
CRTAP-Null Osteoblasts Have Increased Proliferation, Protein Secretion, and Skeletal Morphogenesis Gene Expression with Downregulation of Cellular Adhesion

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. Human Subjects and Cell Culture
2.3. Genomic DNA Preparation and Sequencing
2.4. Steady State Collagen Biochemical Analysis
2.5. RNA Preparation and Real-Time qPCR
2.6. RIPA Lysates and Western Blotting
2.7. Electron Microscopy
2.8. Mass Spectrometry and Amino Acid Analysis
2.9. Immunofluorescence Microscopy
2.10. Collagen Secretion Assay
2.11. RNA-Seq and Analysis
2.12. BrdU Proliferation Assay
2.13. Statistical Analysis
3. Results
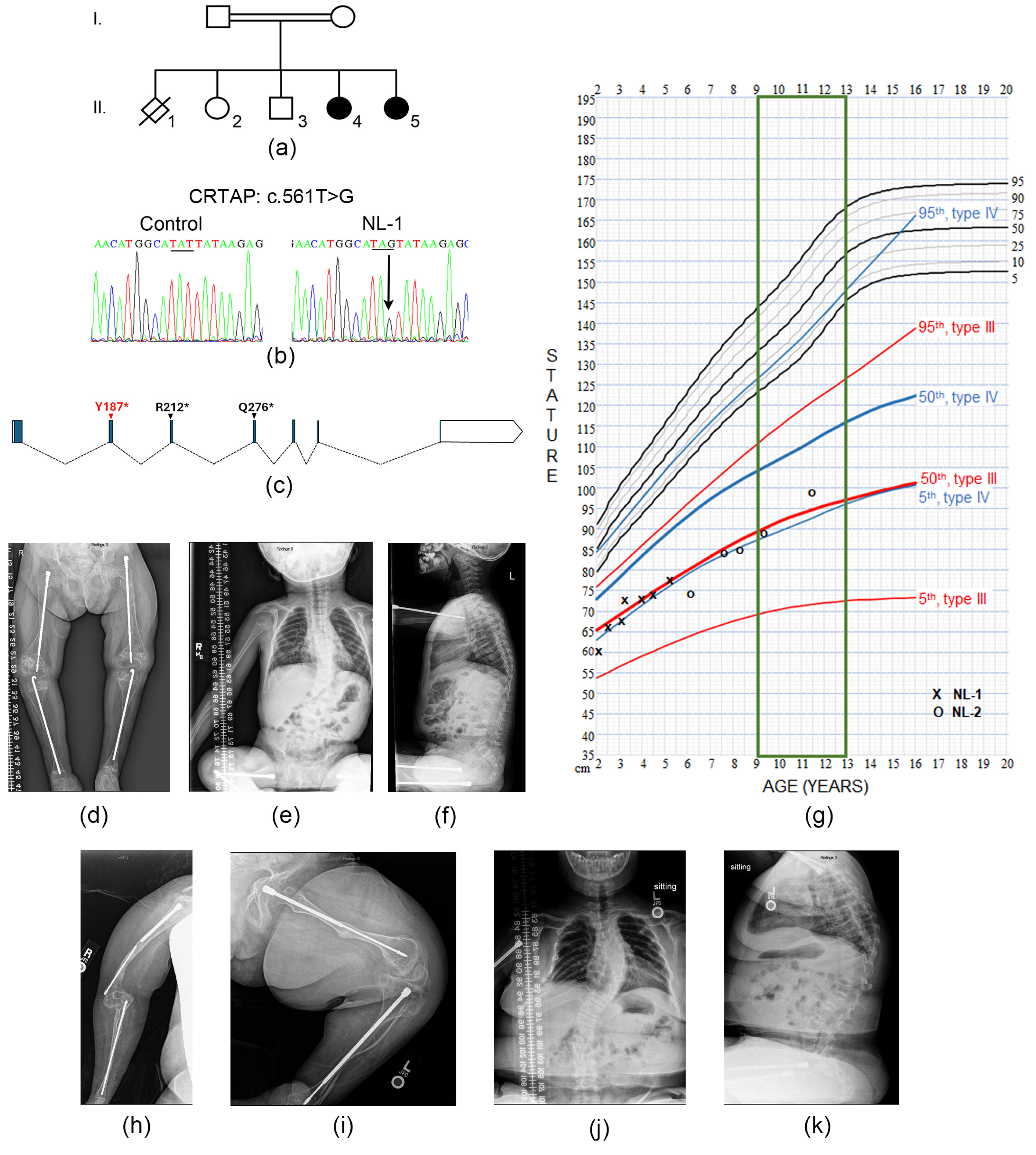
3.1. Clinical Report of Female Siblings with Non-Lethal Type VII OI
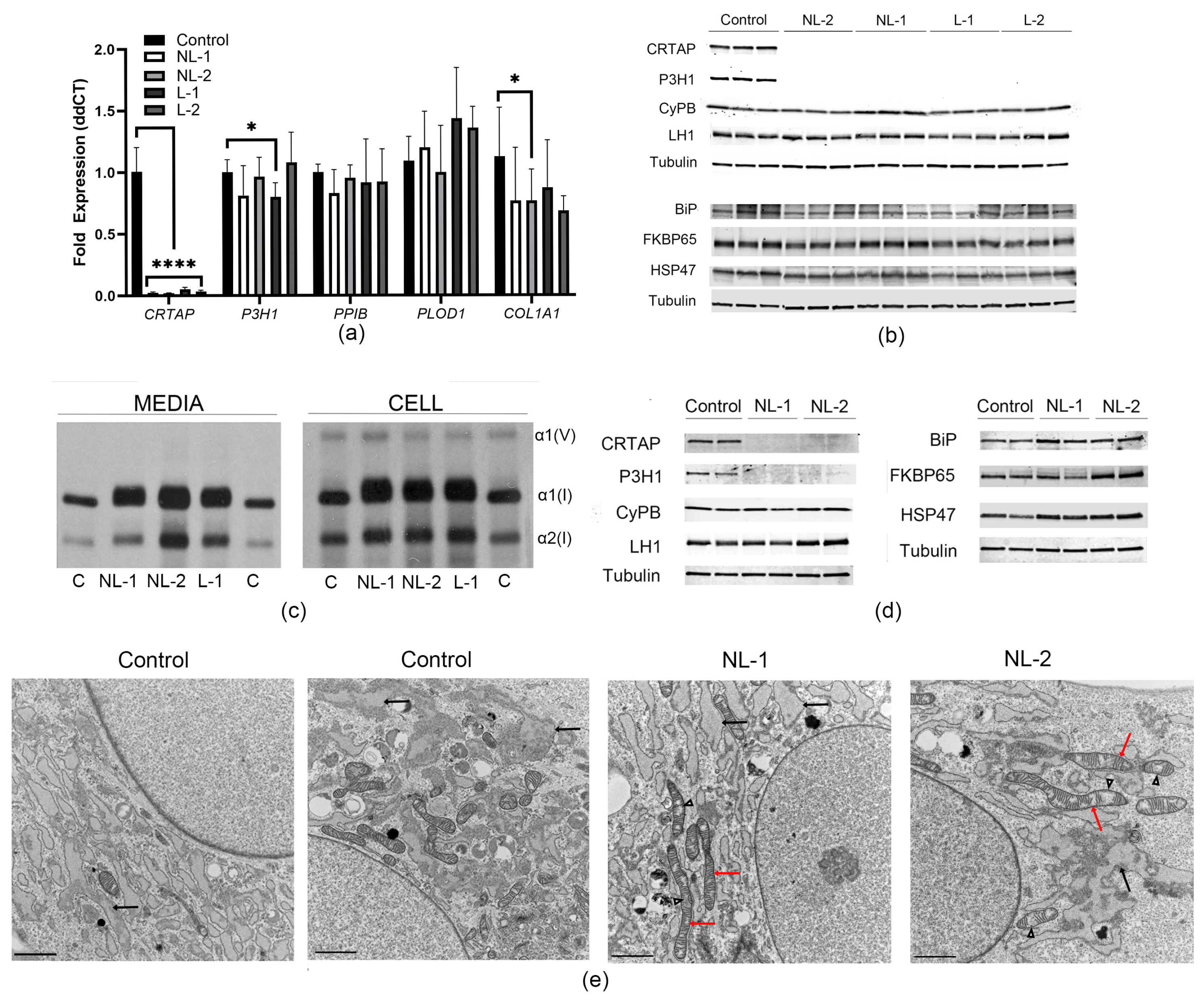
3.2. Fibroblasts from Non-Lethal and Lethal CRTAP-Null Patients Have Similar Chaperone Profiles
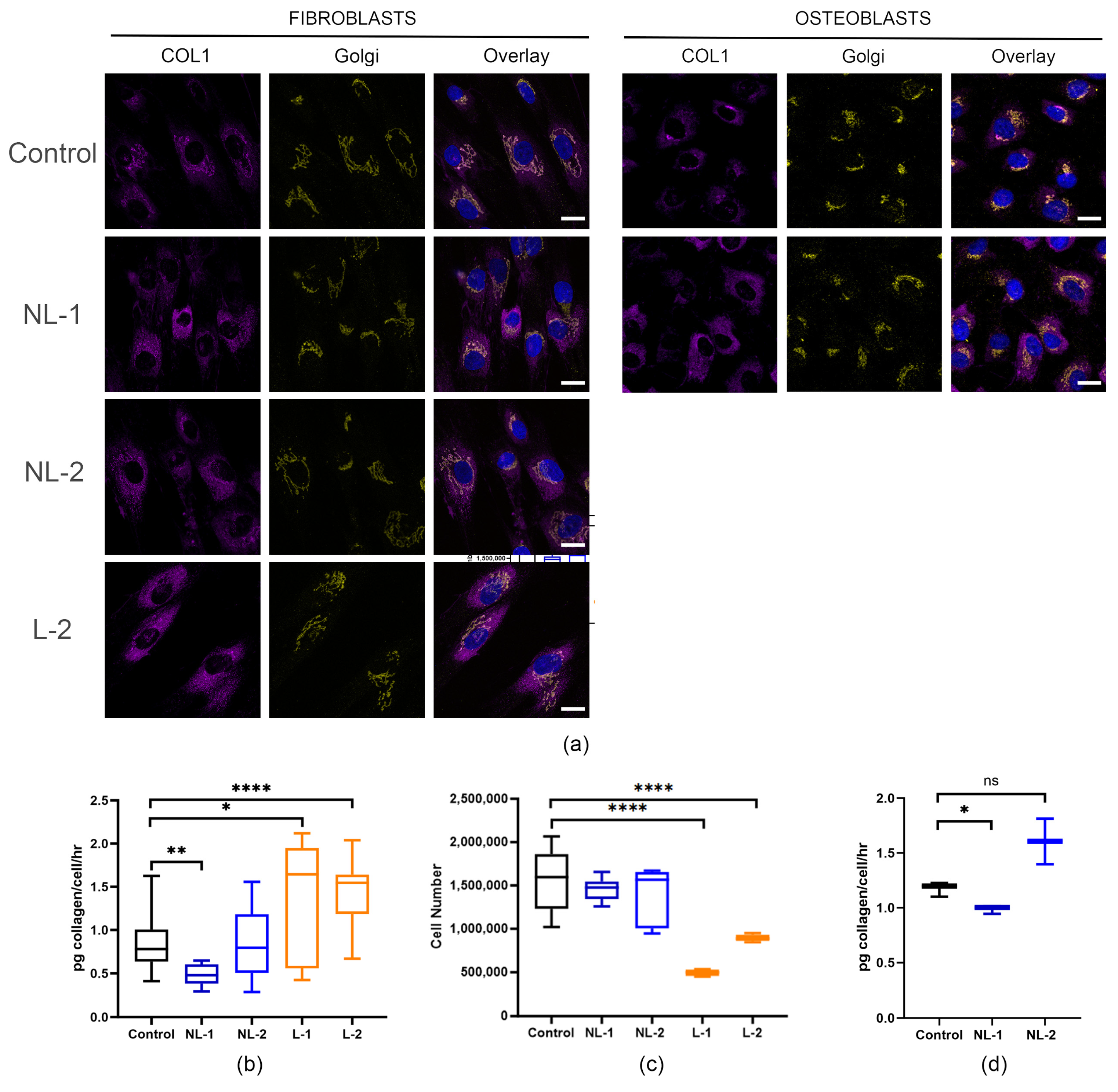
3.3. Collagen Protein Trafficking Is Delayed
3.4. Upregulation of Cell Cycle Genes in CRTAP-Null Osteoblasts
3.5. CRTAP-Null Osteoblasts Upregulate Protein Secretion Early in Differentiation
3.6. Upregulation of Cartilage and Skeletal Developmental Genes
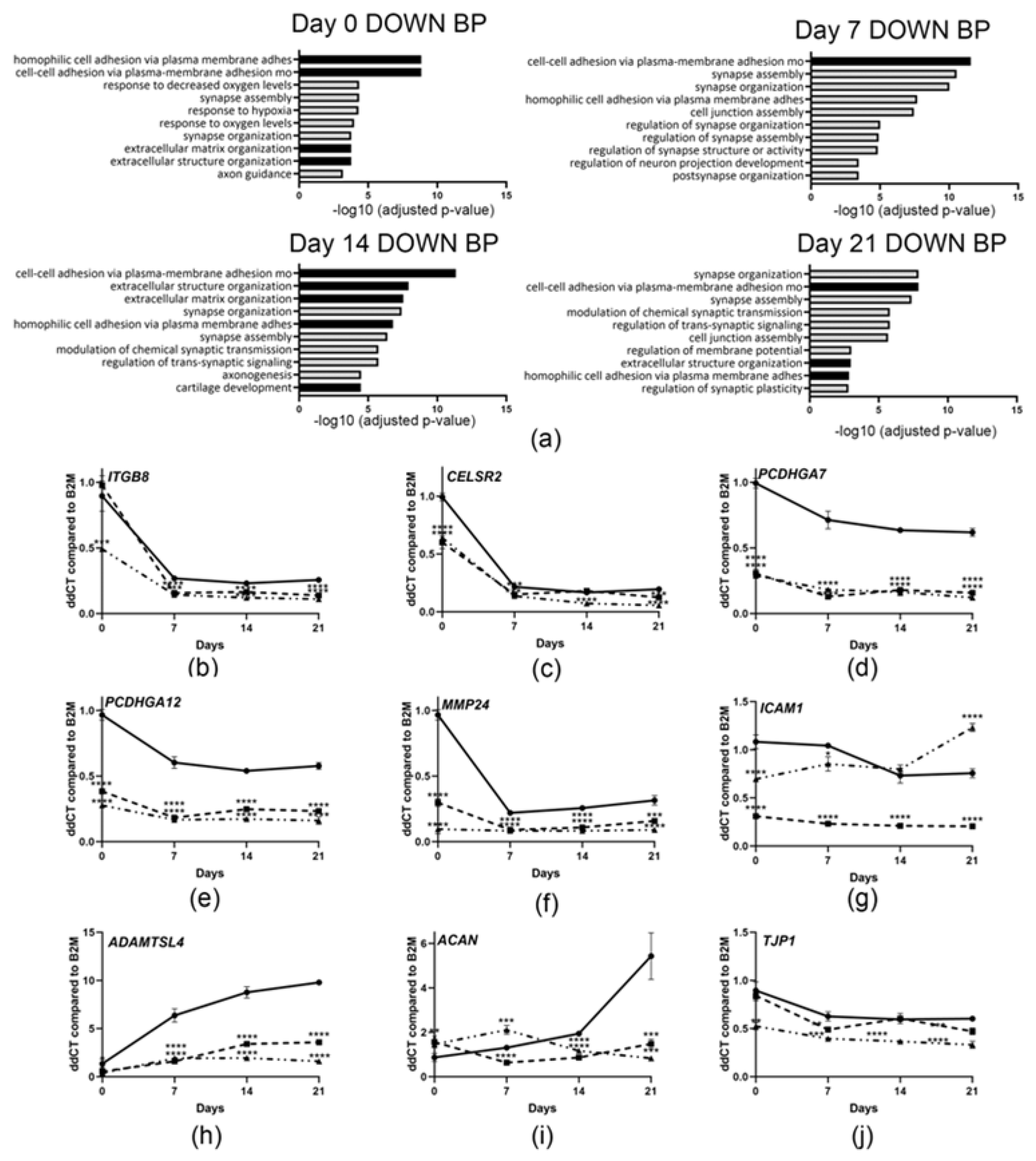
3.7. Downregulation of Genes Involved in Cell Adhesion and Extracellular Matrix Organization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar] [PubMed]
- Ward, L.M.; Rauch, F.; Travers, R.; Chabot, G.; Azouz, E.M.; Lalic, L.; Roughley, P.J.; Glorieux, F.H. Osteogenesis imperfecta type VII: An autosomal recessive form of brittle bone disease. Bone 2002, 31, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.M.; Chang, W.; Morello, R.; Cabral, W.A.; Weis, M.; Eyre, D.R.; Leikin, S.; Makareeva, E.; Kuznetsova, N.; Uveges, T.E.; et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006, 355, 2757–2764. [Google Scholar] [PubMed]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006, 127, 291–304. [Google Scholar]
- Marini, J.C.; Cabral, W.A.; Barnes, A.M.; Chang, W. Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle 2007, 6, 1675–1681. [Google Scholar] [CrossRef]
