
Mitochondrial Dysfunction: A New Hallmark in Hereditable Thoracic Aortic Aneurysm Development

 , ,
, ,
Abstract
:1. Introduction
2. Molecular Mechanisms in Hereditable Thoracic Aortic Aneurysms
3. Mitochondrial Function in Vascular Pathophysiology
3.1. Mitochondrial Biogenesis
3.2. Mitochondrial Fusion–Fission
3.3. Mitophagy
3.4. Mitochondrial Alterations Associated with Reactive Oxygen Species Production
3.5. Mitochondrial Defects Associated with Aging
3.6. Mitochondrial Alterations Associated with Cytoskeleton–ECM Axis
4. Mitochondrial Dysfunction in Marfan Syndrome
5. Mitochondrial Respiratory Dysfunction in Loeys–Dietz Syndrome and Familial Thoracic Aortic Aneurysm and Dissections
6. Clinical Perspectives
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, G.H.; Stansby, G.; Hamilton, G. Suggested Standards for Reporting on Arterial Aneurysms. J. Vasc. Surg. 1992, 15, 452–458. [Google Scholar] [CrossRef]
- Rylski, B.; Schilling, O.; Czerny, M. Acute Aortic Dissection: Evidence, Uncertainties, and Future Therapies. Eur. Heart J. 2023, 44, 813–821. [Google Scholar] [CrossRef]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 607–618. [Google Scholar] [CrossRef]
- Gouveia EMelo, R.; Silva Duarte, G.; Lopes, A.; Alves, M.; Caldeira, D.; Fernandes EFernandes, R.; Mendes Pedro, L. Incidence and Prevalence of Thoracic Aortic Aneurysms: A Systematic Review and Meta-Analysis of Population-Based Studies. Semin. Thorac. Cardiovasc. Surg. 2022, 34, 1–16. [Google Scholar] [CrossRef]
- Kuzmik, G.A.; Sang, A.X.; Elefteriades, J.A. Natural History of Thoracic Aortic Aneurysms. J. Vasc. Surg. 2012, 56, 565–571. [Google Scholar] [CrossRef]
- McClure, R.S.; Brogly, S.B.; Lajkosz, K.; McClintock, C.; Payne, D.; Smith, H.N.; Johnson, A.P. Economic Burden and Healthcare Resource Use for Thoracic Aortic Dissections and Thoracic Aortic Aneurysms-A Population-Based Cost-of-Illness Analysis. J. Am. Heart Assoc. 2020, 9, 14981. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Ramirez, F. Therapies for Thoracic Aortic Aneurysms and Acute Aortic Dissections. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 126–136. [Google Scholar] [CrossRef]
- Salnikova, D.; Orekhova, V.; Grechko, A.; Starodubova, A.; Bezsonov, E.; Popkova, T.; Orekhov, A. Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8990. [Google Scholar] [CrossRef]
- Zhunina, O.A.; Yabbarov, N.G.; Grechko, A.V.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Vascular Disease, Tumorigenesis, and Diabetes. Front. Mol. Biosci. 2021, 8, 671908. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Ruiz-Rodríguez, M.J.; Desdín-Micó, G.; Aranda, J.F.; Rodrigues-Diez, R.; Ballesteros-Martínez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuña, P. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- Nguyen, T.A.V.; Lino, C.A.; Hang, H.T.; Alves, J.V.; Thang, B.Q.; Shin, S.J.; Sugiyama, K.; Matsunaga, H.; Takeyama, H.; Yamashiro, Y. Protective Role of Endothelial Fibulin-4 in Valvulo-Arterial Integrity. J. Am. Heart Assoc. 2023, 12, e026942. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial Dysfunction: Mechanisms and Advances in Therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Prakash, S.K.; Ramirez, F. Therapeutics Targeting Drivers of Thoracic Aortic Aneurysms and Acute Aortic Dissections: Insights from Predisposing Genes and Mouse Models. Annu. Rev. Med. 2017, 68, 51–67. [Google Scholar] [CrossRef]
- Faggion Vinholo, T.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Nonsyndromic Thoracic Aortic Aneurysms and Dissections-Is Screening Possible? Semin. Thorac. Cardiovasc. Surg. 2019, 31, 628–634. [Google Scholar] [CrossRef]
- Rohde, S.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Thoracic aortic aneurysm gene dictionary. Asian Cardiovasc. Thorac. Ann. 2021, 29, 682–696. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Salmasi, M.Y.; Alwis, S.; Cyclewala, S.; Jarral, O.A.; Mohamed, H.; Mozalbat, D.; Nienaber, C.A.; Athanasiou, T.; Morris-Rosendahl, D.; Moore, J., Jr.; et al. The genetic basis of thoracic aortic disease: The future of aneurysm classification? Hell. J. Cardiol. 2023, 69, 41–50. [Google Scholar] [CrossRef]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef]
