; Yam, G.H.-F.; Tay, H.G.; Mehta, J.S.; Ng, X.Y.; Peh, G.S.L.; Yam, G.H.-F.; Tay, H.G.; Mehta, J.S. Corneal Endothelial-like Cells Derived from Induced Pluripotent Stem Cells for Cell Therapy. Int. J. Mol. Sci. 2023, 24, 12433. [Google Scholar] [CrossRef]
- Rizwan, M.; Peh, G.S.L.; Ang, H.P.; Lwin, N.C.; Adnan, K.; Mehta, J.S.; Tan, W.S.; Yim, E.K.F. Sequentially-crosslinked bioactive hydrogels as nano-patterned substrates with customizable stiffness and degradation for corneal tissue engineering applications. Biomaterials 2017, 120, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Beebe, D.C.; Coats, J.M. The lens organizes the anterior segment: Specification of neural crest cell differentiation in the avian eye. Dev. Biol. 2000, 220, 424–431. [Google Scholar] [CrossRef]
- Peh, G.S.; Toh, K.P.; Wu, F.Y.; Tan, D.T.; Mehta, J.S. Cultivation of human corneal endothelial cells isolated from paired donor corneas. PLoS ONE 2011, 6, e28310. [Google Scholar] [CrossRef]
- Zhu, Y.T.; Chen, H.C.; Chen, S.Y.; Tseng, S.C. Nuclear p120 catenin unlocks mitotic block of contact-inhibited human corneal endothelial monolayers without disrupting adherent junctions. J. Cell Sci. 2012, 125 Pt 15, 3636–3648. [Google Scholar] [CrossRef]
- McGowan, S.L.; Edelhauser, H.F.; Pfister, R.R.; Whikehart, D.R. Stem cell markers in the human posterior limbus and corneal endothelium of unwounded and wounded corneas. Mol. Vis. 2007, 13, 1984–2000. [Google Scholar]
- Peh, G.S.; Toh, K.P.; Ang, H.P.; Seah, X.Y.; George, B.L.; Mehta, J.S. Optimization of human corneal endothelial cell culture: Density dependency of successful cultures in vitro. BMC Res. Notes 2013, 6, 176. [Google Scholar] [CrossRef]
- Wongvisavavit, R.; Parekh, M.; Ahmad, S.; Daniels, J.T. Challenges in corneal endothelial cell culture. Regen. Med. 2021, 16, 871–891. [Google Scholar] [CrossRef]
- Bertolin, M.; Lamon, M.; Franco, E.; Barbaro, V.; Ferrari, S.; Bovone, C.; Yu, A.C.; Parekh, M.; Ponzin, D.; Busin, M. Culture of corneal endothelial cells obtained by descemetorhexis of corneas with Fuchs endothelial corneal dystrophy. Exp. Eye Res. 2021, 211, 108748. [Google Scholar] [CrossRef]
- Qureshi, S.; Lee, S.; Steidl, W.; Ritzer, L.; Parise, M.; Chaubal, A.; Kumar, V. Endoplasmic Reticulum Stress Disrupts Mitochondrial Bioenergetics, Dynamics and Causes Corneal Endothelial Cell Apoptosis. Investig. Ophthalmol. Vis. Sci. 2023, 64, 18. [Google Scholar] [CrossRef]
- Ceravolo, I.; Mannino, F.; Irrera, N.; Squadrito, F.; Altavilla, D.; Ceravolo, G.; Pallio, G.; Minutoli, L. Health Potential of Aloe vera against Oxidative Stress Induced Corneal Damage: An “In Vitro” Study. Antioxidants 2021, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Ceravolo, I.; Mannino, F.; Irrera, N.; Minutoli, L.; Arcoraci, V.; Altavilla, D.; Cavallini, G.M.; Guarini, S.; Squadrito, F.; Pallio, G. Beneficial Effects of Polydeoxyribonucleotide (PDRN) in an In Vitro Model of Fuchs Endothelial Corneal Dystrophy. Pharmaceuticals 2022, 15, 447. [Google Scholar] [CrossRef]
- Yan, J.; Mehta, S.; Patel, K.; Dhupar, N.; Little, N.; Tone, S.O. Transcription factor 4 promotes increased corneal endothelial cellular migration by altering microtubules in Fuchs endothelial corneal dystrophy. Sci. Rep. 2024, 14, 10276. [Google Scholar] [CrossRef]
- Ashraf, S.; Deshpande, N.; Cheung, Q.; Asabere, J.B.; Wong, R.J.; Gauthier, A.G.; Parekh, M.; Adhikari, Y.; Melangath, G.; Jurkunas, U.V. Modulation of ATM enhances DNA repair in G2/M phase of cell cycle and averts senescence in Fuchs endothelial corneal dystrophy. Commun. Biol. 2024, 7, 1482. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Efthymiou, S.; Facchini, S.; Dominik, N.; Bhattacharyya, N.; Liu, S.; Costa, M.A.; Szabo, A.; Sadan, A.N.; Jun, A.S.; et al. Tissue-specific TCF4 triplet repeat instability revealed by optical genome mapping. eBioMedicine 2024, 108, 105328. [Google Scholar] [CrossRef]