- Cabral, W.A.; Chang, W.; Barnes, A.M.; Weis, M.; Scott, M.A.; Leikin, S.; Makareeva, E.; Kuznetsova, N.V.; Rosenbaum, K.N.; Tifft, C.J.; et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007, 39, 359–365. [Google Scholar]
- Barnes, A.M.; Carter, E.M.; Cabral, W.A.; Weis, M.; Chang, W.; Makareeva, E.; Leikin, S.; Rotimi, C.N.; Eyre, D.R.; Raggio, C.L.; et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 2010, 362, 521–528. [Google Scholar]
- Ishikawa, Y.; Wirz, J.; Vranka, J.A.; Nagata, K.; Bächinger, H.P. Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein.cyclophilin B complex. J. Biol. Chem. 2009, 284, 17641–17647. [Google Scholar] [CrossRef]
- Chang, W.; Barnes, A.M.; Cabral, W.A.; Bodurtha, J.N.; Marini, J.C. Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex. Hum. Mol. Genet. 2010, 19, 223–234. [Google Scholar]
- Baldridge, D.; Lennington, J.; Weis, M.; Homan, E.P.; Jiang, M.M.; Munivez, E.; Keene, D.R.; Hogue, W.R.; Pyott, S.; Byers, P.H.; et al. Generalized connective tissue disease in Crtap-/- mouse. PLoS ONE 2010, 5, e10560. [Google Scholar]
- Grol, M.W.; Haelterman, N.A.; Lim, J.; Munivez, E.M.; Archer, M.; Hudson, D.M.; Tufa, S.F.; Keene, D.R.; Lei, K.; Park, D.; et al. Tendon and motor phenotypes in the Crtap(-/-) mouse model of recessive osteogenesis imperfecta. eLife 2021, 10, e63488. [Google Scholar]
- Alanay, Y.; Avaygan, H.; Camacho, N.; Utine, G.E.; Boduroglu, K.; Aktas, D.; Alikasifoglu, M.; Tuncbilek, E.; Orhan, D.; Bakar, F.T.; et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 86, 551–559. [Google Scholar]
- Amor, I.M.; Rauch, F.; Gruenwald, K.; Weis, M.; Eyre, D.R.; Roughley, P.; Glorieux, F.H.; Morello, R. Severe osteogenesis imperfecta caused by a small in-frame deletion in CRTAP. Am. J. Med. Genet. A 2011, 155A, 2865–2870. [Google Scholar] [PubMed]
- Baldridge, D.; Schwarze, U.; Morello, R.; Lennington, J.; Bertin, T.K.; Pace, J.M.; Pepin, M.G.; Weis, M.; Eyre, D.R.; Walsh, J.; et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008, 29, 1435–1442. [Google Scholar] [PubMed]
- Holtz, A.P.; Souza, L.T.; Ribeiro, E.M.; Acosta, A.X.; Lago, R.M.; Simoni, G.; Llerena, J.C., Jr.; Félix, T.M. Genetic analysis of osteogenesis imperfecta in a large Brazilian cohort. Bone 2023, 169, 116683. [Google Scholar]