- De Cario, R.; Giannini, M.; Cassioli, G.; Kura, A.; Gori, A.M.; Marcucci, R.; Nistri, S.; Pepe, G.; Giusti, B.; Sticchi, E. Tracking an Elusive Killer: State of the Art of Molecular-Genetic Knowledge and Laboratory Role in Diagnosis and Risk Stratification of Thoracic Aortic Aneurysm and Dissection. Diagnostics 2022, 12, 1785. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Braverman, A.C.; De Backer, J.; Morris, S.A.; Boileau, C.; Maumenee, I.H.; Jondeau, G.; Evangelista, A.; Pyeritz, R.E. Marfan syndrome. Nat. Rev. Dis. Primers 2021, 7, 64. [Google Scholar] [CrossRef]
- Zeigler, S.M.; Sloan, B.; Jones, J.A. Pathophysiology and Pathogenesis of Marfan Syndrome. Prog. Heritable Soft Connect. Tissue Dis. 2021, 1348, 185–206. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef]
- Bitterman, A.D.; Sponseller, P.D. Marfan Syndrome: A Clinical Update. J. Am. Acad. Orthop. Surg. 2017, 25, 603–609. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef]
- Keane, M.G.; Pyeritz, R.E. Medical Management of Marfan Syndrome. Circulation 2008, 117, 2802–2813. [Google Scholar] [CrossRef]
- Chen, M.; Cavinato, C.; Hansen, J.; Tanaka, K.; Ren, P.; Hassab, A.; Li, D.S.; Youshao, E.; Tellides, G.; Iyengar, R.; et al. FN (Fibronectin)-Integrin α5 Signaling Promotes Thoracic Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Arterioscler. Thromb. Vasc. Biol. 2023, 43, e132–e150. [Google Scholar] [CrossRef]
- von Kodolitsch, Y.; De Backer, J.; Schüler, H.; Bannas, P.; Behzadi, C.; Bernhardt, A.M.; Hillebrand, M.; Fuisting, B.; Sheikhzadeh, S.; Rybczynski, M.; et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl. Clin. Genet. 2015, 137, 137–155. [Google Scholar] [CrossRef]
- Du, Q.; Zhang, D.; Zhuang, Y.; Xia, Q.; Wen, T.; Jia, H. The Molecular Genetics of Marfan Syndrome. Int. J. Med. Sci. 2021, 18, 2752–2766. [Google Scholar] [CrossRef]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-β–induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef]
- Yu, C.; Jeremy, R.W. Angiotensin, transforming growth factor β and aortic dilatation in Marfan syndrome: Of mice and humans. Int. J. Cardiol. Heart Vasc. 2018, 18, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Arshad, F.; Quayle, L.A.; George, C.N.; Lefley, D.V.; Kalajzic, I.; Balsubramanian, M.; Cebe, T.; Reilly, G.; Bishop, N.J.; et al. Losartan alters osteoblast differentiation and increases bone mass through inhibition of TGFB signalling in vitro and in an OIM mouse model. Bone Rep. 2024, 22, 101795. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gonzalez, M.; Lubian-Gutierrez, M.; Cascales-Poyatos, H.M.; Perez-Reviriego, A.A.; Castellano-Martinez, A. Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy. Int. J. Mol. Sci. 2020, 22, 356. [Google Scholar] [CrossRef] [PubMed]
- Tingting, T.; Wenjing, F.; Qian, Z.; Hengquan, W.; Simin, Z.; Zhisheng, J.; Shunlin, Q. The TGF-β pathway plays a key role in aortic aneurysms. Clin. Chim. Acta 2020, 501, 222–228. [Google Scholar] [CrossRef]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 Antagonist, Prevents Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef]
- Teixido-Tura, G.; Forteza, A.; Rodríguez-Palomares, J.; González Mirelis, J.; Gutiérrez, L.; Sánchez, V.; Ibáñez, B.; García-Dorado, D.; Evangelista, A. Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients with Marfan Syndrome. J. Am. Coll. Cardiol. 2018, 72, 1613–1618. [Google Scholar] [CrossRef]
- Hofmann Bowman, M.A.; Eagle, K.A.; Milewicz, D.M. Update on Clinical Trials of Losartan with and Without β-Blockers to Block Aneurysm Growth in Patients With Marfan Syndrome. JAMA Cardiol. 2019, 4, 702. [Google Scholar] [CrossRef]
- Pepe, G.; Giusti, B.; Sticchi, E.; Abbate, R.; Gensini, G.; Nistri, S. Marfan Syndrome: Current Perspectives. Appl. Clin. Genet. 2016, 9, 55–65. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Roldan-Montero, R.; Ruiz-Rodríguez, M.J.; Redondo, J.M.; Martín-Ventura, J.L.; Mittelbrunn, M. Rewiring Vascular Metabolism Prevents Sudden Death due to Aortic Ruptures—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 462–469. [Google Scholar] [CrossRef]
