Inferring Novel Autophagy Regulators Based on Transcription Factors and Non-Coding RNAs Coordinated Regulatory Network

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of a Comprehensive TNCR Network
2.2. Collection of Known Autophagy Regulators
2.3. Prioritization of Novel Autophagy Regulators with the RWR Method
2.4. Functional Analysis for Predicted Autophagy Regulators
3. Results
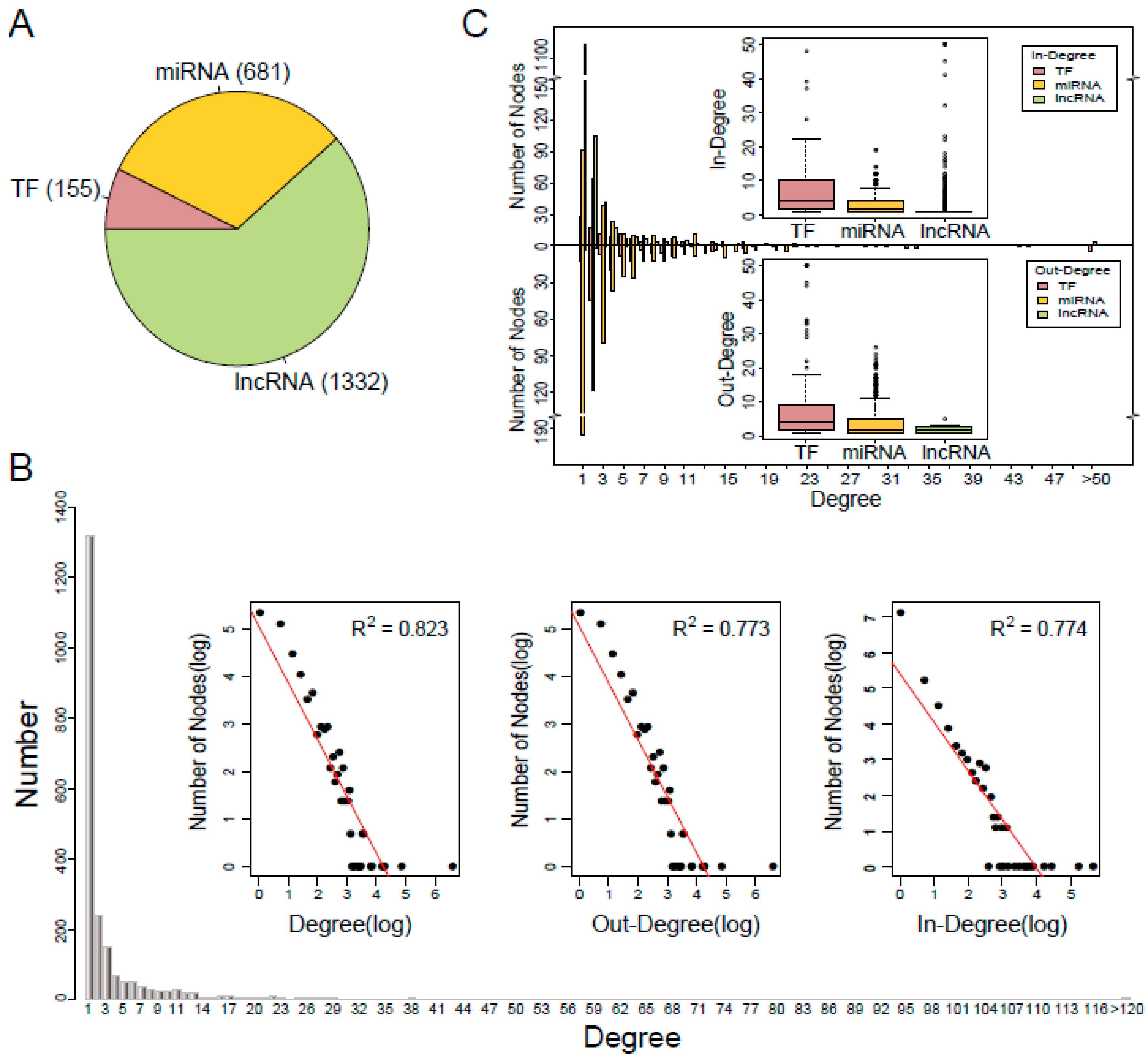
3.1. Characteristics of the TNCR Network
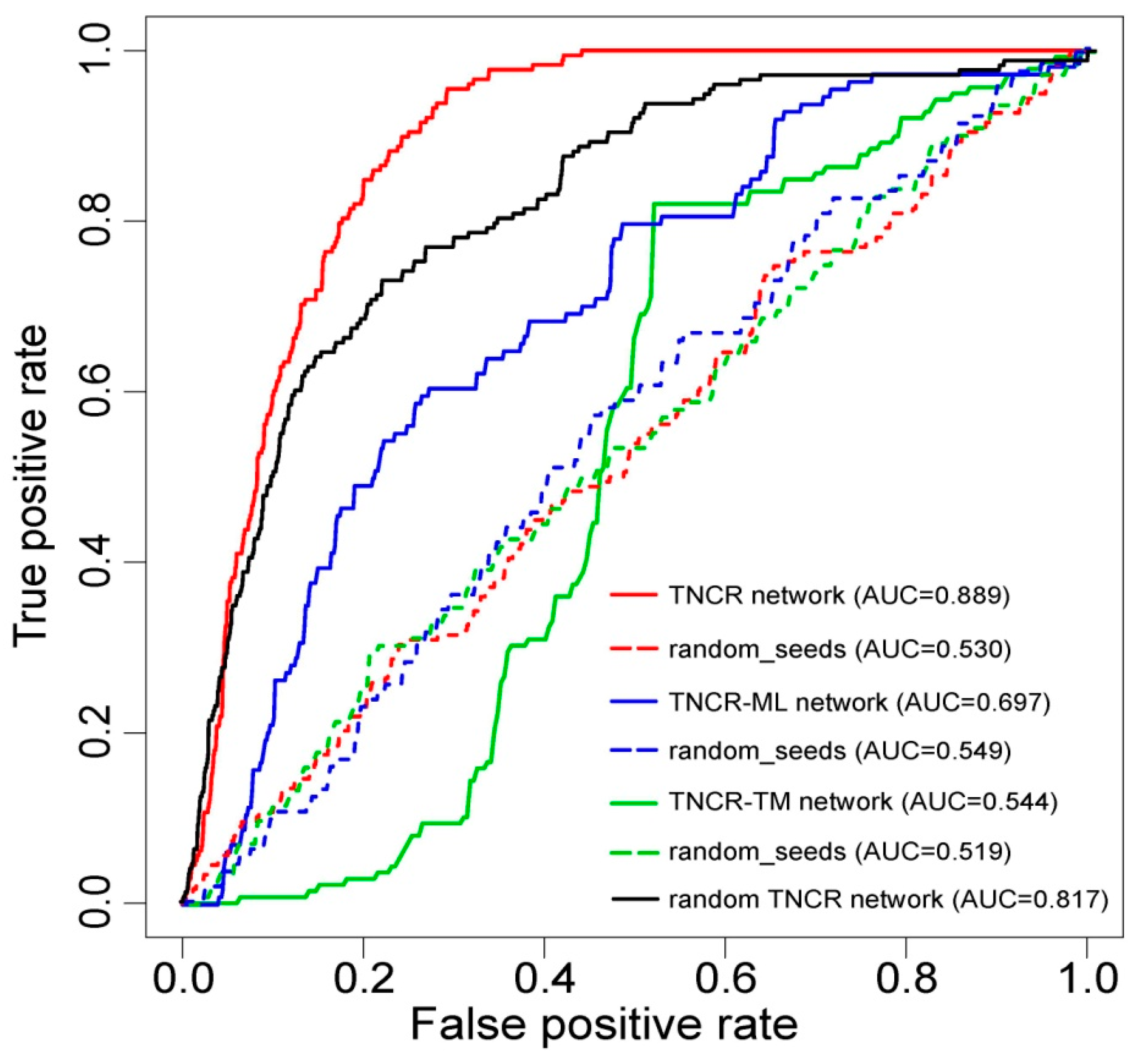
3.2. Performance Evaluation of the Proposed Method
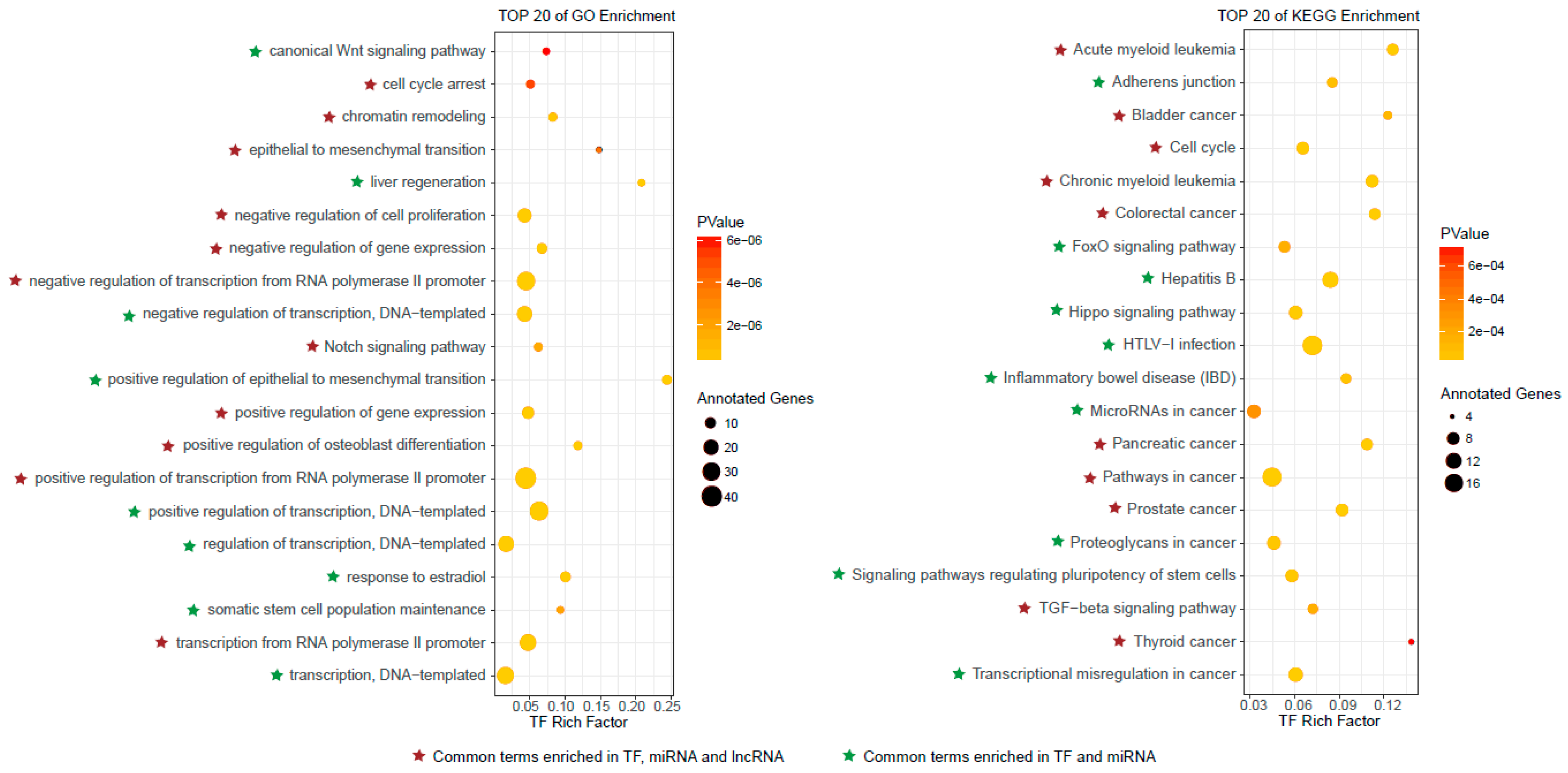
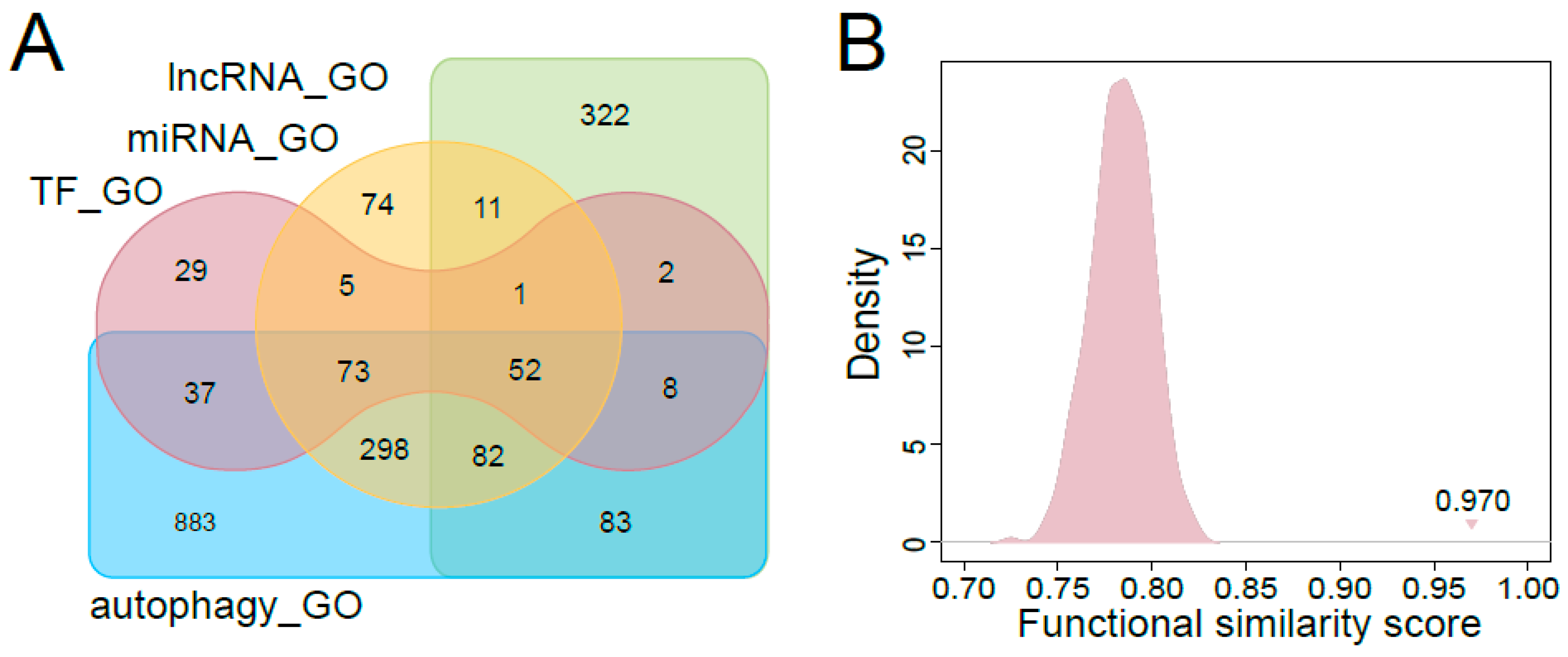
3.3. Functional Characteristics of Predicted Autophagy Regulators
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mizushima:, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-dependent production of secreted factors facilitates oncogenic ras-driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Homma, K.; Suzuki, K.; Sugawara, H. The autophagy database: An all-inclusive information resource on autophagy that provides nourishment for research. Nucleic Acids Res. 2011, 39, D986–D990. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huang, Y.; Kang, J.; Li, K.; Bi, X.; Zhang, T.; Jin, N.; Hu, Y.; Tan, P.; Zhang, L.; et al. Ncrdeathdb: A comprehensive bioinformatics resource for deciphering network organization of the ncrna-mediated cell death system. Autophagy 2015, 11, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Petrini, M. Temsirolimus in the treatment of relapsed and/or refractory mantle cell lymphoma. Cancer Manag. Res. 