- Kuptanon, C.; Thamkunanon, V.; Srichomthong, C.; Theerapanon, T.; Suphapeetiporn, K.; Porntaveetus, T.; Shotelersuk, V. Novel BMP1, CRTAP, and SERPINF1 variants causing autosomal recessive osteogenesis imperfecta. Clin. Genet. 2022, 102, 242–243. [Google Scholar] [PubMed]
- Madhuri, V.; Selina, A.; Loganathan, L.; Kumar, A.; Kumar, V.; Raymond, R.; Ramesh, S.; Vincy, N.; Joel, G.; James, D.; et al. Osteogenesis imperfecta: Novel genetic variants and clinical observations from a clinical exome study of 54 Indian patients. Ann. Hum. Genet. 2021, 85, 37–46. [Google Scholar]
- Marulanda, J.; Ludwig, K.; Glorieux, F.; Lee, B.; Sutton, V.R.; Members of the BBD Consortium; Retrouvey, J.M.; Rauch, F. Craniofacial and dental phenotype of two girls with osteogenesis imperfecta due to mutations in CRTAP. Bone 2022, 164, 116516. [Google Scholar]
- Tang, Y.A.; Wang, L.Y.; Chang, C.M.; Lee, I.W.; Tsai, W.H.; Sun, H.S. Novel Compound Heterozygous Mutations in CRTAP Cause Rare Autosomal Recessive Osteogenesis Imperfecta. Front. Genet. 2020, 11, 897. [Google Scholar]
- Tuysuz, B.; Elkanova, L.; Alkaya, D.U.; Güleç, Ç.; Toksoy, G.; Güneş, N.; Yazan, H.; Bayhan, A.I.; Yıldırım, T.; Yeşil, G.; et al. Osteogenesis imperfecta in 140 Turkish families: Molecular spectrum and, comparison of long-term clinical outcome of those with COL1A1/A2 and biallelic variants. Bone 2022, 155, 116293. [Google Scholar]
- Van Dijk, F.S.; Nesbitt, I.M.; Nikkels, P.G.; Dalton, A.; Bongers, E.M.; Van De Kamp, J.M.; Hilhorst-Hofstee, Y.; Den Hollander, N.S.; Lachmeijer, A.; Marcelis, C.L.; et al. CRTAP mutations in lethal and severe osteogenesis imperfecta: The importance of combining biochemical and molecular genetic analysis. Eur. J. Hum. Genet. 2009, 17, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Gao, P.; Hu, J.; Lin, X.; Sun, L.; Zhang, Q.; Jiang, Y.; Wang, O.; Xia, W.; Xing, X.; et al. Genetic analysis, phenotype spectrum and functional study of rare osteogenesis imperfecta caused by CRTAP Variants. J. Clin. Endocrinol. Metab. 2024, 109, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Robey, P.G.; Termine, J.D. Human bone cells in vitro. Calcif. Tissue Int. 1985, 37, 453–460. [Google Scholar] [CrossRef]
