
Strategies for p53 Activation and Targeted Inhibitors of the p53-Mdm2/MdmX Interaction


Abstract
:1. Introduction
2. The Regulatory Circuit Between p53 and Mdm2/MdmX
3. Strategies for Activating p53
3.1. Activate the Activity of mutp53
- (a)
- Hsp90 inhibitors: These inhibitors prevent the binding of Hsp90 chaperones to mutp53, thereby facilitating its degradation [38];
- (b)
- HDAC inhibitors: These agents suppress transcriptional activity regulated by HDAC and disrupt the HDAC6/Hsp90/mutp53 complex [39];
- (c)
- Statins: Statins inhibit the interaction between mutp53 and DNAJA1, inducing CHIP-dependent mutp53 degradation [40];
- (d)
- Gambogic acid: This compound enhances wtp53 levels, disrupts the mutp53/Hsp90 complex, and promotes CHIP-mediated degradation of mutp53 [41];
- (e)
- Spautin-1: Spautin-1 inhibits macro autophagy and induces mutp53 degradation through partner-mediated autophagy [42].
3.2. Unleash the Tumor-Suppressive Activity of wtp53
3.2.1. Inhibitors Targeting the p53-Mdm2 Interaction
3.2.2. Monotherapy p53-MdmX Inhibitors and Dual p53-Mdm2/MdmX Inhibitors
3.3. Other Emerging p53-Based Therapeutic Strategies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harris, C.C. Structure and function of the p53 tumor suppressor gene: Clues for rational cancer therapeutic strategies. J. Natl. Cancer. Inst. 1996, 88, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Hernández, B.L.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta-Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Paraskeva, C.; Markowitz, S.; Willson, J.K.; Hamilton, S.; Vogelstein, B. P53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990, 50, 7717–7722. [Google Scholar] [PubMed]
- Kawai, H.; Wiederschain, D.; Yuan, Z.M. Critical contribution of the mdm2 acidic domain to p53 ubiquitination. Mol. Cell. Biol. 2003, 23, 4939–4947. [Google Scholar] [CrossRef]
- Yu, G.W.; Rudiger, S.; Veprintsev, D.; Freund, S.; Fernandez-Fernandez, M.R.; Fersht, A.R. The central region of hdm2 provides a second binding site for p53. Proc. Natl. Acad. Sci. USA 2006, 103, 1227–1232. [Google Scholar] [CrossRef]
- Poyurovsky, M.V.; Katz, C.; Laptenko, O.; Beckerman, R.; Lokshin, M.; Ahn, J.; Byeon, I.J.; Gabizon, R.; Mattia, M.; Zupnick, A.; et al. The c terminus of p53 binds the n-terminal domain of mdm2. Nat. Struct. Mol. Biol. 2010, 17, 982–989. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein mdm2 is a ubiquitin ligase e3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: mdm2 and mdmx set the tone. Trends Cell Biol. 2010, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.E.; Piette, J.; Zawadzki, J.A.; Harvey, D.; Levine, A.J. The mdm-2 gene is induced in response to UV light in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1993, 90, 11623–11627. [Google Scholar] [CrossRef]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of p53 inhibits its ubiquitination by mdm2. J. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar] [CrossRef]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef]
- Jenkins, L.M.; Durell, S.R.; Mazur, S.J.; Appella, E. P53 n-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef]