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Turei, D.; Foldvari-Nagy, L.; Fazekas, D.; Modos, D.; Kubisch, J.; Kadlecsik, T.; Demeter, A.; Lenti, K.; Csermely, P.; Vellai, T.; et al. Autophagy regulatory network-a systems-level bioinformatics resource for studying the mechanism and regulation of autophagy. Autophagy 2015, 11, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Zhang, Y.; Meng, F.; Lian, B.; Chen, X.; Yu, X.; Dai, E.; Wang, S.; Liu, X.; Li, X.; et al. Identification of active transcription factor and mirna regulatory pathways in Alzheimer’s disease. Bioinformatics 2013, 29, 2596–2602. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Mitra, R.; Lin, C.C.; Wang, Q.; Cheng, F.; Zhao, Z. Systematic dissection of dysregulated transcription factor-mirna feed-forward loops across tumor types. Brief. Bioinform. 2016, 17, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Wang, J.; Yang, F.; Zhou, X.; Song, Q.; Wang, S.; Yu, X.; Liu, D.; Yang, Q.; Dai, H.; et al. Accurate prediction and elucidation of drug resistance based on the robust and reproducible chemoresponse communities. Int. J. Cancer 2018, 142, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wu, H.; Lei, R.; Chong, R.A.; Wei, Y.; Lu, X.; Tagkopoulos, I.; Kung, S.Y.; Yang, Q.; Hu, G.; et al. Transcriptional network analysis identifies bach1 as a master regulator of breast cancer bone metastasis. J. Biol. Chem. 2012, 287, 33533–33544. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Lian, B.; Liu, X.; Zhang, Y.; Dai, E.; Yu, X.; Meng, F.; Jiang, W.; Li, X. Tmrec: A database of transcription factor and mirna regulatory cascades in human diseases. PloS. ONE 2015, 10, e0125222. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.A.; Laprade, L.; Winston, F. Intergenic transcription is required to repress the saccharomyces cerevisiae ser3 gene. Nature 2004, 429, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense rna controls uchl1 translation through an embedded sineb2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, M.; Qiu, C.; Cui, Q. Transmir: A transcription factor-microrna regulation database. Nucleic Acids Res. 2010, 38, D119–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Li, J.H.; Jiang, S.; Zhou, H.; Qu, L.H. Chipbase: A database for decoding the transcriptional regulation of long non-coding rna and microrna genes from chip-seq data. Nucleic Acids Res. 2013, 41, D177–D187. [Google Scholar] [CrossRef] [PubMed]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. Transfac and its module transcompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. Mirecords: An integrated resource for microrna-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. Mirtarbase update 2018: A resource for experimentally validated microrna-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Vlachos, I.S.; Karagkouni, D.; Georgakilas, G.; Kanellos, I.; Vergoulis, T.; Zagganas, K.; Tsanakas, P.; Floros, E.; Dalamagas, T.; et al. Diana-lncbase v2: Indexing microrna targets on non-coding transcripts. Nucleic Acids Res. 2016, 44, D231–D238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shen, Y.; Khan, M.R.; Li, A. Lncreg: A reference resource for lncrna-associated regulatory networks. Database (Oxford) 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, J.; Wu, X.; Ma, R.; Zhang, T.; Jin, S.; Han, Z.; Tan, R.; Peng, J.; Liu, G.; et al. Lncrna2target: A database for differentially expressed genes after lncrna knockdown or overexpression. Nucleic Acids Res. 2015, 43, D193–D196. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Patra, J.C. Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics 2010, 26, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, H.; Yang, F.; Yang, L.; Lv, Y.; Wang, S.; Dai, E.; Sun, D.; Jiang, W. Large-scale identification of adverse drug reaction-related proteins through a random walk model. Sci. Rep. 2016, 6, 36325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Li, F.; Qin, Y.; Bo, X.; Wu, Y.; Wang, S. Gosemsim: An r package for measuring semantic similarity among go terms and gene products. Bioinformatics 2010, 26, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating myc proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zu, Y.; Fu, X.; Deng, Y. Knockdown of lncrna-xist enhances the chemosensitivity of nsclc cells via suppression of autophagy. Oncol. Rep. 2017, 38, 3347–3354. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548–563. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, T.S.; Tesselaar, M.H.; Morreau, H.; Corssmit, E.P.; Willemsen, B.K.; Kusters, B.; van Engen-van Grunsven, A.C.; Smit, J.W.; Netea-Maier, R.T. Autophagy activity is associated with membranous sodium iodide symporter expression and clinical response to radioiodine therapy in non-medullary thyroid cancer. Autophagy 2016, 12, 1195–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Autophagy and its effects: Making sense of double-edged swords. PLoS biology 2014, 12, e1001967. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, F.; Mendell, J.T. Functional classification and experimental dissection of long noncoding rnas. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of cerna crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.W.; Shao, D.D.; Zhang, S.Y.; Wang, Y.B. Prioritization of candidate disease genes by enlarging the seed set and fusing information of the network topology and gene expression. Mol. Biosyst. 2014, 10, 1400–1408. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Wang, W.; Meng, Q.; Zhou, S.; Liu, H.; Ma, X.; Zhou, X.; Liu, H.; Chen, X.; Jiang, W. Inferring Novel Autophagy Regulators Based on Transcription Factors and Non-Coding RNAs Coordinated Regulatory Network. Cells 2018, 7, 194. https://doi.org/10.3390/cells7110194
Wang S, Wang W, Meng Q, Zhou S, Liu H, Ma X, Zhou X, Liu H, Chen X, Jiang W. Inferring Novel Autophagy Regulators Based on Transcription Factors and Non-Coding RNAs Coordinated Regulatory Network. Cells. 2018; 7(11):194. https://doi.org/10.3390/cells7110194
Chicago/Turabian StyleWang, Shuyuan, Wencan Wang, Qianqian Meng, Shunheng Zhou, Haizhou Liu, Xueyan Ma, Xu Zhou, Hui Liu, Xiaowen Chen, and Wei Jiang. 2018. "Inferring Novel Autophagy Regulators Based on Transcription Factors and Non-Coding RNAs Coordinated Regulatory Network" Cells 7, no. 11: 194. https://doi.org/10.3390/cells7110194
APA StyleWang, S., Wang, W., Meng, Q., Zhou, S., Liu, H., Ma, X., Zhou, X., Liu, H., Chen, X., & Jiang, W. (2018). Inferring Novel Autophagy Regulators Based on Transcription Factors and Non-Coding RNAs Coordinated Regulatory Network. Cells, 7(11), 194. https://doi.org/10.3390/cells7110194