- Bonadio, J.; Byers, P.H. Subtle structural alterations in the chains of type I procollagen produce osteogenesis imperfecta type II. Nature 1985, 316, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.; Zheng, J.; Parmiter, D.; Patri, A.K. Biological tissue and cell culture specimen preparation for TEM nanoparticle characterization. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; Volume 697, pp. 83–91. [Google Scholar]
- Seref-Ferlengez, Z.; Kennedy, O.D.; Schaffler, M.B. Bone microdamage, remodeling and bone fragility: How much damage is too much damage? Bonekey Rep. 2015, 4, 644. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 January 2025).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Shaheen, R.; Alazami, A.M.; Alshammari, M.J.; Faqeih, E.; Alhashmi, N.; Mousa, N.; Alsinani, A.; Ansari, S.; Alzahrani, F.; Al-Owain, M.; et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J. Med. Genet. 2012, 49, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Gochuico, B.R.; Hossain, M.; Talvacchio, S.K.; Zuo, M.X.; Barton, M.; Do, A.N.; Marini, J.C. Pulmonary function and structure abnormalities in children and young adults with osteogenesis imperfecta point to intrinsic and extrinsic lung abnormalities. J. Med. Genet. 2023, 60, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Valli, M.; Barnes, A.M.; Gallanti, A.; Cabral, W.A.; Viglio, S.; Weis, M.A.; Makareeva, E.; Eyre, D.; Leikin, S.; Antoniazzi, F.; et al. Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix. Clin. Genet. 2012, 82, 453–459. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Flanders, T.Y.; Pollock, P.M.; Hayward, N.K. The CDKN2A (p16) gene and human cancer. Mol. Med. 1997, 3, 5–20. [Google Scholar] [CrossRef]
- Pines, J.; Hunter, T. Isolation of a human cyclin cDNA: Evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell 1989, 58, 833–846. [Google Scholar] [CrossRef]
- Amano, K.; Ichida, F.; Sugita, A.; Hata, K.; Wada, M.; Takigawa, Y.; Nakanishi, M.; Kogo, M.; Nishimura, R.; Yoneda, T. MSX2 stimulates chondrocyte maturation by controlling Ihh expression. J. Biol. Chem. 2008, 283, 29513–29521. [Google Scholar] [CrossRef]