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159. [Google Scholar] [CrossRef]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural basis of DNA recognition by p53 tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Lozano, G. Restoring p53 in cancer: The promises and the challenges. J. Mol. Cell Biol. 2019, 11, 615–619. [Google Scholar] [CrossRef]
- Zhang, S.; Carlsen, L.; Hernandez, B.L.; Seyhan, A.A.; Tian, X.; El-Deiry, W.S. Advanced strategies for therapeutic targeting of wild-type and mutant p53 in cancer. Biomolecules 2022, 12, 548. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer. Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef]
- Rippin, T.M.; Bykov, V.J.; Freund, S.M.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug cp-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef]
- Zache, N.; Lambert, J.M.; Rökaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J. Mutant p53 targeting by the low molecular weight compound stima-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef]
- Rökaeus, N.; Shen, J.; Eckhardt, I.; Bykov, V.J.N.; Wiman, K.G.; Wilhelm, M.T. Prima-1met/apr-246 targets mutant forms of p53 family members p63 and p73. Oncogene 2010, 29, 6442–6451. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Prima-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound apr-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase i dose-finding study of apr-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef]
- David, A.; Sallman, M.; Amy, E.; Dezern, M.; Guillermo Garcia-Manero, M.; David, P.; Steensma, M.; Gail, J.; Roboz, M.; Mikkael, A.; et al. Eprenetapopt (apr-246) and azacitidine in tp53-mutant myelodysplastic syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar]
- Xie, X.; Fan, C.; Luo, B.; Zhang, J.; Jensen, L.D.; Burman, J.; Jönsson, C.; Ljusberg, A.; Larsson, P.; Zhao, Z.; et al. Apr-246 enhances colorectal cancer sensitivity to radiotherapy. Mol. Cancer Ther. 2023, 22, 947–961. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh, N.A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-function (gof) mutant p53 as actionable therapeutic target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tsai, M.; Shiao, Y.; Chen, L.; Wei, M.; Lv, X.; Gius, D.; Little, J.B.; Mitchell, J.B.; Chuang, E.Y. Dna (cytosine-5)-methyltransferase 1 as a mediator of mutant p53-determined p16(ink4a) down-regulation. J. Biomed. Sci. 2008, 15, 163–168. [Google Scholar] [CrossRef]
- Subramanian, M.; Francis, P.; Bilke, S.; Li, X.L.; Hara, T.; Lu, X.; Jones, M.F.; Walker, R.L.; Zhu, Y.; Pineda, M.; et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene 2015, 34, 1094–1104. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by mdm2 or p16ink4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Yallowitz, A.R.; Marchenko, N.D.; Xu, S.; Moll, U.M. Blockade of hsp90 by 17aag antagonizes mdmx and synergizes with nutlin to induce p53-mediated apoptosis in solid tumors. Cell Death Dis. 2011, 2, e156. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. Saha shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the hdac6-hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, Q.; Qi, Q.; Gu, H.; Rong, J.; Mu, R.; Zou, M.; Tao, L.; You, Q.; Guo, Q. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to chip. J. Cell. Biochem. 2011, 112, 509–519. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes. Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Targeting the mdm2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Wang, S. Therapeutic strategies to activate p53. Pharmaceuticals 2022, 16, 24. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of mdm2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Li, Y.; Mu, K.; Li, Z.; Meng, Q.; Wu, X.; Wang, Y.; Li, L. Amplification of mdmx and overexpression of mdm2 contribute to mammary carcinogenesis by substituting for p53 mutations. Diagn. Pathol. 2014, 9, 71. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the mdm2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53-mdm2 interaction by small-molecule inhibitors: Learning from mdm2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 91. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Wang, G.; Yang, Y.; Yuan, Y.; Ouyang, L. The past, present and future of potential small-molecule drugs targeting p53-mdm2/mdmx for cancer therapy. Eur. J. Med. Chem. 2019, 176, 92–104. [Google Scholar] [CrossRef]
- Beloglazkina, A.; Zyk, N.; Majouga, A.; Beloglazkina, E. Recent small-molecule inhibitors of the p53-mdm2 protein-protein interaction. Molecules 2020, 25, 1211. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.P.; Blay, J.; Italiano, A.; Gutierrez, M.; Le Cesne, A.; Gomez-Roca, C.A.; Gouw, L.G.; von Mehren, M.; Wagner, A.; Maki, R.G.; et al. Phase ib study of rg7112 with doxorubicin (d) in advanced soft tissue sarcoma (asts). J. Clin. Oncol. 2013, 31, 10514. [Google Scholar] [CrossRef]
- Patnaik, A.; Tolcher, A.; Beeram, M.; Nemunaitis, J.; Weiss, G.J.; Bhalla, K.; Agrawal, M.; Nichols, G.; Middleton, S.; Beryozkina, A.; et al. Clinical pharmacology characterization of rg7112, an mdm2 antagonist, in patients with advanced solid tumors. Cancer. Chemother. Pharmacol. 2015, 76, 587–595. [Google Scholar] [CrossRef]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the mdm2 inhibitor amg 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef]
- Hong, Y.; Ishizuka, T.; Watanabe, A.; Tachibana, M.; Lee, M.; Ishizuka, H.; Lacreta, F.; Abutarif, M. Model-based assessments of cyp3a-mediated drug-drug interaction risk of milademetan. CTS-Clin. Transl. Sci. 2021, 14, 2220–2230. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Demetri, G.D.; Halilovic, E.; Dummer, R.; Meille, C.; Tan, D.; Guerreiro, N.; Jullion, A.; Ferretti, S.; Jeay, S.; et al. Pharmacokinetic-pharmacodynamic guided optimisation of dose and schedule of cgm097, an hdm2 inhibitor, in preclinical and clinical studies. Br. J. Cancer 2021, 125, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.; Papayannidis, C.; Vey, N.; Dickinson, M.J.; Kelly, K.R.; Assouline, S.; Kasner, M.; Seiter, K.; Drummond, M.W.; Yoon, S.S.; et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study. Leuk. Res. 2021, 100, 106489. [Google Scholar] [CrossRef]
- Italiano, A.; Miller, W.J.; Blay, J.Y.; Gietema, J.A.; Bang, Y.J.; Mileshkin, L.R.; Hirte, H.W.; Higgins, B.; Blotner, S.; Nichols, G.L.; et al. Phase i study of daily and weekly regimens of the orally administered mdm2 antagonist idasanutlin in patients with advanced tumors. Investig. New Drugs 2021, 39, 1587–1597. [Google Scholar] [CrossRef]