- Nicholson, M.W.; Ting, C.-Y.; Chan, D.Z.H.; Cheng, Y.-C.; Lee, Y.-C.; Hsu, C.-C.; Huang, C.-Y.; Hsieh, P.C.H. Utility of iPSC-Derived Cells for Disease Modeling, Drug Development, and Cell Therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef]
- Brejchova, K.; Dudakova, L.; Skalicka, P.; Dobrovolny, R.; Masek, P.; Putzova, M.; Moosajee, M.; Tuft, S.J.; Davidson, A.E.; Liskova, P. IPSC-Derived Corneal Endothelial-like Cells Act as an Appropriate Model System to Assess the Impact of SLC4A11 Variants on Pre-mRNA Splicing. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3084–3090. [Google Scholar] [CrossRef]
- Teo, B.K.K.; Goh, K.J.; Ng, Z.J.; Koo, S.; Yim, E.K.F. Functional reconstruction of corneal endothelium using nanotopography for tissue-engineering applications. Acta Biomater. 2012, 8, 2941–2952. [Google Scholar] [CrossRef]
- Koo, S.; Muhammad, R.; Peh, G.S.L.; Mehta, J.S.; Yim, E.K.F. Micro- and nanotopography with extracellular matrix coating modulate human corneal endothelial cell behavior. Acta Biomater. 2014, 10, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, Q.; Yang, Y.; Guo, X.; Lian, R.; Li, S.; Wang, C.; Zhang, S.; Chen, J. The Effects of ROCK Inhibitor Y-27632 on Injectable Spheroids of Bovine Corneal Endothelial Cells. Cell. Reprogram. 2015, 17, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, B.C.; Hellwinkel, O.J.C.; Klameth, C.; Wenzel, D.; Bartz-Schmidt, K.U.; Spitzer, M.S.; Schultheiss, M. Establishment of a porcine corneal endothelial organ culture model for research purposes. Cell Tissue Bank. 2018, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Proulx, S.; Bourget, J.M.; Gagnon, N.; Martel, S.; Deschambeault, A.; Carrier, P.; Giasson, C.J.; Auger, F.A.; Brunette, I.; Germain, L. Optimization of culture conditions for porcine corneal endothelial cells. Mol. Vis. 2007, 13, 524–533. [Google Scholar]
- Meekins, L.C.; Rosado-Adames, N.; Maddala, R.; Zhao, J.J.; Rao, P.V.; Afshari, N.A. Corneal Endothelial Cell Migration and Proliferation Enhanced by Rho Kinase (ROCK) Inhibitors in In Vitro and In Vivo Models. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6731–6738. [Google Scholar] [CrossRef]
- Kassumeh, S.; von Studnitz, A.; Priglinger, S.G.; Fuchshofer, R.; Luft, N.; Moloney, G.; Dirisamer, M.; Ohlmann, A. Ex vivo excimer laser ablation of cornea guttata and ROCK inhibitor-aided endothelial recolonization of ablated central cornea. Acta Ophthalmol. 2020, 98, e773–e780. [Google Scholar] [CrossRef]
- Goyer, B.; Theriault, M.; Gendron, S.P.; Brunette, I.; Rochette, P.J.; Proulx, S. Extracellular Matrix and Integrin Expression Profiles in Fuchs Endothelial Corneal Dystrophy Cells and Tissue Model. Tissue Eng. Part A 2018, 24, 607–615. [Google Scholar] [CrossRef]
- Conconi, M.T.; Borgio, L.; Di Liddo, R.; Sartore, L.; Dalzoppo, D.; Amistà, P.; Lora, S.; Parnigotto, P.P.; Grandi, C. Evaluation of vascular grafts based on polyvinyl alcohol cryogels. Mol. Med. Rep. 2014, 10, 1329–1334. [Google Scholar] [CrossRef]
- Goyal, A.; Wang, Y.; Su, H.; Dobrucki, L.W.; Brennan, M.; Fong, P.; Dardik, A.; Tellides, G.; Sinusas, A.; Pober, J.S.; et al. Development of a model system for preliminary evaluation of tissue-engineered vascular conduits. J. Pediatr. Surg. 2006, 41, 787–791. [Google Scholar] [CrossRef]
- Rabiee, B.; Bheri, A.; Farooq, A.V.; Yazdanpanah, G.; Jabbehdari, S.; Ghassemi, M.; Djalilian, A.R. The Effect of Mesenchymal Stem Cell Secretome on Corneal Endothelial Cell Preservation in an Oxidative Injury Model. Cornea 2020, 39, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Ogita, Y.; Higuchi, S.; Kani, K. Cell movements in a living mammalian tissue: Long-term observation of individual cells in wounded corneal endothelia of cats. J. Morphol. 1982, 174, 25–39. [Google Scholar] [CrossRef]