- Muttigi, M.S.; Han, I.; Park, H.K.; Park, H.; Lee, S.H. Matrilin-3 Role in Cartilage Development and Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 590. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Trehan, S.K.; Guan, Y.; Sun, C.; Moore, D.C.; Jayasuriya, C.T.; Chen, Q. Matrilin-3 inhibits chondrocyte hypertrophy as a bone morphogenetic protein-2 antagonist. J. Biol. Chem. 2014, 289, 34768–34779. [Google Scholar] [CrossRef]
- Cambier, S.; Gline, S.; Mu, D.; Collins, R.; Araya, J.; Dolganov, G.; Einheber, S.; Boudreau, N.; Nishimura, S.L. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: An angiogenic control switch. Am. J. Pathol. 2005, 166, 1883–1894. [Google Scholar] [CrossRef]
- Fratzl-Zelman, N.; Morello, R.; Lee, B.; Rauch, F.; Glorieux, F.H.; Misof, B.M.; Klaushofer, K.; Roschger, P. CRTAP deficiency leads to abnormally high bone matrix mineralization in a murine model and in children with osteogenesis imperfecta type VII. Bone 2010, 46, 820–826. [Google Scholar] [CrossRef]
- Arslan, M.A.; Chikina, M.; Csermely, P.; Sőti, C. Misfolded proteins inhibit proliferation and promote stress-induced death in SV40-transformed mammalian cells. FASEB J. 2012, 26, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Mitra, A.; Besio, R.; Contento, B.M.; Wong, K.W.; Derkyi, A.; To, M.; Forlino, A.; Dale, R.K.; Marini, J.C. Absence of TRIC-B from type XIV Osteogenesis Imperfecta osteoblasts alters cell adhesion and mitochondrial function—A multi-omics study. Matrix Biol. 2023, 121, 127–148. [Google Scholar] [PubMed]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar]
- Gremminger, V.L.; Jeong, Y.; Cunningham, R.P.; Meers, G.M.; Rector, R.S.; Phillips, C.L. Compromised Exercise Capacity and Mitochondrial Dysfunction in the Osteogenesis Imperfecta Murine (oim) Mouse Model. J. Bone Miner. Res. 2019, 34, 1646–1659. [Google Scholar] [CrossRef]
- Dobson, P.F.; Dennis, E.P.; Hipps, D.; Reeve, A.; Laude, A.; Bradshaw, C.; Stamp, C.; Smith, A.; Deehan, D.J.; Turnbull, D.M.; et al. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Sci. Rep. 2020, 10, 11643. [Google Scholar]