- Abdul, R.A.; Miller, W.J.; Uy, G.L.; Blotner, S.; Young, A.M.; Higgins, B.; Chen, L.C.; Gore, L. A phase 1 study of the mdm2 antagonist ro6839921, a pegylated prodrug of idasanutlin, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1156–1165. [Google Scholar] [CrossRef]
- Rasco, D.W.; Lakhani, N.J.; Li, Y.; Men, L.; Wang, H.; Ji, J.; Tang, Y.; Liang, Z.; Amaya, A.; Estkowski, K.; et al. A phase i study of a novel mdm2 antagonist apg-115 in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3126. [Google Scholar] [CrossRef]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local activation of p53 in the tumor microenvironment overcomes immune suppression and enhances antitumor immunity. Cancer Res. 2017, 77, 2292–2305. [Google Scholar] [CrossRef]
- Arkin, M. Protein-protein interactions and cancer: Small molecules going in for the kill. Curr. Opin. Chem. Biol. 2005, 9, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of rg7112: A small-molecule mdm2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the mdm2 antagonist rg7112 on the p53 pathway in patients with mdm2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. Mdm2 small-molecule antagonist rg7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef]
- Rew, Y.; Sun, D. Discovery of a small molecule mdm2 inhibitor (amg 232) for treating cancer. J. Med. Chem. 2014, 57, 6332–6341. [Google Scholar] [CrossRef]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of amg 232, a potent, selective, and orally bioavailable mdm2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The mdm2 inhibitor amg 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, M.; Ma, T.; Ning, H.; Liu, L. Amg232 inhibits angiogenesis in glioma through the p53-rbm4-vegfr2 pathway. J. Cell Sci. 2023, 136, jcs260270. [Google Scholar] [CrossRef]
- Gluck, W.L.; Gounder, M.M.; Frank, R.; Eskens, F.; Blay, J.Y.; Cassier, P.A.; Soria, J.C.; Chawla, S.; de Weger, V.; Wagner, A.J.; et al. Phase 1 study of the mdm2 inhibitor amg 232 in patients with advanced p53 wild-type solid tumors or multiple myeloma. Investig. New Drugs 2020, 38, 831–843. [Google Scholar] [CrossRef]
- Verstovsek, S.; Al-Ali, H.K.; Mascarenhas, J.; Perkins, A.; Vannucchi, A.M.; Mohan, S.R.; Scott, B.L.; Woszczyk, D.; Koschmieder, S.; García-Delgado, R.; et al. Boreas: A global, phase iii study of the mdm2 inhibitor navtemadlin (krt-232) in relapsed/refractory myelofibrosis. Future Oncol. 2022, 18, 4059–4069. [Google Scholar] [CrossRef]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and schedule determine distinct molecular mechanisms underlying the efficacy of the p53-mdm2 inhibitor hdm201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. Discovery of potent and selective p53-mdm2 protein-protein interaction inhibitors as anticancer drugs. Chimia 2017, 71, 716–721. [Google Scholar] [CrossRef]