- Méthot, S.; Proulx, S.; Brunette, I.; Rochette, P.J. The Presence of Guttae in Fuchs Endothelial Corneal Dystrophy Explants Correlates with Cellular Markers of Disease Progression. Investig. Ophthalmol. Vis. Sci. 2023, 64, 13. [Google Scholar] [CrossRef]
- Soh, Y.Q.; Peh, G.; George, B.L.; Seah, X.Y.; Primalani, N.K.; Adnan, K.; Mehta, J.S. Predicative Factors for Corneal Endothelial Cell Migration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 338–348. [Google Scholar] [CrossRef]
- Roy, O.; Leclerc, V.B.; Bourget, J.M.; Theriault, M.; Proulx, S. Understanding the process of corneal endothelial morphological change in vitro. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1228–1237. [Google Scholar] [CrossRef]
- Van Horn, D.L.; Sendele, D.D.; Seideman, S.; Buco, P.J. Regenerative capacity of the corneal endothelium in rabbit and cat. Investig. Ophthalmol. Vis. Sci. 1977, 16, 597–613. [Google Scholar]
- Park, S.; Leonard, B.C.; Raghunathan, V.K.; Kim, S.; Li, J.Y.; Mannis, M.J.; Murphy, C.J.; Thomasy, S.M. Animal models of corneal endothelial dysfunction to facilitate development of novel therapies. Ann. Transl. Med. 2021, 9, 1271. [Google Scholar] [CrossRef]
- Liu, C.; Miyajima, T.; Melangath, G.; Miyai, T.; Vasanth, S.; Deshpande, N.; Kumar, V.; Ong Tone, S.; Gupta, R.; Zhu, S.; et al. Ultraviolet A light induces DNA damage and estrogen-DNA adducts in Fuchs endothelial corneal dystrophy causing females to be more affected. Proc. Natl. Acad. Sci. USA 2020, 117, 573–583. [Google Scholar] [CrossRef]
- White, T.L.; Deshpande, N.; Kumar, V.; Gauthier, A.G.; Jurkunas, U.V. Cell cycle re-entry and arrest in G2/M phase induces senescence and fibrosis in Fuchs Endothelial Corneal Dystrophy. Free. Radic. Biol. Med. 2021, 164, 34–43. [Google Scholar] [CrossRef]
- Meng, H.; Matthaei, M.; Ramanan, N.; Grebe, R.; Chakravarti, S.; Speck, C.L.; Kimos, M.; Vij, N.; Eberhart, C.G.; Jun, A.S. L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1887–1897. [Google Scholar] [CrossRef]
- Han, S.B.; Ang, H.P.; Poh, R.; Chaurasia, S.S.; Peh, G.; Liu, J.; Tan, D.T.; Vithana, E.N.; Mehta, J.S. Mice with a targeted disruption of Slc4a11 model the progressive corneal changes of congenital hereditary endothelial dystrophy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6179–6189. [Google Scholar] [CrossRef]
- Murugan, S.; de Campos, V.S.; Ghag, S.A.; Ng, M.; Shyam, R. Characterization of a Novel Mouse Model for Fuchs Endothelial Corneal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2024, 65, 18. [Google Scholar] [CrossRef]
- Mimura, T.; Yamagami, S.; Usui, T.; Ishii, Y.; Ono, K.; Yokoo, S.; Funatsu, H.; Araie, M.; Amano, S. Long-term outcome of iron-endocytosing cultured corneal endothelial cell transplantation with magnetic attraction. Exp. Eye Res. 2005, 80, 149–157. [Google Scholar] [CrossRef]
- Mimura, T.; Yamagami, S.; Yokoo, S.; Yanagi, Y.; Usui, T.; Ono, K.; Araie, M.; Amano, S. Sphere therapy for corneal endothelium deficiency in a rabbit model. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3128–3135. [Google Scholar] [CrossRef]
- Mimura, T.; Yamagami, S.; Usui, T.; Seiichi; Honda, N.; Amano, S. Necessary prone position time for human corneal endothelial precursor transplantation in a rabbit endothelial deficiency model. Curr. Eye Res. 2007, 32, 617–623. [Google Scholar] [CrossRef]
- Worner, C.H.; Olguin, A.; Ruiz-Garcia, J.L.; Garzon-Jimenez, N. Cell pattern in adult human corneal endothelium. PLoS ONE 2011, 6, e19483. [Google Scholar] [CrossRef]
- Vedana, G.; Villarreal, G., Jr.; Jun, A.S. Fuchs endothelial corneal dystrophy: Current perspectives. Clin. Ophthalmol. 2016, 10, 321–330. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, X.; Tan, J.; Darling, D.S.; Kaplan, H.J.; Dean, D.C. Zeb1 mutant mice as a model of posterior corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1843–1849. [Google Scholar] [CrossRef]