- Khan, A.U.; Qu, R.; Fan, T.; Ouyang, J.; Dai, J. A glance on the role of actin in osteogenic and adipogenic differentiation of mesenchymal stem cells. Stem Cell Res. Ther. 2020, 11, 283. [Google Scholar] [CrossRef]
- Bianchi, L.; Gagliardi, A.; Maruelli, S.; Besio, R.; Landi, C.; Gioia, R.; Kozloff, K.M.; Khoury, B.M.; Coucke, P.J.; Symoens, S. Altered cytoskeletal organization characterized lethal but not surviving Brtl+/− mice: Insight on phenotypic variability in osteogenesis imperfecta. Hum. Mol. Genet. 2015, 24, 6118–6133. [Google Scholar]
- Zimmerman, S.M.; Dimori, M.; Heard Lipsmeyer, M.E.; Morello, R. The Osteocyte Transcriptome Is Extensively Dysregulated in Mouse Models of Osteogenesis Imperfecta. J. Bone Miner. Res. Plus 2019, 3, e10171. [Google Scholar]
- Shapiro, F.; Maguire, K.; Swami, S.; Zhu, H.; Flynn, E.; Wang, J.; Wu, J.Y. Histopathology of osteogenesis imperfecta bone. Supramolecular assessment of cells and matrices in the context of woven and lamellar bone formation using light polarization and ultrastructural microscopy. Bone Rep. 2021, 14, 100734. [Google Scholar]
- Chen, G.; Deng, C.; Li, Y.P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar]
- Beederman, M.; Lamplot, J.D.; Nan, G.; Wang, J.; Liu, X.; Yin, L.; Li, R.; Shui, W.; Zhang, H.; Kim, S.H. BMP signaling in mesenchymal stem cell differentiation and bone formation. J. Biomed. Sci. Eng. 2013, 6, 32–52. [Google Scholar] [PubMed]
- Shu, B.; Zhang, M.; Xie, R.; Wang, M.; Jin, H.; Hou, W.; Tang, D.; Harris, S.E.; Mishina, Y.; O’Keefe, R.J.; et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 2011, 124 Pt 20, 3428–3440. [Google Scholar] [PubMed]
- Grafe, I.; Yang, T.; Alexander, S.; Homan, E.P.; Lietman, C.; Jiang, M.M.; Bertin, T.; Munivez, E.; Chen, Y.; Dawson, B.; et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014, 20, 670–675. [Google Scholar] [CrossRef]
- Markmann, A.; Hausser, H.; Schönherr, E.; Kresse, H. Influence of decorin expression on transforming growth factor-beta-mediated collagen gel retraction and biglycan induction. Matrix Biol. 2000, 19, 631–636. [Google Scholar] [PubMed]
- Castagnola, P.; Gennari, M.; Morello, R.; Tonachini, L.; Marin, O.; Gaggero, A.; Cancedda, R. Cartilage associated protein (CASP) is a novel developmentally regulated chick embryo protein. J. Cell Sci. 1997, 110 Pt 12, 1351–1359. [Google Scholar]