- Bartolák-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of mitochondrial structure and dynamics by the cytoskeleton and mechanical factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Baixauli, F.; Acín-Pérez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabandé-Rodriguez, E.; Ledesma, M.D.; Blázquez, A.; Martin, M.A.; Falcón-Pérez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Escrig-Larena, J.I.; Delgado-Pulido, S.; Mittelbrunn, M. Mitochondria during T cell aging. Semin. Immunol. 2023, 69, 101808. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, W.; Cheng, J.; Qu, H.; Xu, M.; Wang, L. Effects of mitochondrial dysfunction on cellular function: Role in atherosclerosis. Biomed. Pharmacother. 2024, 174, 116587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Weng, J.; Huan, L.; Sheng, S.; Xu, F. Mitophagy in atherosclerosis: From mechanism to therapy. Front. Immunol. 2023, 14, 1165507. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, W.; Zhang, K.; Ke, X.; Wang, Z. New insights into the relationship of mitochondrial metabolism and atherosclerosis. Cell Signal 2024, 127, 111580. [Google Scholar] [CrossRef]
- Glanz, V.Y.; Sobenin, I.A.; Grechko, A.V.; Yet, S.-F.; Orekhov, A.N. The role of mitochondria in cardiovascular diseases related to atherosclerosis. Front. Biosci. 2020, 12, 102–112. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41, bnaa005. [Google Scholar] [CrossRef]
- Mendez-Barbero, N.; Oller, J.; Sanz, A.B.; Ramos, A.M.; Ortiz, A.; Ruiz-Ortega, M.; Rayego-Mateos, S. Mitochondrial Dysfunction in the Cardio-Renal Axis. Int. J. Mol. Sci. 2023, 24, 8209. [Google Scholar] [CrossRef]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, UK, 2000. [Google Scholar]
- Bernard, K.; Logsdon, N.J.; Ravi, S.; Xie, N.; Persons, B.P.; Rangarajan, S.; Zmijewski, J.W.; Mitra, K.; Liu, G.; Darley-Usmar, V.M.; et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem. 2015, 290, 25427–25438. [Google Scholar] [CrossRef]
- Wei, Z.; Chong, H.; Jiang, Q.; Tang, Y.; Xu, J.; Wang, H.; Shi, Y.; Cui, L.; Li, J.; Zhang, Y.; et al. Smooth Muscle Overexpression of PGC1α Attenuates Atherosclerosis in Rabbits. Circ. Res. 2021, 129, e72–e86. [Google Scholar] [CrossRef] [PubMed]
- Tavris, B.S.; Peters, A.S.; Böckler, D.; Dihlmann, S. Mitochondrial Dysfunction and Increased DNA Damage in Vascular Smooth Muscle Cells of Abdominal Aortic Aneurysm (AAA-SMC). Oxid. Med. Cell Longev. 2023, 2023, 6237960. [Google Scholar] [CrossRef]
- Yap, C.; Wanga, S.; Wüst, R.C.I.; van Os, B.W.; Pijls, M.M.E.; Keijzer, S.; van Zanten, E.; Koolbergen, D.R.; Driessen, A.H.G.; Balm, R.; et al. Doxycycline induces mitochondrial dysfunction in aortic smooth muscle cells. Vascul. Pharmacol. 2024, 154, 107279. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Luo, W.; Wang, Y.; Zhang, L.; Ren, P.; Zhang, C.; Li, Y.; Azares, A.R.; Zhang, M.; Guo, J.; Ghaghada, K.B.; et al. Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture. Circulation 2020, 141, 42–66. [Google Scholar] [CrossRef]
- Ishida, M. Mitochondrial fusion and fission in vascular disease. Hypertens. Res. 2024, 47, 2935–2938. [Google Scholar] [CrossRef]
- Ouyang, M.; Wang, M.; Yu, B. Aberrant Mitochondrial Dynamics: An Emerging Pathogenic Driver of Abdominal Aortic Aneurysm. Cardiovasc. Ther. 2021, 2021, 6615400. [Google Scholar] [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [CrossRef]
- Li, W.; Jiang, W.-S.; Su, Y.-R.; Tu, K.-W.; Zou, L.; Liao, C.-R.; Wu, Q.; Wang, Z.-H.; Zhong, Z.-M.; Chen, J.-T.; et al. PINK1/Parkin-mediated mitophagy inhibits osteoblast apoptosis induced by advanced oxidation protein products. Cell Death Dis. 2023, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Liu, Y.; Li, S.; Li, X.-J.; Yang, W. PINK1-PRKN mediated mitophagy: Differences between in vitro and in vivo models. Autophagy 2023, 19, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Valente, E.M. PINK1 in the limelight: Multiple functions of an eclectic protein in human health and disease. J. Pathol. 2017, 241, 251–263. [Google Scholar] [CrossRef]
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.-F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef]