- Stein, E.M.; Deangelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a first-in-human phase i study of siremadlin (hdm201) in patients with advanced wild-type tp53 solid tumors and acute leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; Mceachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. Sar405838: An optimized inhibitor of mdm2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, M.; de Weger, V.A.; Dickson, M.A.; Langenberg, M.; Le Cesne, A.; Wagner, A.J.; Hsu, K.; Zheng, W.; Macé, S.; Tuffal, G.; et al. A phase i study of sar405838, a novel human double minute 2 (hdm2) antagonist, in patients with solid tumours. Eur. J. Cancer 2017, 76, 144–151. [Google Scholar] [CrossRef]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D.; Mceachern, D.; Przybranowski, S.; Li, X.; Luo, R.; Wen, B.; et al. Discovery of 4-((3′r,4′s,5′r)-6″-chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic acid (aa-115/apg-115): A potent and orally active murine double minute 2 (mdm2) inhibitor in clinical development. J. Med. Chem. 2017, 60, 2819–2839. [Google Scholar]
- Yi, H.; Yan, X.; Luo, Q.; Yuan, L.; Li, B.; Pan, W.; Zhang, L.; Chen, H.; Wang, J.; Zhang, Y.; et al. A novel small molecule inhibitor of mdm2-p53 (apg-115) enhances radiosensitivity of gastric adenocarcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 97. [Google Scholar] [CrossRef]
- Zhai, Y.; Tang, Q.; Fang, D.D.; Deng, J.; Zhang, K.; Wang, Q.; Yin, Y.; Fu, C.; Xue, S.L.; Li, N.; et al. Lisaftoclax in combination with alrizomadlin overcomes venetoclax resistance in acute myeloid leukemia and acute lymphoblastic leukemia: Preclinical studies. Clin. Cancer Res. 2023, 29, 183–196. [Google Scholar] [CrossRef]
- Gollner, A.; Rudolph, D.; Arnhof, H.; Bauer, M.; Blake, S.M.; Boehmelt, G.; Cockroft, X.L.; Dahmann, G.; Ettmayer, P.; Gerstberger, T.; et al. Discovery of novel spiro[3h-indole-3,2′-pyrrolidin]-2(1h)-one compounds as chemically stable and orally active inhibitors of the mdm2-p53 interaction. J. Med. Chem. 2016, 59, 10147–10162. [Google Scholar] [CrossRef]
- Gollner, A.; Broeker, J.; Kerres, N.; Kofink, C.; Ramharter, J.; Weinstabl, H.; Gille, A.; Goepper, S.; Henry, M.; Huchler, G. Spiro[3h-indole-3,2′-pyrrolidin]-2(1h)-One Compounds and Derivatives as mdm2-p53 Inhibitors. U.S. Patent 10,138,251, 27 November 2016. [Google Scholar]
- Lorusso, P.; Yamamoto, N.; Patel, M.R.; Laurie, S.A.; Bauer, T.M.; Geng, J.; Davenport, T.; Teufel, M.; Li, J.; Lahmar, M.; et al. The mdm2-p53 antagonist brigimadlin (bi 907828) in patients with advanced or metastatic solid tumors: Results of a phase ia, first-in-human, dose-escalation study. Cancer Discov. 2023, 13, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Bahia, R.K.; Cseh, O.; Bozek, D.A.; Blake, S.; Rinnenthal, J.; Weyer-Czernilofsky, U.; Rudolph, D.; Artee, L.H. Bi-907828, a novel potent mdm2 inhibitor, inhibits glioblastoma brain tumor stem cells in vitro and prolongs survival in orthotopic xenograft mouse models. Neuro-Oncology 2023, 25, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Cornillie, J.; Wozniak, A.; Li, H.; Gebreyohannes, Y.K.; Wellens, J.; Hompes, D.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P. Anti-tumor activity of the mdm2-tp53 inhibitor bi-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring mdm2 amplification. Clin. Transl. Oncol. 2020, 22, 546–554. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the mdm2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef]