- Fratzl-Zelman, N.; Barnes, A.M.; Weis, M.; Carter, E.; Hefferan, T.E.; Perino, G.; Chang, W.; Smith, P.A.; Roschger, P.; Klaushofer, K.; et al. Non-Lethal Type VIII Osteogenesis Imperfecta Has Elevated Bone Matrix Mineralization. J. Clin. Endocrinol. Metab. 2016, 101, 3516–3525. [Google Scholar] [CrossRef]
- Hall, J.G.; Gripp, K.W.; Slavotinek, A.M. Hall’s Handbook of Physical Measurements; Oxford University Press: Oxford, UK, 2006. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Hyp% (FB) | Hyl% (FB) | P986 3-OH (FB) | P986 3-OH (OB) |
|---|---|---|---|---|
| Controls | 42.7–49.4 | 22.5–33.8 | 95–98 | 95 |
| NL-1 | 46.1 | 48.1 | 0 | 0 |
| NL-2 | 44.5 | 49.1 | 4 | ND |
| L-1 | 45.8 | 48.4 | 0 * | ND |
| L-2 | 44.7 | 42.9 | 0 ** | ND |
| GO Pathway | Description | p-adj | Pathway Genes |
|---|---|---|---|
| GO: 0051047 | positive regulation of secretion | 0.000937 | FGR, PTPN22, AIM2, AGT, INHBB, CACNA1D, TRH, SNCA, NPY2R, C1QTNF3, F2R, F2RL1, TSLP, HLA-F, AQP1, ZP3, BLK, CCL19, GATA3, CASP1, IGF1, P2RX7, RPH3AL, ALOX15B, CACNA1G, MYOM1, MBP, NLRP2, OPRL1, ITGB2, APLN |
| GO: 1903532 | positive regulation of secretion by cell | 0.00132 | FGR, PTPN22, AIM2, AGT, INHBB, CACNA1D, TRH, SNCA, NPY2R, C1QTNF3, F2R, F2RL1, TSLP, HLA-F, ZP3, BLK, CCL19, GATA3, CASP1, IGF1, P2RX7, RPH3AL, ALOX15B, CACNA1G, MYOM1, MBP, NLRP2, ITGB2, APLN |
| GO: 0050715 | positive regulation of cytokine secretion | 0.00581 | FGR, PTPN22, AIM2, C1QTNF3, F2R, F2RL1, TSLP, CCL19, GATA3, CASP1, P2RX7, ALOX15B, MBP, NLRP2 |
| GO: 0002791 | regulation of peptide secretion | 0.00581 | FGR, PTPN22, AIM2, INHBB, CACNA1D, TRH, NPY2R, C1QTNF3, F2R, F2RL1, TSLP, CD74, BLK, CHD7, CCL19, GATA3, ADRA2A, CASP1, CARD16, IGF1, P2RX7, RPH3AL, ALOX15B, NR1D1, MYOM1, MBP, C5AR2, NLRP2, CD40, APLN |
| GO: 0002793 | positive regulation of peptide secretion | 0.00581 | FGR, PTPN22, AIM2, TRH, NPY2R, C1QTNF3, F2R, F2RL1, TSLP, BLK, CCL19, GATA3, CASP1, IGF1, P2RX7, RPH3AL, ALOX15B, MYOM1, MBP, NLRP2, APLN |
| GO: 0050663 | cytokine secretion | 0.0096 | FGR, GBP5, PTPN22, AIM2, AGT, C1QTNF3, F2R, F2RL1, TSLP, CCL19, GATA3, CASP1, CARD16, P2RX7, ALOX15B, MBP, C5AR2 |