- Shen, A.L.; O’Leary, K.A.; Dubielzig, R.R.; Drinkwater, N.; Murphy, C.J.; Kasper, C.B.; Bradfield, C.A. The PPCD1 mouse: Characterization of a mouse model for posterior polymorphous corneal dystrophy and identification of a candidate gene. PLoS ONE 2010, 5, e12213. [Google Scholar] [CrossRef]
- Okumura, N.; Matsumoto, D.; Fukui, Y.; Teramoto, M.; Imai, H.; Kurosawa, T.; Shimada, T.; Kruse, F.; Schlotzer-Schrehardt, U.; Kinoshita, S.; et al. Feasibility of cell-based therapy combined with descemetorhexis for treating Fuchs endothelial corneal dystrophy in rabbit model. PLoS ONE 2018, 13, e0191306. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, C.C. Tensile mechanical and creep properties of Descemet’s membrane and lens capsule. Exp. Eye Res. 2004, 79, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Iovieno, A.; Neri, A.; Soldani, A.M.; Adani, C.; Fontana, L. Descemetorhexis Without Graft Placement for the Treatment of Fuchs Endothelial Dystrophy: Preliminary Results and Review of the Literature. Cornea 2017, 36, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Patel, D.V. The pathophysiology of Fuchs’ endothelial dystrophy–a review of molecular and cellular insights. Exp. Eye Res. 2015, 130, 97–105. [Google Scholar] [CrossRef] [PubMed]


| Model Species | Prime Characteristic | Pathological and Physiological Recapitulation | Pros | Cons | Ref. |
|---|---|---|---|---|---|
| Primary HCEnCs | In vitro expansion of primary healthy and FECD cells | Cell genetic mutation, molecular changes in cells | Closest recapitulation of the HCEnCs in vivo | Limited proliferative capacity. Primary FECD CEnCs isolated from old donors are challenging to expand | [16,41,97,98,99,100,101,102,103] |
| Immortalized HCEnCs | Viral oncogenes, Cellular Oncogene, RNA Interference, CRISPR/dCas9 immortalized FECD, and healthy cells | Cell genetic mutation, molecular changes in cells | Induced proliferative ability with active ion transport and full confluence. Less batch variability compared to primary cells | Less accurate biological information, specific to donors’ genetic profiles. Different from in vivo environment. | [44,45,48,59,88,93,103,104,105,106,107,108,109,110,111,112,113,114] |
| iPSC-Derived CEnCs | Cells derived from disease-specific iPSCs | Recapitulate cellular and molecular changes | Suitable for evaluating the precise mechanisms of cell death | Residual undifferentiated iPSCs present, limited reproducibility. | [115,116] |
| Bovine corneal endothelial cells | Cells harvested for in vitro drug testing | Recapitulate cellular and molecular changes | Ease of in vitro maintenance | Xenogeneic cells, do not fully recapitulate HCEnCs | [46] |
| Primary HCEnCs on biomimicking synthetic guttata | Hot embossed topographical patterns | Recapitulate the impaired DM and CEnCs microenvironment in vitro by mimicking guttata structure | Ability to control guttata size, morphology, stiffness, and spacing. Surfaces can be replicated, not reliant upon animal donors. | Not a complete representation of the in vivo environment. | [48,117] |
| Model and Species | Prime Characteristic | Pathological and Physiological Recapitulation | Pros | Cons | Ref. |
|---|---|---|---|---|---|
| FECD ex vivo corneal tissue with cells (human) | DM and CEnCs isolated from Stroma | FECD microenvironment, ECM | A close representation of the FECD in vivo, cells reside in native tissue | Not reusable, limited cell amount | [37,38,82,88,114,139] |
| Normal ex vivo corneal tissue with cells (human) | DM and CEnCs isolated from Stroma | Introducing oxidative stress via chemical or physical approach | A close representation of the normal environment in vivo | Not reusable | [113] |