- Koyama, T.; Shimizu, T.; Kojima, Y.; Sudo, K.; Okuma, H.S.; Shimoi, T.; Ichikawa, H.; Kohsaka, S.; Sadachi, R.; Hirakawa, A.; et al. Clinical activity and exploratory resistance mechanism of milademetan, an mdm2 inhibitor, in intimal sarcoma with mdm2 amplification: An open-label phase ib/ii study. Cancer Discov. 2023, 13, 1814–1825. [Google Scholar] [CrossRef]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to tp53-mdm2 inhibition identified by in vivo piggybac transposon mutagenesis screen in an arf(−/−) mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Ozaki, T.; Yoshida, K.; Hosoda, M.; Todo, S.; Akiyama, S.; Nakagawara, A. P73 and mdm2 confer the resistance of epidermoid carcinoma to cisplatin by blocking p53. Biochem. Biophys. Res. Commun. 2006, 347, 60–66. [Google Scholar] [CrossRef]
- Bista, M.; Smithson, D.; Pecak, A.; Salinas, G.; Pustelny, K.; Min, J.; Pirog, A.; Finch, K.; Zdzalik, M.; Waddell, B.; et al. On the mechanism of action of sj-172550 in inhibiting the interaction of mdm4 and p53. PLoS ONE 2012, 7, e37518. [Google Scholar] [CrossRef]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D.; et al. Identification and characterization of the first small molecule inhibitor of mdmx. J. Biol. Chem. 2010, 285, 10786–10796. [Google Scholar] [CrossRef]
- Karan, G.; Wang, H.; Chakrabarti, A.; Karan, S.; Liu, Z.; Xia, Z.; Gundluru, M.; Moreton, S.; Saunthararajah, Y.; Jackson, M.W.; et al. Identification of a small molecule that overcomes hdmx-mediated suppression of p53. Mol. Cancer Ther. 2016, 15, 574–582. [Google Scholar] [CrossRef]
- Popowicz, G.M.; Czarna, A.; Wolf, S.; Wang, K.; Wang, W.; Dömling, A.; Holak, T.A. Structures of low molecular weight inhibitors bound to mdmx and mdm2 reveal new approaches for p53-mdmx/mdm2 antagonist drug discovery. Cell Cycle 2010, 9, 1104–1111. [Google Scholar] [CrossRef]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α-helical peptide drug development: A potent dual inhibitor of mdm2 and mdmx for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef]
- Pairawan, S.; Zhao, M.; Yuca, E.; Annis, A.; Evans, K.; Sutton, D.; Carvajal, L.; Ren, J.G.; Santiago, S.; Guerlavais, V.; et al. First in class dual mdm2/mdmx inhibitor alrn-6924 enhances antitumor efficacy of chemotherapy in tp53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, K.; Cai, Y. An overview of development in gene therapeutics in china. Gene Ther. 2020, 27, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene therapy leaves a vicious cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The first approved gene therapy product for cancer ad-p53 (gendicine): 12 years in the clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Du, Z.; Wang, X.; Li, X. Treatment of uterine sarcoma with rad-p53 (gendicine) followed by chemotherapy: Clinical study of tp53 gene therapy. Hum. Gene Ther. 2018, 29, 242–250. [Google Scholar] [CrossRef]
- Liu, S.; Chen, P.; Hu, M.; Tao, Y.; Chen, L.; Liu, H.; Wang, J.; Luo, J.; Gao, G. Randomized, controlled phase ii study of post-surgery radiotherapy combined with recombinant adenoviral human p53 gene therapy in treatment of oral cancer. Cancer Gene Ther. 2013, 20, 375–378. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Ingn 201 (advexin): Adenoviral p53 gene therapy for cancer. Expert Opin. Biol. Ther. 2006, 6, 823–832. [Google Scholar] [CrossRef]