- Burger, F.; Baptista, D.; Roth, A.; da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. Int. J. Mol. Sci. 2021, 23, 340. [Google Scholar] [CrossRef]
- He, X.; Bai, Q.; Zhang, X.; Zhang, L. MgCl2 Attenuates ox-LDL-Induced Vascular Smooth Muscle–Derived Foam Cells Pyroptosis by Downregulating the TLR4/NF-κB Signaling Pathway. Biol. Trace Elem. Res. 2023, 201, 5242–5256. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 28821–28835. [Google Scholar] [CrossRef]
- Nahapetyan, H.; Moulis, M.; Grousset, E.; Faccini, J.; Grazide, M.-H.; Mucher, E.; Elbaz, M.; Martinet, W.; Vindis, C. Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell Death Dis. 2019, 10, 119. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, Z.; Huang, L.; Kan, Q.; Wang, Z.; Wu, W.; Yao, C. PINK1 dominated mitochondria associated genes signature predicts abdominal aortic aneurysm with metabolic syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2024, 1870, 166919. [Google Scholar] [CrossRef]
- Wang, H.; Kazaleh, M.; Gioscia-Ryan, R.; Millar, J.; Temprano-Sagrera, G.; Wood, S.; Van Den Bergh, F.; Blin, M.G.; Wragg, K.M.; Luna, A.; et al. Deficiency of mitophagy mediator Parkin in aortic smooth muscle cells exacerbates abdominal aortic aneurysm. BioRxiv 2024. BioRxiv:2024.10.30.621201. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Branchetti, E.; Poggio, P.; Sainger, R.; Shang, E.; Grau, J.B.; Jackson, B.M.; Lai, E.K.; Parmacek, M.S.; Gorman, R.C.; Gorman, J.H.; et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc. Res. 2013, 100, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Irace, F.G.; Cammisotto, V.; Valenti, V.; Forte, M.; Schirone, L.; Bartimoccia, S.; Iaccarino, A.; Peruzzi, M.; Schiavon, S.; Morelli, A.; et al. Role of Oxidative Stress and Autophagy in Thoracic Aortic Aneurysms. JACC Basic. Transl. Sci. 2021, 6, 719–730. [Google Scholar] [CrossRef]
- Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int. J. Mol. Sci. 2018, 19, 2125. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-Modulated mtDNA: A Potential Target for Cardiac Aging. Oxid. Med. Cell Longev. 2020, 2020, 9423593. [Google Scholar] [CrossRef]
- Schreckenberger, Z.J.; Wenceslau, C.F.; Joe, B.; McCarthy, C.G. Mitophagy in Hypertension-Associated Premature Vascular Aging. Am. J. Hypertens. 2020, 33, 804–812. [Google Scholar] [CrossRef]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.-Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26, 101288. [Google Scholar] [CrossRef]
- Huang, T.-H.; Chang, H.-H.; Guo, Y.-R.; Chang, W.-C.; Chen, Y.-F. Vitamin B Mitigates Thoracic Aortic Dilation in Marfan Syndrome Mice by Restoring the Canonical TGF-β Pathway. Int. J. Mol. Sci. 2021, 22, 11737. [Google Scholar] [CrossRef]
- Xia, L.; Sun, C.; Zhu, H.; Zhai, M.; Zhang, L.; Jiang, L.; Hou, P.; Li, J.; Li, K.; Liu, Z.; et al. Melatonin protects against thoracic aortic aneurysm and dissection through SIRT1-dependent regulation of oxidative stress and vascular smooth muscle cell loss. J. Pineal Res. 2020, 69, e12661. [Google Scholar] [CrossRef]
- Liu, W.; Wang, B.; Wang, T.; Liu, X.; He, X.; Liu, Y.; Li, Z.; Zeng, H. Ursodeoxycholic Acid Attenuates Acute Aortic Dissection Formation in Angiotensin II-Infused Apolipoprotein E-Deficient Mice Associated with Reduced ROS and Increased Nrf2 Levels. Cell Physiol. Biochem. 2016, 38, 1391–1405. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Ramírez, J.; Silva-Platas, C.; Jerjes-Sánchez, C.; Ramos-González, M.R.; Vázquez-Garza, E.; Chapoy-Villanueva, H.; Ramírez-Rivera, A.; Zarain-Herzberg, Á.; García, N.; García-Rivas, G. Resveratrol Prevents Right Ventricle Dysfunction, Calcium Mishandling, and Energetic Failure via SIRT3 Stimulation in Pulmonary Arterial Hypertension. Oxid. Med. Cell Longev. 2021, 2021, 9912434. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Jin, X.; Li, D.; Lu, J.; Zhang, X.-N.; Yang, S.-J.; Zhao, Y.-X.; Wu, M. New insights into vascular aging: Emerging role of mitochondria function. Biomed. Pharmacother. 2022, 156, 113954. [Google Scholar] [CrossRef]
- van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef]