| FECD ex vivo corneal tissue with seeded immortalized cells (human) | Decellularized tissue seeded with HCEnC-21T | FECD microenvironment, ECM | A close representation of the FECD in vivo. Studies using FECD DM with healthy cells are possible | HCEnC-21T may not be the closest representation of the diseased cell state; ex vivo tissue is not reusable | [60,140,141,142] |
| Thin porcine corneal buttons | Harvested as ex vivo tissues | Recapitulating human ex vivo corneal tissue | Cell death rates are comparable to HCEnCs. It could be used as organotypic culture ex vivo | Samples may be damaged when placed facing down | [60,113,136,143] |
| Mouse corneal tissue | Whole mount corneas, with CEnCs | Recapitulating human ex vivo corneal tissue | Great potential for early-stage preliminary studies | Different in physiology and limited representation of human FECD phenotype | [11] |
| Model Species | Prime Characteristic | Pathological and Physiological Recapitulation | Pros | Cons | Ref. |
|---|---|---|---|---|---|
| Murine (mouse) | Exposure to ultraviolet A light | Recapitulate cellular DNA damage induced by UVA irradiation | Model for oxidative damage through ROS, in vivo recapitulation of the diseased environment | UVA exposure is considered the sole environmental factor in FECD induction, while many genes are involved in FECD development | [150,151] |
| Mutation in COL8A2 gene (including Q455K and L450W) | Early onset form of FECD | Model for early-onset FECD | Not representative of late-onset FECD | [11,63,66,68,152] | |
| SLC4A11 knockout mouse | Late-onset form of FECD and congenital hereditary endothelia dystrophy | Progressive corneal edema, vacuolated CEnCs, thickened DM with age. | Phenotypic changes are possibly milder than those in human patients | [11,63,66,68,152,153] | |
| Double mutation: SLC4A11 knockdown and Q455k knock-in | Presence of guttata, increased corneal thickness, decreased CEC density, elevated ROS | Model shows important typical FECD phenotypes | Not correspond to a specific genotype associated with FECD in humans. | [154] | |
| Rabbit | Mechanical or cryogenic injury | Microenvironment changes to study CEnC dysfunction and corneal edema | Migration and proliferation model for the regeneration of wounds. Helpful in exploring therapeutics in a shorter time frame. | Human CEnCs do not proliferate, unlike rabbit CEnCs. | [13,21,24,138,155,156,157] |
| Feline | Partial DM removal in vivo | Recapitulate impaired DM and CEnCs | Does not proliferate in vivo. Cell density and corneal thickness are similar to humans. Human xenograft transplantations into felines are well tolerated. | Not a full recapitulation of human DM and CEnCs in vivo | [16,18,148] |
| Cynomolgus monkey | Mechanical or cryogenic injury | Microenvironment changes to study CEnC dysfunction | Most similar and physiological relevant to human CEnCs | Not a full recapitulation of human FECD | [19,21,23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, F.; Xi, L.W.Q.; Luu, W.; Enkhbat, M.; Neo, D.; Mehta, J.S.; Peh, G.S.L.; Yim, E.K.F. Preclinical Models for Studying Fuchs Endothelial Corneal Dystrophy. Cells 2025, 14, 505. https://doi.org/10.3390/cells14070505
Sun F, Xi LWQ, Luu W, Enkhbat M, Neo D, Mehta JS, Peh GSL, Yim EKF. Preclinical Models for Studying Fuchs Endothelial Corneal Dystrophy. Cells. 2025; 14(7):505. https://doi.org/10.3390/cells14070505
Chicago/Turabian StyleSun, Fancheng, Lexie W. Q. Xi, Wesley Luu, Myagmartsend Enkhbat, Dawn Neo, Jodhbir S. Mehta, Gary S. L. Peh, and Evelyn K. F. Yim. 2025. "Preclinical Models for Studying Fuchs Endothelial Corneal Dystrophy" Cells 14, no. 7: 505. https://doi.org/10.3390/cells14070505
APA StyleSun, F., Xi, L. W. Q., Luu, W., Enkhbat, M., Neo, D., Mehta, J. S., Peh, G. S. L., & Yim, E. K. F. (2025). Preclinical Models for Studying Fuchs Endothelial Corneal Dystrophy. Cells, 14(7), 505. https://doi.org/10.3390/cells14070505