- Atencio, I.A.; Grace, M.; Bordens, R.; Fritz, M.; Horowitz, J.A.; Hutchins, B.; Indelicato, S.; Jacobs, S.; Kolz, K.; Maneval, D.; et al. Biological activities of a recombinant adenovirus p53 (sch 58500) administered by hepatic arterial infusion in a phase 1 colorectal cancer trial. Cancer Gene Ther. 2006, 13, 169–181. [Google Scholar] [CrossRef]
- Buller, R.E.; Runnebaum, I.B.; Karlan, B.Y.; Horowitz, J.A.; Shahin, M.; Buekers, T.; Petrauskas, S.; Kreienberg, R.; Slamon, D.; Pegram, M. A phase i/ii trial of rad/p53 (sch 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Ther. 2002, 9, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.E.; Hunger, A.; Fernandes, D.C.; Laurindo, F.R.; Costanzi-Strauss, E.; Strauss, B.E. Induction of oxidants distinguishes susceptibility of prostate carcinoma cell lines to p53 gene transfer mediated by an improved adenoviral vector. Hum. Gene Ther. 2017, 28, 639–653. [Google Scholar] [CrossRef]
- Chen, S.; Rong, L.; Lei, Q.; Cao, P.; Qin, S.; Zheng, D.; Jia, H.; Zhu, J.; Cheng, S.; Zhuo, R.; et al. A surface charge-switchable and folate modified system for co-delivery of proapoptosis peptide and p53 plasmid in cancer therapy. Biomaterials 2016, 77, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.K.; Naz, S.; Kondaiah, P.; Bhattacharya, S. A cationic cholesterol based nanocarrier for the delivery of p53-egfp-c3 plasmid to cancer cells. Biomaterials 2014, 35, 1334–1346. [Google Scholar] [CrossRef]
- Rejeeth, C.; Kannan, S. P53 gene therapy of human breast carcinoma: Using a transferrin-modified silica nanoparticles. Breast Cancer 2016, 23, 101–110. [Google Scholar] [CrossRef]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase i study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.C.; Sun, L.; Clavijo, P.E.; Friedman, J.; Harford, J.B.; Saleh, A.D.; Van Waes, C.; Chang, E.H.; Allen, C.T. Nanocomplex-based tp53 gene therapy promotes anti-tumor immunity through tp53- and sting-dependent mechanisms. OncoImmunology 2018, 7, e1404216. [Google Scholar] [CrossRef]
- Adams, C.M.; Mitra, R.; Xiao, Y.; Michener, P.; Palazzo, J.; Chao, A.; Gour, J.; Cassel, J.; Salvino, J.M.; Eischen, C.M. Targeted mdm2 degradation reveals a new vulnerability for p53-inactivated triple-negative breast cancer. Cancer Discov. 2023, 13, 1210–1229. [Google Scholar] [CrossRef]
- Kong, L.; Meng, F.; Zhou, P.; Ge, R.; Geng, X.; Yang, Z.; Li, G.; Zhang, L.; Wang, J.; Ma, J.; et al. An engineered dna aptamer-based protac for precise therapy of p53-r175h hotspot mutant-driven cancer. Sci. Bull. 2024, 69, 2122–2135. [Google Scholar] [CrossRef]



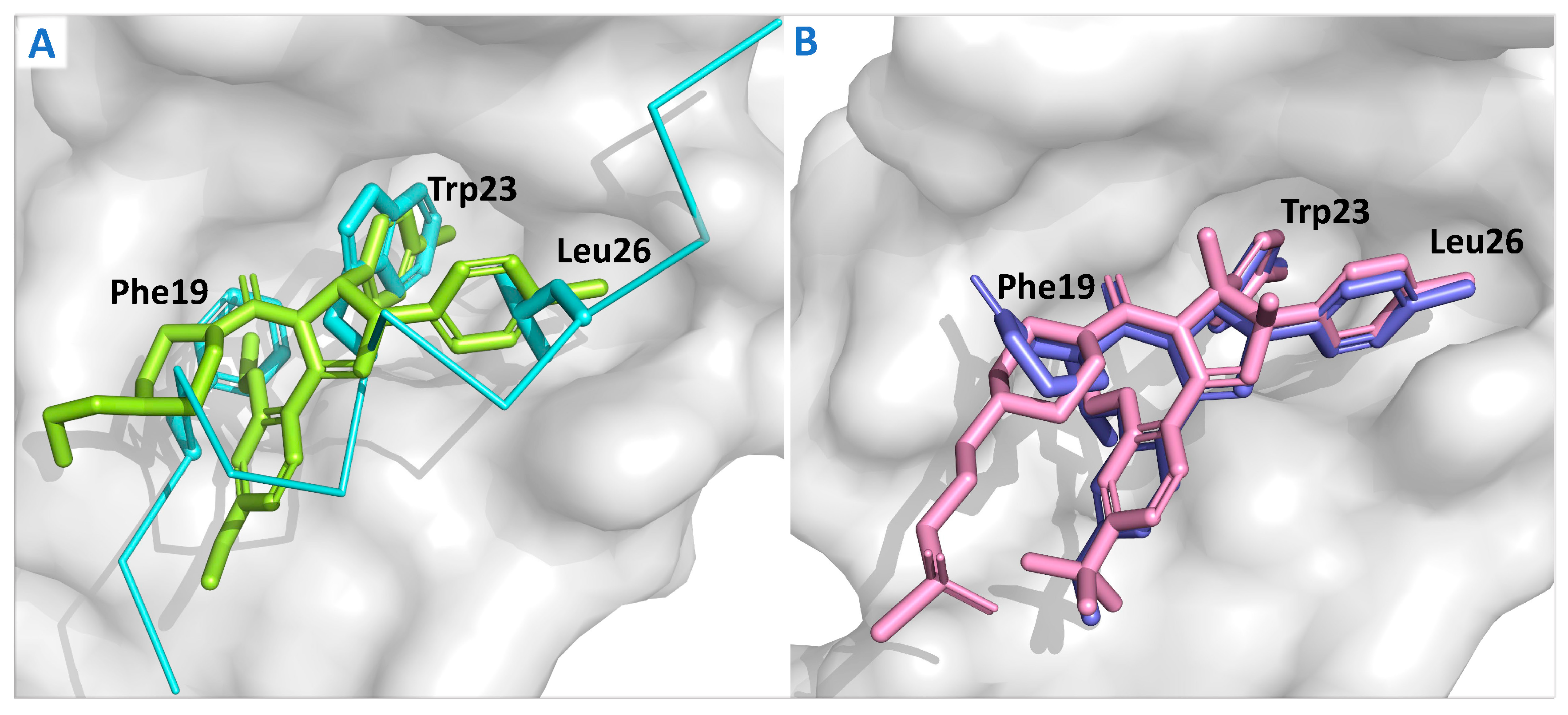
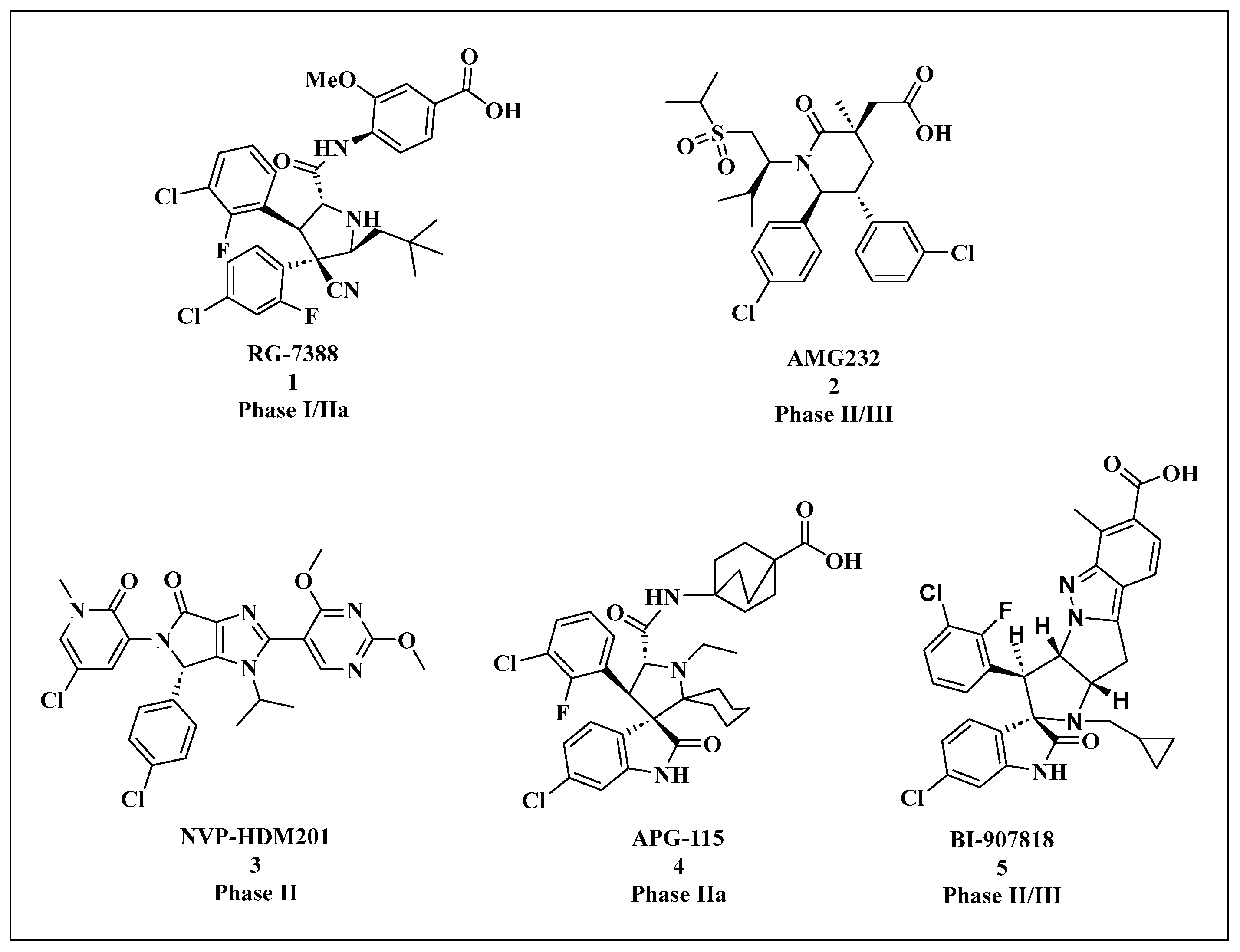


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Structure | Combination | Object | Phase | Result |
|---|---|---|---|---|---|
| RG7112 | imidazoline scaffold | Doxorubicin | Soft tissue sarcoma | Phase I NCT01605526 | Neutropenia (60%) and thrombocytopenia (45%) were observed [52]. |
| RG7112 | imidazoline scaffold | Late-stage solid tumors | Phase I NCT01164033 | High-dose treatment for 3 to 5 days was more effective than prolonged weekly or lower-dose daily treatments [53]. | |