- Bejarano, E.; Weinberg, J.; Clark, M.; Taylor, A.; Rowan, S.; Whitcomb, E.A. Redox Regulation in Age-Related Cataracts: Roles for Glutathione, Vitamin C, and the NRF2 Signaling Pathway. Nutrients 2023, 15, 3375. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.S.; Lokhandwala, M.F.; Banday, A.A. Age-Related Mitochondrial Impairment and Renal Injury Is Ameliorated by Sulforaphane via Activation of Transcription Factor NRF2. Antioxidants 2022, 11, 156. [Google Scholar] [CrossRef]
- Ungvari, Z.; Orosz, Z.; Labinskyy, N.; Rivera, A.; Xiangmin, Z.; Smith, K.; Csiszar, A. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H37–H47. [Google Scholar] [CrossRef]
- Rice, K.M.; Preston, D.L.; Walker, E.M.; Blough, E.R. Aging influences multiple incidices of oxidative stress in the aortic media of the Fischer 344/NNiaxBrown Norway/BiNia rat. Free Radic. Res. 2006, 40, 185–197. [Google Scholar] [CrossRef]
- Wolf, A.M. MtDNA mutations and aging-not a closed case after all? Signal Transduct. Target. Ther. 2021, 6, 56. [Google Scholar] [CrossRef]
- Fedotova, E.I.; Berezhnov, A.V.; Popov, D.Y.; Shitikova, E.Y.; Vinokurov, A.Y. The Role of mtDNA Mutations in Atherosclerosis: The Influence of Mitochondrial Dysfunction on Macrophage Polarization. Int. J. Mol. Sci. 2025, 26, 1019. [Google Scholar] [CrossRef]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Pinto, J.T.; Ballabh, P.; Zhang, H.; Losonczy, G.; Pearson, K.; de Cabo, R.; Pacher, P.; Zhang, C.; et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H13–H20. [Google Scholar] [CrossRef] [PubMed]
- Conlon, N.; Ford, D. A systems-approach to NAD+ restoration. Biochem. Pharmacol. 2022, 198, 114946. [Google Scholar] [CrossRef]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.-I.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheong, J.-H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells 2020, 9, 1691. [Google Scholar] [CrossRef]
- Zhao, X.; Psarianos, P.; Ghoraie, L.S.; Yip, K.; Goldstein, D.; Gilbert, R.; Witterick, I.; Pang, H.; Hussain, A.; Lee, J.H.; et al. Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat. Metab. 2019, 1, 147–157. [Google Scholar] [CrossRef]
- Zhang, H.; Tsui, C.K.; Garcia, G.; Joe, L.K.; Wu, H.; Maruichi, A.; Fan, W.; Pandovski, S.; Yoon, P.H.; Webster, B.M.; et al. The extracellular matrix integrates mitochondrial homeostasis. Cell 2024, 187, 4289–4304.e26. [Google Scholar] [CrossRef]
- Verhagen, J.M.A.; Burger, J.; Bekkers, J.A.; den Dekker, A.T.; von der Thüsen, J.H.; Zajec, M.; Brüggenwirth, H.T.; van der Sterre, M.L.T.; van den Born, M.; Luider, T.M.; et al. Multi-Omics Profiling in Marfan Syndrome: Further Insights into the Molecular Mechanisms Involved in Aortic Disease. Int. J. Mol. Sci. 2021, 23, 438. [Google Scholar] [CrossRef]
- Latham, R.H.; Zeleznik, D.; Minshew, B.H.; Schoenknecht, F.D.; Stamm, W.E. Staphylococcus saprophyticus beta-lactamase production and disk diffusion susceptibility testing for three beta-lactam antimicrobial agents. Antimicrob. Agents Chemother. 1984, 26, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Wan, S.; Qi, J.; Hao, Y.; An, P.; Luo, Y.; Luo, J. Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown. Int. J. Mol. Sci. 2024, 25, 2662. [Google Scholar] [CrossRef]
- van der Pluijm, I.; Burger, J.; van Heijningen, P.M.; IJpma, A.; van Vliet, N.; Milanese, C.; Schoonderwoerd, K.; Sluiter, W.; Ringuette, L.-J.; Dekkers, D.H.W.; et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Muller, J.; Daugherty, A.; Norman, P. Abdominal aortic aneurysm: Pathogenesis and implications for management. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2605–2613. [Google Scholar] [CrossRef]
- Gäbel, G.; Northoff, B.H.; Balboa, A.; Becirovic-Agic, M.; Petri, M.; Busch, A.; Maegdefessel, L.; Mahlmann, A.; Ludwig, S.; Teupser, D. Parallel Murine and Human Aortic Wall Genomics Reveals Metabolic Reprogramming as Key Driver of Abdominal Aortic Aneurysm Progression. J. Am. Heart Assoc. 2021, 10, 20231. [Google Scholar] [CrossRef] [PubMed]
- Roldán-Montero, R.; Pérez-Sáez, J.M.; Cerro-Pardo, I.; Oller, J.; Martinez-Lopez, D.; Nuñez, E.; Maller, S.M.; Gutierrez-Muñoz, C.; Mendez-Barbero, N.; Escola-Gil, J.C. Galectin-1 Prevents Pathological Vascular Remodeling in Atherosclerosis and Abdominal Aortic Aneurysm. Sci. Adv. 2022, 8, 7322. [Google Scholar] [CrossRef]