| GO: 0050702 | interleukin-1 beta secretion | 0.01 | GBP5, AIM2, F2RL1, CCL19, CASP1, CARD16, P2RX7, NLRP2 |
| GO: 0050714 | positive regulation of protein secretion | 0.0108 | FGR, PTPN22, AIM2, TRH, C1QTNF3, F2R, F2RL1, TSLP, BLK, CCL19, GATA3, CASP1, IGF1, P2RX7, RPH3AL, ALOX15B, MYOM1, MBP, NLRP2 |
| GO: 0050707 | regulation of cytokine secretion | 0.0138 | FGR, PTPN22, AIM2, C1QTNF3, F2R, F2RL1, TSLP, CCL19, GATA3, CASP1, CARD16, P2RX7, ALOX15B, MBP, C5AR2, NLRP2 |
| GO: 0050701 | interleukin-1 secretion | 0.018 | GBP5, AIM2, F2RL1, CCL19, CASP1, CARD16, P2RX7, NLRP2 |
| GO: 0050708 | regulation of protein secretion | 0.0229 | FGR, PTPN22, AIM2, INHBB, CACNA1D, TRH, C1QTNF3, F2R, F2RL1, TSLP, BLK, CCL19, GATA3, ADRA2A, CASP1, CARD16, IGF1, P2RX7, RPH3AL, ALOX15B, NR1D1, MYOM1, MBP, C5AR2, NLRP2, CD40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barnes, A.M.; Mitra, A.; Knue, M.M.; Derkyi, A.; Dang Do, A.; Dale, R.K.; Marini, J.C. CRTAP-Null Osteoblasts Have Increased Proliferation, Protein Secretion, and Skeletal Morphogenesis Gene Expression with Downregulation of Cellular Adhesion. Cells 2025, 14, 518. https://doi.org/10.3390/cells14070518
Barnes AM, Mitra A, Knue MM, Derkyi A, Dang Do A, Dale RK, Marini JC. CRTAP-Null Osteoblasts Have Increased Proliferation, Protein Secretion, and Skeletal Morphogenesis Gene Expression with Downregulation of Cellular Adhesion. Cells. 2025; 14(7):518. https://doi.org/10.3390/cells14070518
Chicago/Turabian StyleBarnes, Aileen M., Apratim Mitra, Marianne M. Knue, Alberta Derkyi, An Dang Do, Ryan K. Dale, and Joan C. Marini. 2025. "CRTAP-Null Osteoblasts Have Increased Proliferation, Protein Secretion, and Skeletal Morphogenesis Gene Expression with Downregulation of Cellular Adhesion" Cells 14, no. 7: 518. https://doi.org/10.3390/cells14070518
APA StyleBarnes, A. M., Mitra, A., Knue, M. M., Derkyi, A., Dang Do, A., Dale, R. K., & Marini, J. C. (2025). CRTAP-Null Osteoblasts Have Increased Proliferation, Protein Secretion, and Skeletal Morphogenesis Gene Expression with Downregulation of Cellular Adhesion. Cells, 14(7), 518. https://doi.org/10.3390/cells14070518