| AMG232 | Piperidone class | Trametinib | Relapsed/Refractory AML | Phase I NCT02016729 | The single-dose treatment was 360 mg, and 60 mg when combined with trametinib. Among the 13 patients, 4 (31%) showed a response to the treatment [54]. |
| Milademetan(RAIN-32, DS-3032b) | Spirooxindole | Late-stage solid tumors and lymphomas | Phase I NCT01877382 | The maximum tolerated dose was 160 mg in the once-daily 21/28 schedule and 260 mg in the every day 3/14 × 2 schedule (1 cycle was 28 days) [55] | |
| NVP-CGM097 Series | Dihydro-isoquinolinones | Late-stage solid tumor | Phase I NCT01760525 | The drug was administered at doses of 10–400 mg weekly for 3 weeks, or 300–700 mg weekly for 2 weeks with a 1-week interval. The maximum tolerated dose was not reached. A portion (39%) of patients responded to the treatment, including one case of partial response and 19 patients with stable disease [56]. | |
| HDM201 | Pyrrolidono-imidazole | Late-stage solid tumors and hematological wtp53 tumors | Phase I NCT02143635 | Among patients with solid tumors, the response rate was 10.3%, whereas in patients with acute myeloid leukemia, the response rates varied across different regimens, being 4.2%, 20%, and 22.2%, respectively (NCT02143635). | |
| Idasanutlin (RO5503781, RG-7388) | Pyrrolidine | Cytarabine | wtp53 cancer patients | Phase I NCT01773408 | The remission rate for monotherapy was 18.9%, whereas the remission rate for combination therapy was 35.6% [57]. |
| Idasanutlin (RO5503781, RG-7388) | Pyrrolidine | tumor | Phase I NCT01462175 | The drug was administered at a daily dose of 500 mg for 5 days in a 28-day cycle and showed hematological toxicity [58]. | |
| RO6839921 (RG7775) | Polyethylene glycolation of pyrrolidine | Solid tumor and AML | Phase I NCT02098967 | Among solid tumor patients, 34% had stable disease, whereas the disease control rate for AML patients was 42% [59]. | |
| APG-115 | Spirooxindole | Advanced solid tumors or lymphomas. | Phase I NCT02935907 | The dosing regimen for APG-115 in a 28-day cycle involved administering 100 mg every other day for 21 days [60]. | |
| SAR405838 (MI-77301) | pirooxindole | Late-stage solid tumors | Phase I NCT01636479 | Each treatment involved 300 mg once daily and was associated with elevated plasma macrophage inhibitory cytokine-1 (MIC-1). 56% of patients experienced stable disease, and 32% remained progression-free at 3 months [60]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Li, W.; Zhou, Y.; Bai, J.; Li, N.; Su, Z.; Cheng, X. Strategies for p53 Activation and Targeted Inhibitors of the p53-Mdm2/MdmX Interaction. Cells 2025, 14, 583. https://doi.org/10.3390/cells14080583
Huang Y, Li W, Zhou Y, Bai J, Li N, Su Z, Cheng X. Strategies for p53 Activation and Targeted Inhibitors of the p53-Mdm2/MdmX Interaction. Cells. 2025; 14(8):583. https://doi.org/10.3390/cells14080583
Chicago/Turabian StyleHuang, Ye, Wang Li, Yuke Zhou, Jinping Bai, Ning Li, Zhengding Su, and Xiyao Cheng. 2025. "Strategies for p53 Activation and Targeted Inhibitors of the p53-Mdm2/MdmX Interaction" Cells 14, no. 8: 583. https://doi.org/10.3390/cells14080583
APA StyleHuang, Y., Li, W., Zhou, Y., Bai, J., Li, N., Su, Z., & Cheng, X. (2025). Strategies for p53 Activation and Targeted Inhibitors of the p53-Mdm2/MdmX Interaction. Cells, 14(8), 583. https://doi.org/10.3390/cells14080583