- Marcos-Rios, D.; Rochano-Ortiz, A.; Méndez-Barbero, N.; Oller Pedrosa, J. Defective Mitochondrial Respiration in Hereditary Thoracic Aneurysms. Preprint 2025, 1. [Google Scholar] [CrossRef]
- Rochano-Ortiz, A.; San Sebastian-Jaraba, I.; Zamora, C.; Simó, C.; García-Cañas, V.; Martínez-Albaladejo, S.; Fernandez-Gomez, M.J.; Velho, T.R.; Ruíz-Rodríguez, M.J.; Leal-Zafra, A. Integrated Stress Response Triggered by Excessive Glycosylation Drives Thoracic Aortic Aneurysm. Biorxiv 2024. BioRxiv:2024.05.31.596791. [Google Scholar] [CrossRef]
- Rocha, R.V.; Lindsay, T.F.; Friedrich, J.O.; Shan, S.; Sinha, S.; Yanagawa, B.; Al-Omran, M.; Forbes, T.L.; Ouzounian, M. Sys-Tematic Review of Contemporary Outcomes of Endovascular and Open Thoracoabdominal Aortic Aneurysm Repair. J. Vasc. Surg. 2020, 71, 1396–1412. [Google Scholar] [CrossRef]
- van Andel, M.M.; Groenink, M.; Zwinderman, A.H.; Mulder, B.J.M.; de Waard, V. The potential beneficial effects of resveratrol on cardiovascular complications in marfan syndrome patients–insights from rodent-based animal studies. Int. J. Mol. Sci. 2019, 20, 1122. [Google Scholar] [CrossRef]
- Sandor, G.G.S.; Alghamdi, M.H.; Raffin, L.A.; Potts, M.T.; Williams, L.D.; Potts, J.E.; Kiess, M.; Breemen, C. A Randomized, Double Blind Pilot Study to Assess the Effects of Losartan vs. Atenolol on the Biophysical Properties of the Aorta in Patients with Marfan and Loeys–Dietz Syndromes. Int. J. Cardiol. 2015, 179, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, J.H.; Matsumura, J.S. Pharmacologic Management of Aneurysms. Circ. Res. 2019, 124, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Silverman, D.I.; Burton, K.J.; Gray, J.; Bosner, M.S.; Kouchoukos, N.T.; Roman, M.J.; Boxer, M.; Devereux, R.B.; Tsipouras, P. Life Expectancy in the Marfan Syndrome. Am. J. Cardiol. 1995, 75, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Salim, M.A.; Alpert, B.S.; Ward, J.C.; Pyeritz, R.E. Effect of Beta-Adrenergic Blockade on Aortic Root Rate of Dilation in the Marfan Syndrome. Am. J. Cardiol. 1994, 74, 629–633. [Google Scholar] [CrossRef]
- Groenink, M.; Hartog, A.W.; Franken, R.; Radonic, T.; Waard, V.; Timmermans, J.; Scholte, A.J.; Berg, M.P.; Spijkerboer, A.M.; Marquering, H.A. Losartan Reduces Aortic Dilatation Rate in Adults with Marfan Syndrome: A Ran-Domized Controlled Trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Selamet Tierney, E.S.; Levine, J.C.; Atz, A.M. Atenolol versus Losartan in Children and Young Adults with Marfan’s Syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef]
- Forteza, A.; Evangelista, A.; Sánchez, V.; Teixidó-Turà, G.; Sanz, P.; Gutiérrez, L.; Gracia, T.; Centeno, J.; Rodríguez-Palomares, J.; Rufilanchas, J.J. Efficacy of Losartan vs. Atenolol for the Prevention of Aortic Dilation in Marfan Syndrome: A Ran-Domized Clinical Trial. Eur. Heart J. 2016, 37, 978–985. [Google Scholar] [CrossRef]
- Jun, C.; Fang, B. Current Progress of Fluoroquinolones-Increased Risk of Aortic Aneurysm and Dissection. BMC Cardiovasc. Disord. 2021, 21, 1–10. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.-L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612. [Google Scholar] [CrossRef]
- Daneman, N.; Lu, H.; Redelmeier, D.A. Fluoroquinolones and Collagen Associated Severe Adverse Events: A Longitudinal Cohort Study. BMJ Open 2015, 5, e010077. [Google Scholar] [CrossRef]
- Maumus-Robert, S.; Debette, S.; Bérard, X.; Mansiaux, Y.; Tubert-Bitter, P.; Pariente, A. Risk of Intracranial Aneurysm and Dissection and Fluoroquinolone Use. Stroke 2020, 51, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Luger, A.-L.; Sauer, B.; Lorenz, N.I.; Engel, A.L.; Braun, Y.; Voss, M.; Harter, P.N.; Steinbach, J.P.; Ronellenfitsch, M.W. Doxycycline Impairs Mitochondrial Function and Protects Human Glioma Cells from Hypoxia-Induced Cell Death: Impli-Cations of Using Tet-Inducible Systems. Int. J. Mol. Sci. 2018, 19, 1504. [Google Scholar] [CrossRef] [PubMed]
- Chatzispyrou, I.A.; Held, N.M.; Mouchiroud, L.; Auwerx, J.; Houtkooper, R.H. Tetracycline Antibiotics Impair Mitochondrial Function and Its Experimental Use Confounds Research. Cancer Res. 2015, 75, 4446–4449. [Google Scholar] [CrossRef]
- Meijer, C.A.; Stijnen, T.; Wasser, M.N.J.M.; Hamming, J.F.; Bockel, J.H.; Lindeman, J.H.N.; Group, P.A.S.T.S. Doxycycline for stabilization of abdominal aortic aneurysms: A randomized trial. Ann. Intern. Med. 2013, 159, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Gouveia EMelo, R.; Rodrigues, M.; Caldeira, D.; Alves, M.; Fernandes EFernandes, R.; Mendes Pedro, L. Doxycycline Is Not Effective in Reducing Abdominal Aortic Aneurysm Growth: A Mini Systematic Review and Meta-Analysis of Randomised Controlled Trials. Eur. J. Vasc. Endovasc. Surg. 2021, 61, 863–864. [Google Scholar] [CrossRef]
- Stechmiller, J.; Cowan, L.; Schultz, G. The Role of Doxycycline as a Matrix Metalloproteinase Inhibitor for the Treatment of Chronic Wounds. Biol. Res. Nurs. 2010, 11, 336–344. [Google Scholar] [CrossRef]
- Tan, Q.; Yan, X.; Song, L.; Yi, H.; Li, P.; Sun, G.; Yu, D.; Li, L.; Zeng, Z.; Guo, Z. Induction of Mitochondrial Dysfunction and Oxidative Damage by Antibiotic Drug Doxycycline Enhances the Responsiveness of Glioblastoma to Chemotherapy. Med. Sci. Monit. 2017, 23, 4117–4125. [Google Scholar] [CrossRef]
- Wegdam-Blans, M.C.A.; Vainas, T.; Sambeek, M.R.; Cuypers, P.W.; Tjhie, H.T.J.; Straten, A.H.M.; Teijink, J.A. Vascular complications of Q-fever infections. Eur. J. Vasc. Endovasc. Surg. 2011, 42, 384–392. [Google Scholar] [CrossRef]
- Hibender, S.; Franken, R.; Roomen, C.; Ter Braake, A.; Made, I.; Schermer, E.E.; Gunst, Q.; Den Hoff, M.J.; Lutgens, E.; Pinto, Y.M. Resveratrol Inhibits Aortic Root Dilatation in the Fbn1 C1039G/+ Marfan Mouse Model. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1618–1626. [Google Scholar] [CrossRef]
- Mieremet, A.; Stoel, M.; Li, S.; Coskun, E.; Krimpen, T.; Huveneers, S.; Waard, V. Endothelial Dysfunction in Marfan Syndrome Mice Is Restored by Resveratrol. Sci. Rep. 2022, 12, 22504. [Google Scholar] [CrossRef]
- Andel, M.M.; Bosshardt, D.; Schrauben, E.M.; Merton, R.; Kimmenade, R.R.L.; Scholte, A.; Dickinson, M.G.; Rob-bers-Visser, D.; Zwinderman, A.H.; Mulder, B. Effects of Resveratrol on Aortic Growth in Patients with Marfan Syndrome: A Single-Arm Open-Label Multicentre Trial. Heart 2024, 111, 11–17. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Prolla, T.A.; Denu, J.M. NAD+ Deficiency in Age-Related Mitochondrial Dysfunction. Cell Metab. 2014, 19, 178–180. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide Riboside Is Uniquely and Orally Bioavailable in Mice and Humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Nong, Z.; Yin, H.; O’Neil, C.; Fox, S.; Balint, B.; Guo, L.; Leo, O.; Chu, M.W.A.; Gros, R. Nicotinamide Phosphoribosyltransferase in Smooth Muscle Cells Maintains Genome Integrity, Resists Aortic Medial Degeneration, and Is Suppressed in Human Thoracic Aortic Aneurysm Disease. Circ. Res. 2017, 120, 1889–1902. [Google Scholar] [CrossRef]
- Nollet, E.E.; Duursma, I.; Rozenbaum, A.; Eggelbusch, M.; Wüst, R.C.I.; Schoonvelde, S.A.C.; Michels, M.; Jansen, M.; Wel, N.N.; Bedi, K.C. Mitochondrial Dysfunction in Human Hypertrophic Cardiomyopathy Is Linked to Cardiomyocyte Architecture Disruption and Corrected by Improving NADH-Driven Mitochondrial Respiration. Eur. Heart J. 2023, 44, 1170–1185. [Google Scholar] [CrossRef]
- Seeburun, S.; Wu, S.; Hemani, D.; Pham, L.; Ju, D.; Xie, Y.; Kata, P.; Li, L. Insights into Elastic Fiber Fragmentation: Mechanisms and Treatment of Aortic Aneurysm in Marfan Syndrome. Vasc. Pharmacol. 2023, 153, 107215. [Google Scholar] [CrossRef] [PubMed]
- Muiño-Mosquera, L.; Backer, J. Angiotensin-II receptor blockade in Marfan syndrome. Lancet 2019, 394, 2206–2207. [Google Scholar] [CrossRef]
- Stefens, S.J.M.; Vliet, N.; IJpma, A.; Burger, J.; Li, Y.; Heijningen, P.M.; Lindeman, J.H.N.; Majoor-Krakauer, D.; Ver-hagen, H.J.M.; Kanaar, R. Increased Vascular Smooth Muscle Cell Senescence in Aneurysmal Fibulin-4 Mutant Mice. Npj Aging 2024, 10, 31. [Google Scholar] [CrossRef]
- Malashicheva, A.; Kostina, D.; Kostina, A.; Irtyuga, O.; Voronkina, I.; Smagina, L.; Ignatieva, E.; Gavriliuk, N.; Uspensky, V.; Moiseeva, O. Phenotypic and Functional Changes of Endothelial and Smooth Muscle Cells in Thoracic Aortic Aneu-Rysms. Int. J. Vasc. Med. 2016, 3107879. [Google Scholar] [CrossRef]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian Target of Rapamycin Complex 2 (MTORC2. In Coordinates Pulmonary Artery Smooth Muscle Cell Metabolism, Proliferation, and Survival in Pulmonary Arterial Hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, H.; Li, Y.; Luo, Y.; He, Y.; Shui, X.; Lei, W. Glycolysis Modulation: New Therapeutic Strategies to Improve Pul-Monary Hypertension (Review). Int. J. Mol. Med. 2024, 54, 115. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Pyruvate Kinase and Warburg Metabolism in Pulmonary Arterial Hypertension. Circulation 2017, 136, 2486–2490. [Google Scholar] [CrossRef] [PubMed]
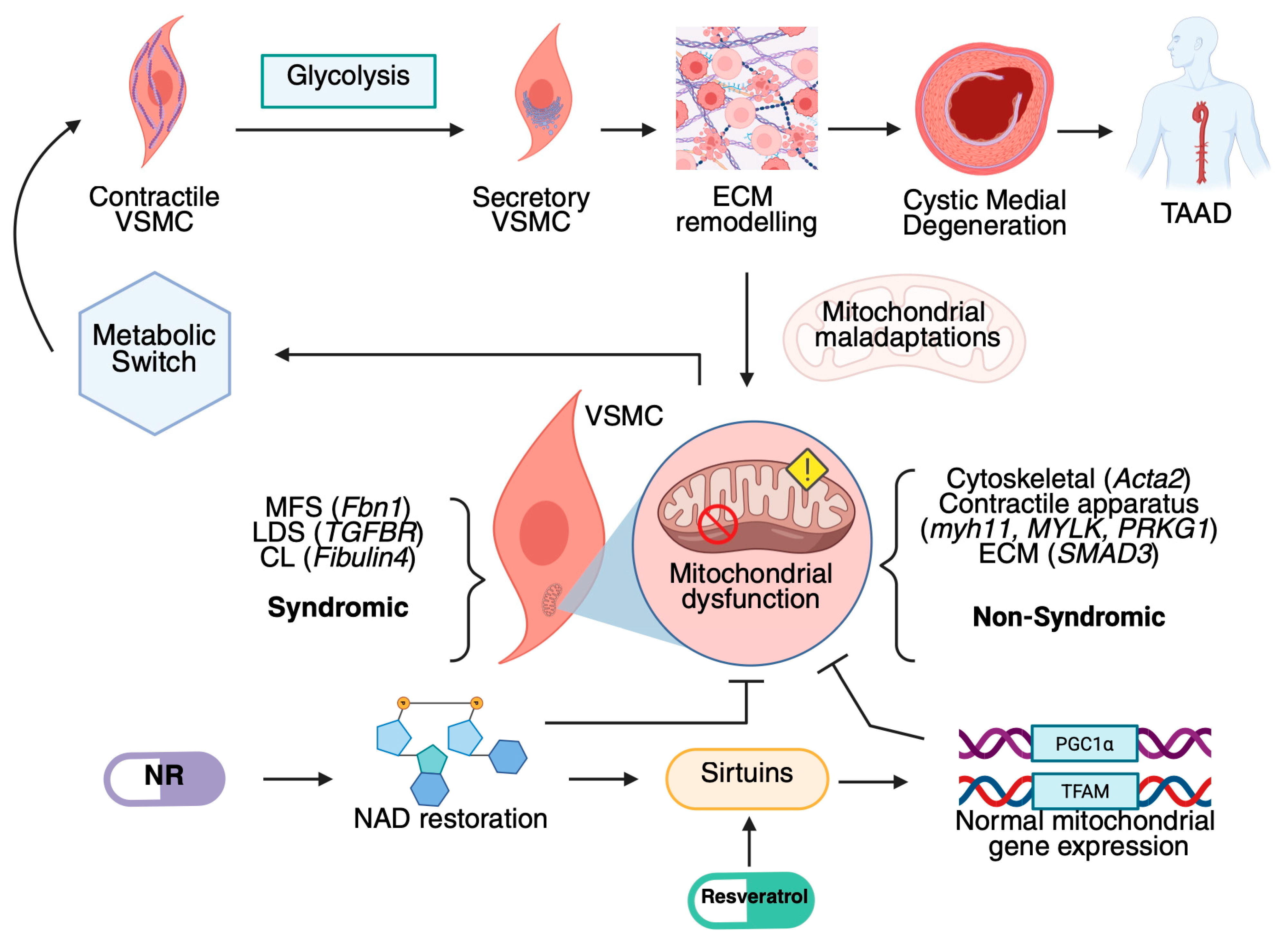
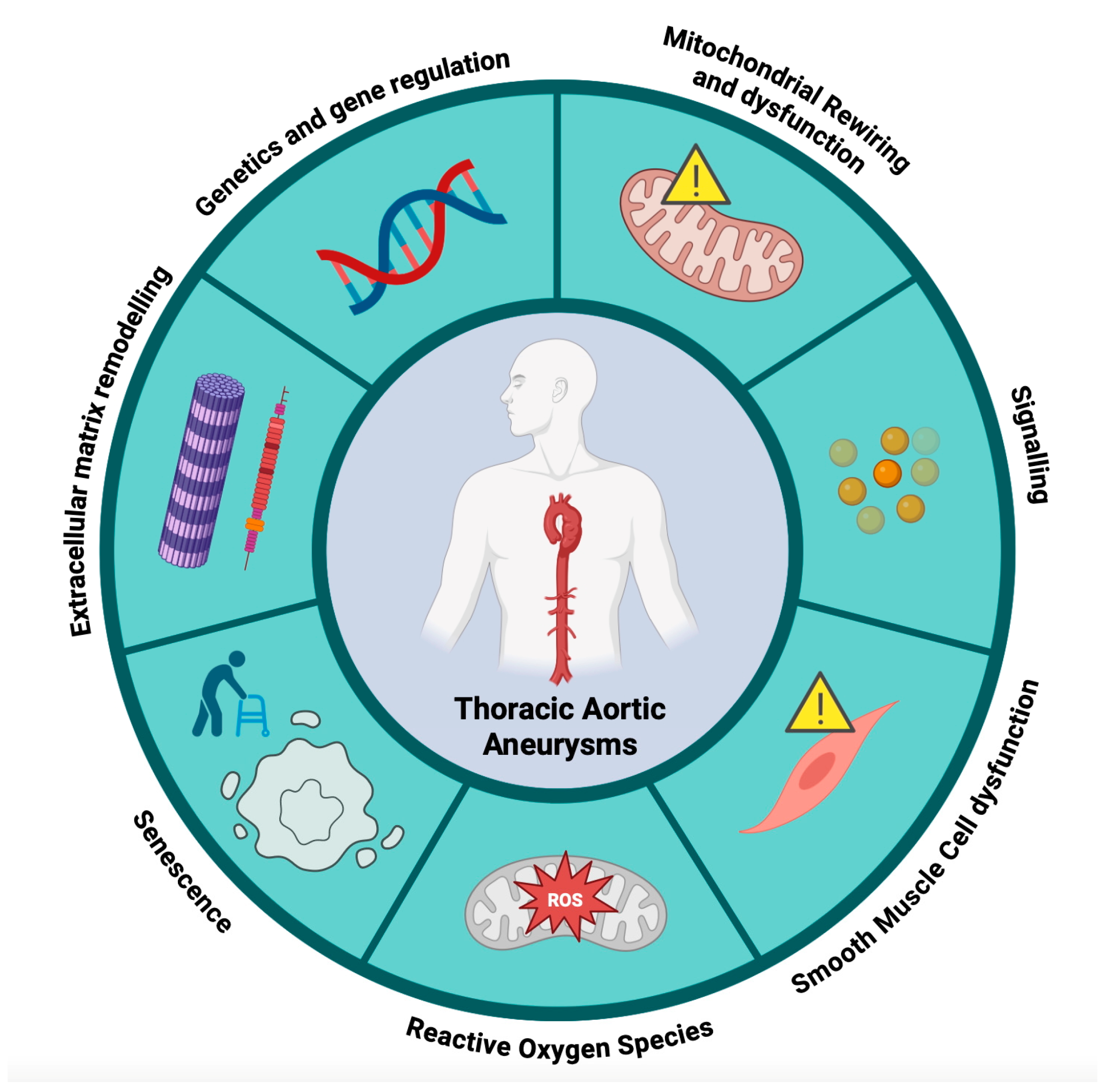

{kind=link}
{kind=link}
| Treatment | Target | Mitocondrial Defects Targeted | Impact on VSMCs | Preclinical Model | Human Patients/Sample | Reference |
|---|---|---|---|---|---|---|
| NR (NAD+ precursor) | NADH/NAD Imbalance SIrt1/Pgc1a/Tfam axis | Decrease Oxphos Decrease mitochondrial complexes Mitochondrial dysfunction Decrease Tfam/mtDNA Decrease Pgc1a | Improve Oxphos Decrease ECM synthesis Improve contractile phenotype Improve transcriptomic profile Decrease aortic diameter Decrease elastin breaks | Fbn1C1041G/+ | Marfan patient’s fibroblasts Aortic samples from Marfan patients | [10] |
| NR (NAD+ precursor) | Improve Oxphos Decrease ECM synthesis Improve contractile phenotype | VSMCs lentiviral-transduced with ACTA2R178H VSMCs lentiviral-transduced with TGFBR2G357W | [108] | |||
| Coenzyme Q10 | Mitochondrial Oxphos | Decrease Oxphos Decrease Tfam/mtDNA Decrease mitochondrial mass | Improve Oxphos Decrease ECM synthesis Increase contractile phenotype | Fbn1 silencing in human VSMCs | [103] | |
| Forskolin | PGC1A | Mitochondrial dysfuncion Decrease Oxphos Decrease Pgc1a Increase ROS | Improve Oxphos | Hypomorphic Fibulin-4 mice (Fibulin-4R/R) Fibulin-4 VSMCsConditional deficient mice | Marfan patient’s fibroblasts LDS patient’s fibroblasts (TRGFR2 mutant, SMAD3 mutant) | [104] |
| Resveratrol SRT1720 | Sirtuins | Sirtuin activity decrease | Decrease aortic growth Decrease elastin breaks | Fbn1C1041G/+ | [129] | |
| Sirt1 | Aging, senescence | Increase Sirt1 nuclear staining Decrease senescence Decrease medial area Decrease apoptosis | ||||
| Resveratrol | Sirtuins | No impact: no significant differences in aortic diameter (1 year) | 60 Adults aged 18–50 years with MFS (1 year treatment) | [131] | ||
| Amlexanox TBK1 Inhibitor | CGAS/STING pathway | Presence of mtDNA | Inflammation Aortic degeneration Aortic dissection Apoptosis Decrease ECM synthesis Increase contractile phenotype | C57/BL6J HFD + AngII STING-deficient (STINGgt/gt) mice | Non-genetic TAA human aortic samples | [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcos-Ríos, D.; Rochano-Ortiz, A.; San Sebastián-Jaraba, I.; Fernández-Gómez, M.J.; Méndez-Barbero, N.; Oller, J. Mitochondrial Dysfunction: A New Hallmark in Hereditable Thoracic Aortic Aneurysm Development. Cells 2025, 14, 618. https://doi.org/10.3390/cells14080618
Marcos-Ríos D, Rochano-Ortiz A, San Sebastián-Jaraba I, Fernández-Gómez MJ, Méndez-Barbero N, Oller J. Mitochondrial Dysfunction: A New Hallmark in Hereditable Thoracic Aortic Aneurysm Development. Cells. 2025; 14(8):618. https://doi.org/10.3390/cells14080618
Chicago/Turabian StyleMarcos-Ríos, Daniel, Antonio Rochano-Ortiz, Irene San Sebastián-Jaraba, María José Fernández-Gómez, Nerea Méndez-Barbero, and Jorge Oller. 2025. "Mitochondrial Dysfunction: A New Hallmark in Hereditable Thoracic Aortic Aneurysm Development" Cells 14, no. 8: 618. https://doi.org/10.3390/cells14080618
APA StyleMarcos-Ríos, D., Rochano-Ortiz, A., San Sebastián-Jaraba, I., Fernández-Gómez, M. J., Méndez-Barbero, N., & Oller, J. (2025). Mitochondrial Dysfunction: A New Hallmark in Hereditable Thoracic Aortic Aneurysm Development. Cells, 14(8), 618. https://doi.org/10.3390/cells14080618